
Abstract
There has been a resurgence in gene therapy efforts that is partly fueled by the identification and understanding of new gene delivery vectors. Adeno-associated virus (AAV) is a non-enveloped virus that can be engineered to deliver DNA to target cells, and has attracted a significant amount of attention in the field, especially in clinical-stage experimental therapeutic strategies. The ability to generate recombinant AAV particles lacking any viral genes and containing DNA sequences of interest for various therapeutic applications has thus far proven to be one of the safest strategies for gene therapies. This review will provide an overview of some important factors to consider in the use of AAV as a vector for gene therapy.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Adeno-associated virus (AAV) is a versatile viral vector technology that can be engineered for very specific functionality in gene therapy applications. |
To date, AAV has been shown to be safe and effective in preclinical and clinical settings. |
AAV can be used in a wide range of clinical applications in multiple diseases due its unique biological and biophysical properties. |
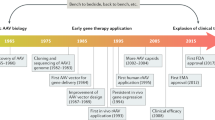
1 Introduction
The discovery of DNA as the biomolecule of genetic inheritance and disease opened up the prospect of therapies in which mutant, damaged genes could be altered for the improvement of the human condition. The recent ability to rapidly and affordably perform human genetics on hundreds of thousands of people, and to sequence complete genomes, has resulted in an explosion of nucleic acid sequence information and has allowed us to identify the gene, or genes, that might be driving a particular disease state. If the mutant gene(s) could be ‘fixed’, or if the expression of overactive/underactive genes could be normalized, the disease could be treated at the molecular level, and, in best case scenarios, potentially be cured. This concept seems particularly true for the treatment of monogenic diseases, i.e. those diseases caused by mutations in a single gene. This seemingly simple premise has been the goal of gene therapy for over 40 years.
Until relatively recently, that simple goal was very elusive as technologies to safely deliver nucleic acid cargo inside cells have lagged behind those used to identify disease-associated genes. One of the earliest approaches investigated was the use of viruses, naturally occurring biological agents that have evolved to do one thing, i.e. deliver their nucleic acid (DNA or RNA) into a host cell for replication. There are numerous viral agents that could be selected for this purpose, each with some unique attributes that would make them more or less suitable for the task, depending on the desired profile [1]. However, the undesired properties of some viral vectors, including their immunogenic profiles or their propensity to cause cancer have resulted in serious clinical adverse events and, until recently, limited their current use in the clinic to certain applications, for example, vaccines and oncolytic strategies [2]. More artificial delivery technologies, such as nanoparticles, i.e. chemical formulations meant to encapsulate the nucleic acid, protect it from degradation, and get through the cell membrane, have also achieved some levels of preclinical and clinical success. Not surprisingly, they also have encountered some unwanted safety signals that need to be better understood and controlled [3].
Adeno-associated virus (AAV) is one of the most actively investigated gene therapy vehicles. It was initially discovered as a contaminant of adenovirus preparations [4, 5], hence its name. Simply put, AAV is a protein shell surrounding and protecting a small, single-stranded DNA genome of approximately 4.8 kilobases (kb). AAV belongs to the parvovirus family and is dependent on co-infection with other viruses, mainly adenoviruses, in order to replicate. Initially distinguished serologically, molecular cloning of AAV genes has identified hundreds of unique AAV strains in numerous species. Its single-stranded genome contains three genes, Rep (Replication), Cap (Capsid), and aap (Assembly). These three genes give rise to at least nine gene products through the use of three promoters, alternative translation start sites, and differential splicing. These coding sequences are flanked by inverted terminal repeats (ITRs) that are required for genome replication and packaging. The Rep gene encodes four proteins (Rep78, Rep68, Rep52, and Rep40), which are required for viral genome replication and packaging, while Cap expression gives rise to the viral capsid proteins (VP; VP1/VP2/VP3), which form the outer capsid shell that protects the viral genome, as well as being actively involved in cell binding and internalization [6]. It is estimated that the viral coat is comprised of 60 proteins arranged into an icosahedral structure with the capsid proteins in a molar ratio of 1:1:10 (VP1:VP2:VP3) [6]. The aap gene encodes the assembly-activating protein (AAP) in an alternate reading frame overlapping the cap gene. This nuclear protein is thought to provide a scaffolding function for capsid assembly [7]. While AAP is essential for nucleolar localization of VP proteins and capsid assembly in AAV2, the subnuclear localization of AAP varies among 11 other serotypes recently examined, and is nonessential in AAV4, AAV5, and AAV11 [8].
Although there is much more to the biology of wild-type AAV, much of which is not fully understood, this is not the form that is used to generate gene therapeutics. Recombinant AAV (rAAV), which lacks viral DNA, is essentially a protein-based nanoparticle engineered to traverse the cell membrane, where it can ultimately traffic and deliver its DNA cargo into the nucleus of a cell. In the absence of Rep proteins, ITR-flanked transgenes encoded within rAAV can form circular concatemers that persist as episomes in the nucleus of transduced cells [9]. Because recombinant episomal DNA does not integrate into host genomes, it will eventually be diluted over time as the cell undergoes repeated rounds of replication. This will eventually result in the loss of the transgene and transgene expression, with the rate of transgene loss dependent on the turnover rate of the transduced cell. These characteristics make rAAV ideal for certain gene therapy applications. Following is an overview of the practical considerations for the use of rAAV as a gene therapy agent, based on our current understanding of viral biology and the state of the platform. The final section provides an overview for how rAAV has been incorporated into clinical-stage gene therapy candidates, as well as the lessons learned from those studies that can be applied to future therapeutic opportunities.
2 Adeno-Associated Virus (AAV) Vector Designs
The main point of consideration in the rational design of an rAAV vector is the packaging size of the expression cassette that will be placed between the two ITRs. As a starting point, it is generally accepted that anything under 5 kb (including the viral ITRs) is sufficient [10]. Attempts at generating rAAV vectors exceeding packaging cassettes in excess of 5 kb results in a considerable reduction in viral production yields or transgene recombination (truncations) [11]. As a result, large coding sequences, such as full-length dystrophin, will not be effectively packaged in AAV vectors. Therefore, the use of dual, overlapping vector strategies (reviewed by Chamberlain et al.) [12], should be considered in these cases. An additional consideration relates to the biology of the single-stranded AAV-delivered transgenes. After delivery to the nucleus, the single-stranded transgene needs to be converted into a double-stranded transgene, which is considered a limiting step in the onset of transgene expression [13]. An alternative is to use self-complementary AAV, in which the single-stranded packaged genome complements itself to form a double-stranded genome in the nucleus, thereby bypassing that process [13, 14]. Although the onset of expression is more rapid, the packaging capacity of the vector will be reduced to approximately 3.3 kb [13, 14].
AAV2 was one of the first AAV serotypes identified and characterized, including the sequence of its genome. As a result of the detailed understanding of AAV2 biology from this early work, most rAAV vectors generated today utilize the AAV2 ITRs in their vector designs. The sequences placed between the ITRs will typically include a mammalian promoter, gene of interest, and a terminator (Fig. 1). In many cases, strong, constitutively active promoters are desired for high-level expression of the gene of interest. Commonly used promoters of this type include the CMV (cytomegalovirus) promoter/enhancer, EF1a (elongation factor 1a), SV40 (simian virus 40), chicken β-actin and CAG (CMV, chicken β-actin, rabbit β-globin) [15]. All of these promoters provide constitutively active, high-level gene expression in most cell types. Some of these promoters are subject to silencing in certain cell types, therefore this consideration needs to be evaluated for each application [16]. For example, the CMV promoter has been shown to be silenced in the central nervous system (CNS) [16]. It has been observed that the chicken β-actin and CAG promoters are the strongest of these constitutive promoters in most cell types; however, the CAG promoter is significantly larger than the others (1.7 kb vs. 800 bp for CMV), a consideration to take into account when packaging larger gene inserts [15].

Schematic representation of the basic components of a gene insert packaged inside recombinant AAV gene transfer vector. AAV adeno-associated virus, ITR inverted terminal repeat
Although many therapeutic strategies involve systemic delivery, it is often desirable to have cell- or tissue-specific expression. Likewise, for local delivery strategies, undesired systemic leakage of the AAV particle can result in transduction and expression of the gene of interest in unwanted cells or tissues. The muscle creatine kinase and desmin promoters have been used to achieve high levels of expression, specifically in skeletal muscle, whereas the α-myosin heavy chain promoter can significantly restrict expression to cardiac muscle [15, 17]. Likewise, the neuron-specific enolase promoter can attain high levels of neuron-specific expression [18, 19]. Often is the case, systemic delivery of AAV results in a significant accumulation in the liver. While this may be desirable for some applications, AAV can also efficiently transduce other cells and tissues types. Thus, in order to restrict expression to only the liver, a common approach is to use the α1-antitrypsin promoter [20, 21]. Finally, there are now technologies that have the ability to generate novel, tissue-specific promoters, based on DNA regulatory element libraries [22].
Over the course of the past 10–15 years, much work has been done to understand the correlation between codon usage and protein expression levels. Although bacterial expression systems seem to be most affected by codon choice, there are now many examples of the effects of codon engineering on mammalian expression [23]. Many groups have developed their own codon optimization strategies, and there are many free services that can similarly provide support for codon choice. Codon usage has also been shown to contribute to tissue-specific expression, and play a role in the innate immune response to foreign DNA [24, 25]. With regard to the gene of interest, codon engineering to support maximal, tissue-specific expression should be performed.
Additionally, terminator/polyadenylation signal choices, the inclusion of post-transcriptional regulator elements and messenger RNA (mRNA) stability elements, and the presence of microRNA (miRNA) target sequence in the gene cassette can all have effects on gene expression [26]. The human factor IX 3′ UTR, for example, was shown to dramatically increase factor IX expression in vivo, especially in the context of additional cis regulatory elements [27]. Likewise, synthetic miRNA target sequences have been engineered into the 3′ UTR of AAV-delivered genes to make them susceptible to miRNA-122-driven suppression in the liver [28]. Although there is much known about these individual components that needs to be considered when designing an AAV vector, the final design will most likely need to be determined empirically. It is not yet possible to know how a particular design will function by just combining the best elements together based on published reports, therefore considerable trial and error will eventually be required for deciding on the final construct. In addition, one also needs to consider the differences between in vitro and in vivo activity. Although it is possible to model rAAV expression in rodents, there is still significant concern about the translatability to humans.
3 AAV Capsid Selection and Optimization
AAV has evolved to enter cells through initial interactions with carbohydrates present on the surface of target cells, typically sialic acid, galactose and heparin sulfate [29, 30]. Subtle differences in sugar-binding preferences, encoded in capsid sequence differences, can influence cell-type transduction preferences of the various AAV variants [31,32,33]. For example, AAV9 has a preference for primary cell binding through galactose as a result of unique amino acid differences in its capsid sequence [34]. It has been postulated that this preferential galactose binding could confer AAV9 with the unique ability to cross the blood–brain barrier (BBB) and infect cells of the CNS, including primary neurons [35, 36].
In addition to the primary carbohydrate interactions, secondary receptors have been identified that also play a role in viral transduction and contribute to cell and tissue selectivity of viral variants. AAV2 uses the fibroblast/hepatocyte growth factor receptor and the integrins αVβ5 and α5β1; AAV6 utilizes the epidermal growth factor receptor; and AAV5 utilizes the platelet-derived growth factor receptor. Recently, an uncharacterized type I membrane protein, AAVR (KIAA0319L), was identified as a critical receptor for AAV cell binding and internalization [37].
As a result of these subtle variations in primary and secondary receptor interactions for the various AAV variants, one can choose a variant that possesses a particular tropism and preferentially infects one cell or tissue type over others (Table 1). For example, AAV8 has been shown to effectively transduce and deliver genes to the liver of rodents and non-human primates, and is currently being explored in clinical trials to deliver genes for hemoglobinopathies and other diseases [38]. Likewise, AAV1 and AAV9 have been shown to be very effective at delivering genes to skeletal and cardiac muscle in various animal models [39,40,41,42,43,44,45,46]. Engineered AAV1 is currently being explored as the gene transfer factor in clinical trials for heart failure, and has been approved for the treatment of lipoprotein lipase deficiency [47]. However, although different AAV vectors have been identified that preferentially transduce many different cell types, there are still cell types for which AAV has proven difficult to transduce.
With the strong desire to utilize AAV to deliver genes to very selective cell and tissue types, efforts to clone novel AAV variants from human and primate tissues have identified a number of unique capsid sequences that are now being studied for tropism specificities [48]. In addition, recombinant techniques involving capsid shuffling, directed evolution, and random peptide library insertions are being utilized to derive variants of known AAVs with unique attributes [49,50,51]. In vivo-directed evolution has been successfully used to identify novel AAV variants that preferentially transduce the retinal cells of the eye, as well as other cell populations, including those in the CNS [50, 52, 53]. In addition, these techniques have been employed to identify novel AAV variants with reduced sensitivities to neutralizing antibodies (NAbs) [54,55,56,57].
Alternatively, other investigators have inserted larger binding proteins into different regions of AAV capsid proteins to confer selectivity. For example, DARPins (designed ankyrin repeat proteins), portions of protein A, and cytokines, have all been engineered into the capsid of AAV for the purpose of greater cell specificity and targeting [58, 59]. Employing this concept, others have been able to selectively target AAV to tumors and CD4+ T cells, as examples of engineered tropism [60, 61].
As we continue to learn more about the biology of AAV with regard to the mechanisms involved in membrane translocation, endosomal escape, and nuclear entry, we will undoubtedly find opportunities to engineer unique properties into viral vectors through modulating one or more of these functions. For example, it has been hypothesized that surface-exposed serine and tyrosine residues could be phosphorylated upon viral cell entry, resulting in their ubiquitination and proteolytic degradation [62,63,64]. Studies have shown that mutation of tyrosine to phenylalanine, which prevents this phosphorylation, results in dramatically improved transduction efficiencies [63]. Similar efforts have been made in attempts to limit the effects of NAbs, as discussed below.
The choice of a particular AAV to use as a gene transfer vector is heavily reliant on several critically important criteria: (1) which cell/tissue types are being targeted; (2) the safety profile associated with the delivered gene; (3) the choice of systemic versus local delivery; and (4) the use of tissue-specific or constitutively active promoters. As one gives careful consideration to these selection criteria, it is possible to narrow the choices of which AAVs (natural or engineered) to profile. Alternatively, one can begin the path of exploring fully engineered versions of AAV for truly selective cell targeting and optimized transduction. Because our understanding of AAV biology is in relative infancy, many of these efforts will remain empirical for quite some time as optimization for one activity could have a negative impact on another. Nonetheless, the future looks promising for this highly adaptable platform.
4 AAV Immunogenicity
One of the appealing aspects of using rAAV as a gene transfer vector is that it is composed of biomolecules, i.e. proteins and nucleic acids. Fortunately, a full-package virus lacks engineered lipids or other chemical components that could contribute to unwanted toxicities or immunogenicities that may not be predictable or fully understood. In general, AAV has been shown to be less immunogenic than other viruses. Although not completely understood, one possible reason for this may hinge on the observation that certain AAVs do not efficiently transduce antigen-presenting cells (APCs) [65]. Additionally, unlike previous viral delivery strategies, rAAV does not contain any viral genes, therefore there will be no active viral gene expression to amplify the immune response [66]. Although AAV has been shown to be poorly immunogenic compared with other viruses (i.e. adenovirus), the capsid proteins, as well as the nucleic acid sequence delivered, can trigger the various components of our immune system. This is further complicated by the fact that most people have already been exposed to AAV and have already developed an immune response against the particular variants to which they had previously been exposed, resulting in a pre-existing adaptive response. This can include NAbs and T cells that could diminish the clinical efficacy of subsequent re-infections with AAV and/or the elimination of cells that have been transduced. It should be of no surprise that the formidable challenge is how to deliver a therapeutically efficacious dose of rAAV to a patient population that already contains a significant amount of circulating NAbs and immunological memory against the virus [67]. Whether administered locally or systemically, the virus will be seen as a foreign protein, hence the adaptive immune system will attempt to eliminate it.
The humoral response to AAV is driven by the uptake of the virus by professional APCs, and their presentation of AAV capsid peptides in the context of class II major histocompatibility proteins (MHCs) to B cells and CD4+ T cells [68, 69]. This leads to plasma cell and memory cell development that has the capacity to secrete antibodies to the AAV capsid. These antibodies can either be neutralizing, which has the potential to prevent subsequent AAV infection, or non-neutralizing. Non-NAbs are thought to opsonize the viral particles and facilitate their removal through the spleen [70].
Upon entry of the virus into target cells during the course of the natural infection process, the virus is internalized through clathrin-mediated uptake into endosomes [71]. After escape from the endosome, the virus is transported to the nucleus where the ITR-flanked transgene is uncoated from the capsid [72]. The pathway and mechanism of AAV intracellular transport and processing is not fully understood, and there are quite a few areas of debate with regard to current understanding. The most current hypothesis is that following endosomal escape, capsid breakdown and uncoating occurs after subsequent nuclear translocation. However, it is thought that cytosolic ubiquitination of the intact virus can occur during transport to the nucleus [73]. This would be a critical step in directing capsid proteins to the proteasome for proteolytic processing into peptides for class I MHC presentation. This hypothesis is supported by data in which proteasome inhibitors, or mutations in capsid residues that are sites for ubiquitination, can limit class I presentation and T-cell activation [73,74,75,76]. However, apparent differences have been observed for T-cell activation to different AAV variants with significant sequence identity. At this time, it is unclear whether this is due to subtle capsid sequence differences and susceptibility to MHC I presentation or differential cellular processing that is innate to the different AAV variants, or simply due to contaminants in vector preparations [76].
In addition to an adaptive immunological reaction to the capsid of AAV, the transgene can elicit both an adaptive and an innate response. If the transgene encodes a protein that can be recognized as foreign, it too can generate a similar B- and T-cell response. For example, in replacement therapy applications in which the protein to be replaced is the consequence of a null genotype, the immune system will have never selected against precursor B and T cells to that protein [70, 77]. Likewise, if the transgene is an engineered variant, the engineered sequence can be recognized as foreign. Even the variable regions of antibodies can activate an adaptive response that can result in deletion of target cells that are expressing transgene as a result of AAV delivery. Finally, a transgene with a significant number of CpG dinucleotides can activate innate responses through toll-like receptor (TLR) molecular pattern receptors [78].
Pre-existing immunity to AAV, especially the presence of circulating NAb, can have a dramatic effect on AAV clinical efficacy. To date, this represents one of the biggest therapeutic challenges to the use of systemically delivered AAV, and is thought to be one of the factors in early clinical failures [79]. Pre-existing immunity to AAV can often be overcome by selecting a particular AAV variant that has not circulated throughout the human population, and, therefore, does not have any memory responses elicited against it, including NAbs and T cells [80]. Additionally, some of the AAV evolution technologies discussed above have been used to identify AAVs that are resistant to the effects of NAbs [50, 57]. Although not optimal, it is possible to prescreen subjects for the presence of NAbs to the particular AAV variant to be used. In addition, the impact of this immunological response can sometimes be minimized by the particular route of administration employed for the particular therapeutic strategy, as discussed in Sect. 6 [80].
5 Manufacturing for Clinical Use
Like most biotherapeutics, AAV needs to be produced in a living system (Fig. 2). The parallels with recombinant antibody production during the 1990s and 2000s, with regard to the upstream challenges of robust production levels, are important to understand where the industry currently is, and where we need to strive to be.

Overview of AAV production/purification. Cell platform: HEK-293T, Sf9, or other suitable cell system can be grown on a small scale on 150 mm tissue culture-treated culture dish, hyperflasks, or shake flasks. Cells are then transfected with adenovirus helper virus, rep/cap, and ITR-transgene plasmids for 293T, or infected with baculovirus for Sf9. Producer lines with integrated expression of rep/cap and ITR-transgene can be infected with adenovirus and grown to scale. Scale-up: For larger-scale culture volumes, virus can be produced in roller bottles, continuous perfusion, or WAVE Bioreactor systems. Purification/polishing: Affinity or heparin chromatography are optimal for isolation of virus from culture supernatants with or without cell pellet harvesting. Benzonase/DNAse treatment of eluted virus is required for removal of extraviral DNA contamination, followed by anion-exchange chromatography to fractionate ‘empty’ vs. ‘full’ AAV particles. QC/release: Upper left of far right panel: image depicts a silver stain analysis of culture FT next to affinity/anion exchange purified AAV (pure). The three bands represent the viral capsid proteins VP1, VP2, and VP3. Upper right of far right panel: Dynamic light scattering analysis of purified AAV1 indicates a uniform particle distribution of approximately 25–30 nM. Bottom half of far right panel: Analytical ultracentrifugation can resolve the proportion of ‘empty’ vs, ‘full’ particles of purified material. Additional assays that should be employed are digital drop polymerase chain reaction for determining titer in GC/mL, cryo or transmission electron microscopy for visual representation of purified particles, endotoxin testing, and other assays to evaluate the presence of residual host-cell protein contamination. AAV adeno-associated virus, FT flow-through, GC genome copies, rep/cap replication/capsid, QC quality control
5.1 Transient Transfection Platforms
Current methods to produce rAAV are still expensive despite years of research (Table 2). The most widely used platform for producing rAAV involves transfecting HEK293 cells with either two or three plasmids; one encoding the gene of interest, one carrying the AAV rep/cap genes, and another containing helper genes provided by either adeno or herpes viruses [6]. While most robust production rates have been achieved with adherent cells in either roller bottles or cell stacks, similar rates are now achievable in suspension-adapted HEK293 cells (Table 2). Production rates of approximately 105 genome copies (GC)/cell are now common, resulting in 1014 GC/L [81]. While this has proven to be sufficient to support early clinical trials, and could supply marketed product for small patient population indications, the deficiencies in scalability with this platform are a significant limitation [82, 83]. As one could surmise, successfully delivering three plasmids to one cell is a relatively inefficient process. For larger-scale manufacturing efforts, transient delivery of plasmid requires excess quantities of DNA, adding to the overall cost of production and purification. Moreover, transient delivery of rep/cap genes in the presence of helper genes can also contribute to product heterogeneity, including AAV vectors lacking a transgene. These ‘empty capsids’ represent a significant proportion of virus produced in transient transfection assays. Thus, it is critically important to develop robust analytical quality control (QC) methods that are able to distinguish between these viral variants in order to ensure similarities between production lots [82, 83].
5.2 Producer Mammalian Cell Lines
In three other AAV manufacturing platforms, one or more genetic components for the AAV manufacturing has been integrated into the genome of mammalian or insect production cell lines. While most viral helper genes needed for AAV production cannot be stably transfected, the adenoviral E1a and E1b genes are exceptions. These genes have been used to transform HEK293 cells, however they induce expression of the AAV rep gene, which is toxic to mammalian and insect cells [84, 85]. Hence, two different approaches have been used to develop mammalian cell lines. The first uses co-infection of BHK cells with two replication-defective HSVs engineered to encode the ITR-flanked transgene and the rep/cap genes. The second is based on stable producer cell lines in HeLa cells carrying the ITR-flanked transgene and the rep/cap genes. Rep proteins are not expressed in these cells since HeLa carries no adenoviral genes. However, infection with wild-type adenovirus is required for AAV production. The inclusion of replication-competent viral agents into a production process is a concern that needs to be addressed and also requires additional steps during the downstream processing [82, 83].
5.3 Producer Insect Cell Lines
More recently, the Sf9 insect cell system in combination with baculovirus infection has been utilized to produce bulk quantities of rAAV. In this system, two or three baculovirus particles may be used to infect the Sf9 cells and initiate AAV production. In one example, one virus contains the rep gene, a second contains the cap gene, and the final virus carries the ITR-flanked gene of interest. In an alternative system, the Sf9 cells can be engineered to have the ITR-flanked gene of interest integrated into their genome, upon which production is initiated with only two baculovirus preps [81, 82]. A further improvement has recently been shown whereby the rep/cap genes are stably integrated into the Sf9 cell line genome, but are under the control of a promoter/enhancer that is induced by subsequent baculovirus infection. In this system, infection can occur, with only one baculovirus containing the ITR-flanked gene of interest, simplifying the system significantly [86, 87].
Production levels of approximately 105 GC/cell and 1015 GC/L have routinely been achieved with these Sf9 systems. Because of their ease of manipulation and their ability to grow to very high cell densities, the Sf9 system is rapidly becoming the platform of choice for AAV manufacturing. Concerns regarding baculovirus instability and differences in post-translational modifications between mammalian and insect cell systems are now beginning to be understood and controlled. These concerns are offset by the fact that baculovirus cannot efficiently infect mammalian cells which makes it inherently safer then other viral-based production systems [81,82,83, 86, 87].
5.4 Purification and Downstream Processing
Unlike antibody manufacturing that relied on a single protein A-based purification platform early in the development of the downstream process, AAV is still rapidly evolving in that area. The products of an AAV production run will contain not only cellular debris (protein/lipids/nucleic acids) but also two main populations of AAV particles: particles that contain (full capsids) or those lacking (empty capsids) the ITR-flanked transgene. Although still widely debated in the field, the presence of empty capsids represents another contaminant that must be removed or controlled. Initial attempts to separate these two populations originally relied on the cumbersome and non-scalable method of density ultracentrifugation. In addition to the scalability issue, there are also concerns about the physiochemical effects of this method on the particles. Regardless, this method is still employed by many organizations as either a primary or secondary step in AAV purification [83].
Current technologies utilizing various affinity resins and/or ion exchange chromatography are being adopted by the industry. As mentioned above, AAV uses cell membrane-associated carbohydrates as the primary cell receptor for transduction. This affinity for carbohydrates can be exploited as an initial capture step in AAV purification. Indeed, heparin columns are frequently used in many downstream processing steps for AAV [88]. However, because of the lack of specificity, alternative affinity columns based on AAV-specific binding proteins such as scFvs and antibody single domains from llamas (camelids) have started to dominate the field. Improvements in generating these AAV-specific resins confers many advantages in downstream purification. These resins have the ability to bind to more than one AAV variant, have very high binding capacities (>1014 GC/mL resin), and are stable against harsh clean-in-place and regeneration methods, making them suitable for use multiple times. Some of these commercial resins are already Good Manufacturing Practice (GMP) compliant, making them ideal for downstream manufacturing at commercial scales. Polishing steps using anion exchange chromatography are now routinely included after affinity capture steps, and can efficiently separate full capsids from empty capsids [89,90,91,92].
As with any new therapeutic platform, and, again, similar to antibody-based therapeutic evolution, details on product specification and regulatory requirements are still evolving. With still very limited clinical experience, the impact of empty particles, host-cell impurities, post-translational modifications from different production platforms, fidelity of the packaged transgene, capsid ratio integrity, and probably many other specifications are still not known. However, over time, and as more clinical experience is gained, the field will be able to better relate these details to product performance and safety [83].
The use of rAAV as a delivery vector for gene therapies has been rapidly gaining interest over the past 3–5 years. As approvals begin to increase (see Sect. 6), efforts to optimize and maximize clinical manufacturing technologies will see a burst of activity. This will most likely mirror what occurred with antibody therapeutics in the 1990s and 2000s, in which early technologies were quickly overcome by next-generation technologies, resulting in significant cost savings and increased clinical supplies.
6 Delivery Strategies for Recombinant AAV Therapeutics and Clinical Candidates
AAV has been shown to be a very stable vector able to withstand wide temperature and pH changes with little to no loss in activity [93]. To date, the only limitation seems to be the concentration with which it can be formulated, currently maximized around 5 × 1013 particles per milliliter [83]. With the resurgence in clinical use, this formulation limit will most likely be overcome in the near future. However, the robust stability of these vectors provides ample opportunities to attempt different routes of administration and specialized delivery strategies (Table 3).
Other than the European Medicines Agency (EMA)-approved AAV-based product alipogene tiparvovec (Glybera®), the most advanced current clinical trial using AAV is sponsored by Spark Therapeutics and utilizes local injection of AAV2 into the eye for inherited retinal diseases (voretigene neparvovec-RPE65) (Table 3) [94]. Phase III studies have just been completed on this candidate and a Biologics License Application (BLA) submission is expected this year. This type of local delivery has proven to be safe and efficacious, but requires specialized surgical techniques and/or devices to deliver the vector [94, 95]. Similar strategies are being conducted by Applied Genetic Technologies Corporation (AGTC), targeting X-linked retinoschisis and achromatopsia, X-linked retinitis pigmentosa, and age-related macular degeneration. These programs are at various stages of development, with the most advanced for X-linked retinoschisis and achromatopsia in phase I safety studies (http://www.AGTC.com) (Table 3).
6.1 Systemic Delivery
Several clinical trials are being run in which systemic administration is being used to target the liver, a tissue that is readily accessible through this route of administration and a tissue type that is readily transduced by many well-understood AAV variants [96]. These trials are mostly for monogenic, inherited diseases, in which the goal is gene replacement for defective genes, including those mutated in hemophilia A and B. Currently, these trials are in phase I/II, and are sponsored by academic groups, as well as biopharmaceutical companies such as Spark Therapeutics (SPK-9001, SPK-8011), Sangamo Therapeutics (SB-525), UniQure (AMT-060), Dimension Therapeutics (DTX101, DTX201), and Biomarin (BMN 270) (Table 3) [97]. Unlike local administration to the eye, which is considered an immune-privileged site that might not be affected by the existence of NAbs, systemic administration will require patient stratification for patient NAb levels. In addition, the possibility for re-administration becomes very difficult, should the need arise [80]. Although rare, there have been reports of rAAV vector integration into animal model genomes with subsequent genotoxicities [98, 99]. In addition, AAV genome sequences have been found in human hepatocellular carcinoma samples near known cancer driver genes, although at a low frequency [100]. There is an ongoing debate on these findings regarding cause and effect, and mouse/human translation. Regardless, hepatocellular, as well as other tissue genotoxicity, will need to be monitored in the course of AAV clinical development.
6.2 Intramuscular Delivery
Another common delivery strategy is direct intramuscular injections. The only approved AAV gene therapy in Europe (Glybera®) is an AAV1 encoding the gene for lipoprotein lipase deficiency [47, 101]. Skeletal muscle has been shown to be a target tissue type that is efficiently transduced by many AAV variants [39]. Once transduced, the muscle cells serve as a production site for protein products that can act locally or systemically, as is the case with Glybera®. As a result of the low cellular turnover rate of the muscle cells, the transduced AAV gene product will be maintained in these cells as an episome for years, as has been shown in many studies in non-human primates [39]. Consequently, a single-dose regimen of an intramuscularly-delivered product may never need to be readministered unless there is significant damage or immune clearance of the transduced cells. This strategy is also being employed by Adverum and AGTC for α1-antitrypsin deficiency, as well as for certain muscular dystrophies (Table 3) [97].
6.3 Central Nervous System Delivery
Direct CNS administration is being utilized for Parkinson’s disease, as well as various inherited diseases such as Batten disease, Canavan disease, and mucopolysaccharidosis (MPS) IIA and IIB, as well as MPS IIIa and MPS IIIb (Sanfilippo syndromes type A and type B, respectively). Phase I/II studies for these diseases using a variety of AAV variants, including AAV2, AAVrh10, and AAV9, are currently ongoing by various academic groups and biopharmaceutical companies, such as Abeona Therapeutics (ABO-101, ABO-102, ABO-201, ABO-202) [97, 102, 103]. Delivery strategies range from direct intraparenchymal administration into particular areas of the brain, intracerebroventricular, and cisternal and lumbar intrathecal routes [102]. The decision on the best route of administration is intimately related to the disease and affected areas. For example, for Parkinson’s disease, according to our current understanding of disease pathogenesis and therapeutic strategies, direct injection into the putamen, substantia nigra or striatum is thought to be required. Similarly, for diseases that affect larger areas of the brain, such as Canavan disease or MPS, direct injection into the cerebellum is thought to be most beneficial [102, 103].
Alternatively, administration directly into the cerebrospinal fluid through an intrathecal route can result in wide CNS biodistribution, which is thought to be necessary for diseases such as spinal muscular atrophy (SMA) and Alzheimer’s disease [102,103,104,105,106]. An alternative to cerebral spinal fluid (CSF)-based routes is the use of systemic administration of AAV variants that have been shown to cross the BBB. AAV9 has been shown to transcytosis across the BBB and transduce large sections of the CNS [36, 104, 107, 108]. This approach is currently being explored in the clinic for the treatment of SMA by AveXis (AVXS-101).
Neurodegenerative diseases represent a particular devastating health problem for which there is significant unmet medical need. These diseases of the CNS have proven to be very difficult to treat as a result of our poor understanding of their etiology and difficulty getting efficacious agents across the BBB. With regard to Alzheimer’s disease, although there is still some disagreement in the field, idiopathic amyloid plaque formation or generation of neurofibrillary tau tangles (NFTs), both of which are thought to be neurotoxic, are still the prevailing hypotheses behind the mechanism of many of these neuropathologies. Attempts to clear these plaques with plaque-specific antibodies have shown signs of limiting this process in animals and early-stage clinical trials [109, 110]; However, larger studies have all shown to be inconclusive at best, or failures at worst. It is unclear if these failures were because the plaque hypothesis is wrong, or if there was inefficient CNS exposure to the antibody therapeutic [110, 111]. Alternative strategies taking advantage of the safety and persistence of AAV would utilize either local administration of antibody-encoding AAVs directly to the CNS, or systemic delivery of AAVs that can cross the BBB, resulting in significantly higher CNS exposure levels of the antibody [112].
6.4 Cardiac Delivery
Local delivery of AAV to cardiac muscle for heart failure has been attempted in various clinical trials. In one case, Celladon failed in their attempt to deliver SERCA2A directly to the heart, and, in a second case, there is an ongoing program sponsored by UniQure to deliver S100A directly to the heart that is currently still in preclinical development [46, 113,114,115]. Although it is not thoroughly clear why Celladon failed in the clinic, and why one would expect UniQure/BMS to succeed, there are significant differences in the delivery methods used by the two programs and the target gene delivered. Celladon used intracoronary infusion to deliver their AAV1 SERCA2A gene product, whereas UniQure is using retroinfusion and left anterior descending (LAD) coronary occlusion [41, 115]. This procedure is thought to better localize and restrict the delivered AAV9 S100A gene product to better target the heart tissue of interest. The reality of this suspected benefit will be realized in the clinic in the coming years.
6.5 Pulmonary Delivery
Aerosolized AAV for inhaled pulmonary delivery was utilized in some of the earliest trials for cystic fibrosis (CF). Although none of these trials resulted in significant benefit or showed much of a pharmacodynamic response, they did help to show the safety of AAV when administered via this route [116,117,118]. More importantly, the pathophysiology of CF, molecular biology of the CF transmembrane conductance regulator (CFTR) gene, and the target cell population for this type of indication exposed some key considerations when using AAV [117]. Congestion of the airways in these patients can limit AAV biodistribution after delivery, thus attenuating robust transduction [118]. In addition, the CFTR gene is over 4 kb in size, putting it at the upper limit of the packaging capacity of AAV after also considering a required promoter and terminator. Finally, CFTR is expressed by the submucosal glands, which may be difficult to target efficiently [116, 117]. Nonetheless, these early efforts proved that AAV can safely deliver genes to the lung, which might be an ideal strategy for other diseases, such as influenza and other infectious diseases of the lung [119].
The field is just beginning to explore localized delivery of AAV for gene therapy applications. The stability of the virus and broad tropism for many different cell and tissue types make them ideal for most applications. There appears to be at least one AAV variant option for every tissue type of interest, with engineering and novel AAV discovery efforts sure to identify and create AAV variants with very specialized functions on demand. These efforts will undoubtedly result in new therapeutic strategies for many new indications.
7 Concluding Remarks
The transfer of genes and other nucleic acids into cells has been a research tool in the laboratory for more than four decades. However, it was our growing understanding of the genetic components underlying certain diseases that has driven the search for true gene therapies. Progressively, research in other areas have identified other potential opportunities in which gene delivery could be applied therapeutically. In addition, limitations with current small molecule and protein therapeutic platforms have also driven the search for alternative therapeutic platforms that accommodate those limitations [120, 121]. Gene therapies accommodate all of those limitations, especially around target accessibility. As a result, the search for safe and effective gene delivery technologies has been a major focus in pharmaceutical research and development, and will hopefully represent a paradigm shift in how we approach disease-state intervention.
AAV was discovered over 50 years ago and has since become one of the leading gene delivery vectors in clinical development. As a result of its unique biology, simple structure, and no known disease associations, AAV could become the vector of choice for most gene therapy applications. Gene therapy using rAAV has been demonstrated to be safe and well-tolerated in virtually every clinical setting in which it has been used. These studies, along with basic research on its biology, have revealed many facets of this vector that can be applied to future efforts.
Among the critical parameters to be considered are vector design, capsid selection, desired target cell and tissue type, and route of administration. The transgene to be delivered optimized for expression, the right AAV variant with an appropriate capsid for target cell transduction and immunoreactivity profile, and the appropriate delivery approach to maximize target tissue exposure while limiting off-tissue exposure are key focal points for AAV-based therapies.
All of these variables will be dictated by the overall therapeutic strategy which will be influenced by our understanding of the pathobiology of the disease to be treated. Will the transgene have the desired effect? Is the target cell driving the disease state? Is the turnover rate of the target cell high, requiring repeat dosing? This cannot be emphasized enough; without a strong understanding of the mechanisms driving the disease state, it will not be possible to design, discover, and develop the right gene therapeutic. Better designed trials, optimized vector construction, and novel AAV variants will certainly result in future regulatory approvals and improvements on patient outcomes and health.
References
Ni R, Zhou J, Hossain N, Chau Y. Virus-inspired nucleic acid delivery system: linking virus and viral mimicry. Adv Drug Deliv Rev. 2016;106(Pt A):3–26. doi:10.1016/j.addr.2016.07.005.
Cotter MJ, Muruve DA. The induction of inflammation by adenovirus vectors used for gene therapy. Front Biosci. 2005;10:1098–105.
Chen J, Guo Z, Tian H, Chen X. Production and clinical development of nanoparticles for gene delivery. Mol Ther Methods Clin Dev. 2016;3:16023. doi:10.1038/mtm.2016.23.
Hastie E, Samulski RJ. Adeno-associated virus at 50: a golden anniversary of discovery, research, and gene therapy success—a personal perspective. Hum Gene Ther. 2015;26(5):257–65. doi:10.1089/hum.2015.025.
Rose JA, Hoggan MD, Shatkin AJ. Nucleic acid from an adeno-associated virus: chemical and physical studies. Proc Natl Acad Sci USA. 1966;56(1):86–92.
Samulski RJ, Muzyczka N. AAV-mediated gene therapy for research and therapeutic purposes. Annu Rev Virol. 2014;1(1):427–51. doi:10.1146/annurev-virology-031413-085355.
Naumer M, Sonntag F, Schmidt K, Nieto K, Panke C, Davey NE, et al. Properties of the adeno-associated virus assembly-activating protein. J Virol. 2012;86(23):13038–48. doi:10.1128/jvi.01675-12.
Earley LF, Powers JM, Adachi K, Baumgart JT, Meyer NL, Xie Q et al. Adeno-associated Virus (AAV) assembly-activating protein is not an essential requirement for capsid assembly of AAV serotypes 4, 5, and 11. J Virol. 2017;91(3):1–21. doi:10.1128/jvi.01980-16.
Choi VW, McCarty DM, Samulski RJ. Host cell DNA repair pathways in adeno-associated viral genome processing. J Virol. 2006;80(21):10346–56. doi:10.1128/jvi.00841-06.
Dong B, Nakai H, Xiao W. Characterization of genome integrity for oversized recombinant AAV vector. Mol Ther. 2010;18(1):87–92. doi:10.1038/mt.2009.258.
Wu Z, Yang H, Colosi P. Effect of genome size on AAV vector packaging. Mol Ther. 2010;18(1):80–6. doi:10.1038/mt.2009.255.
Chamberlain K, Riyad JM, Weber T. Expressing transgenes that exceed the packaging capacity of adeno-associated virus capsids. Hum Gene Ther Methods. 2016;27(1):1–12. doi:10.1089/hgtb.2015.140.
McCarty DM, Monahan PE, Samulski RJ. Self-complementary recombinant adeno-associated virus (scAAV) vectors promote efficient transduction independently of DNA synthesis. Gene Ther. 2001;8(16):1248–54. doi:10.1038/sj.gt.3301514.
McCarty DM. Self-complementary AAV vectors; advances and applications. Mol Ther. 2008;16(10):1648–56. doi:10.1038/mt.2008.171.
Powell SK, Rivera-Soto R, Gray SJ. Viral expression cassette elements to enhance transgene target specificity and expression in gene therapy. Discov Med. 2015;19(102):49–57.
Gray SJ, Foti SB, Schwartz JW, Bachaboina L, Taylor-Blake B, Coleman J, et al. Optimizing promoters for recombinant adeno-associated virus-mediated gene expression in the peripheral and central nervous system using self-complementary vectors. Hum Gene Ther. 2011;22(9):1143–53. doi:10.1089/hum.2010.245.
Wang B, Li J, Fu FH, Chen C, Zhu X, Zhou L, et al. Construction and analysis of compact muscle-specific promoters for AAV vectors. Gene Ther. 2008;15(22):1489–99. doi:10.1038/gt.2008.104.
Dashkoff J, Lerner EP, Truong N, Klickstein JA, Fan Z, Mu D, et al. Tailored transgene expression to specific cell types in the central nervous system after peripheral injection with AAV9. Mol Ther Methods Clin Dev. 2016;3:16081. doi:10.1038/mtm.2016.81.
Xu R, Janson CG, Mastakov M, Lawlor P, Young D, Mouravlev A, et al. Quantitative comparison of expression with adeno-associated virus (AAV-2) brain-specific gene cassettes. Gene Ther. 2001;8(17):1323–32. doi:10.1038/sj.gt.3301529.
Zhang R, Wang Q, Zhang L, Chen S. Optimized human factor IX expression cassettes for hepatic-directed gene therapy of hemophilia B. Front Med. 2015;9(1):90–9. doi:10.1007/s11684-015-0390-2.
Murillo O, Luqui DM, Gazquez C, Martinez-Espartosa D, Navarro-Blasco I, Monreal JI, et al. Long-term metabolic correction of Wilson’s disease in a murine model by gene therapy. J Hepatol. 2016;64(2):419–26. doi:10.1016/j.jhep.2015.09.014.
Shen SQ, Myers CA, Hughes AE, Byrne LC, Flannery JG, Corbo JC. Massively parallel cis-regulatory analysis in the mammalian central nervous system. Genome Res. 2016;26(2):238–55. doi:10.1101/gr.193789.115.
Carton JM, Sauerwald T, Hawley-Nelson P, Morse B, Peffer N, Beck H, et al. Codon engineering for improved antibody expression in mammalian cells. Protein Expr Purif. 2007;55(2):279–86. doi:10.1016/j.pep.2007.05.017.
Plotkin JB, Robins H, Levine AJ. Tissue-specific codon usage and the expression of human genes. Proc Natl Acad Sci USA. 2004;101(34):12588–91. doi:10.1073/pnas.0404957101.
Quax TE, Claassens NJ, Soll D, van der Oost J. Codon bias as a means to fine-tune gene expression. Mol Cell. 2015;59(2):149–61. doi:10.1016/j.molcel.2015.05.035.
Gray JT, Zolotukhin S. Design and construction of functional AAV vectors. Methods Mol Biol. 2011;807:25–46. doi:10.1007/978-1-61779-370-7_2.
Miao CH, Ohashi K, Patijn GA, Meuse L, Ye X, Thompson AR, et al. Inclusion of the hepatic locus control region, an intron, and untranslated region increases and stabilizes hepatic factor IX gene expression in vivo but not in vitro. Mol Ther. 2000;1(6):522–32. doi:10.1006/mthe.2000.0075.
Geisler A, Fechner H. MicroRNA-regulated viral vectors for gene therapy. World J Exp Med. 2016;6(2):37–54. doi:10.5493/wjem.v6.i2.37.
Asokan A, Schaffer DV, Samulski RJ. The AAV vector toolkit: poised at the clinical crossroads. Mol Ther. 2012;20(4):699–708. doi:10.1038/mt.2011.287.
Wu Z, Asokan A, Samulski RJ. Adeno-associated virus serotypes: vector toolkit for human gene therapy. Mol Ther. 2006;14(3):316–27. doi:10.1016/j.ymthe.2006.05.009.
Agbandje-McKenna M, Kleinschmidt J. AAV capsid structure and cell interactions. Methods Mol Biol. 2011;807:47–92. doi:10.1007/978-1-61779-370-7_3.
DiMattia MA, Nam HJ, Van Vliet K, Mitchell M, Bennett A, Gurda BL, et al. Structural insight into the unique properties of adeno-associated virus serotype 9. J Virol. 2012;86(12):6947–58. doi:10.1128/JVI.07232-11.
Halder S, Van Vliet K, Smith JK, Duong TT, McKenna R, Wilson JM, et al. Structure of neurotropic adeno-associated virus AAVrh.8. J Struct Biol. 2015;192(1):21–36. doi:10.1016/j.jsb.2015.08.017.
Bell CL, Gurda BL, Van Vliet K, Agbandje-McKenna M, Wilson JM. Identification of the galactose binding domain of the adeno-associated virus serotype 9 capsid. J Virol. 2012;86(13):7326–33. doi:10.1128/JVI.00448-12.
Merkel SF, Andrews AM, Lutton EM, Mu D, Hudry E, Hyman BT, et al. Trafficking of adeno-associated virus vectors across a model of the blood–brain barrier; a comparative study of transcytosis and transduction using primary human brain endothelial cells. J Neurochem. 2017;140(2):216–30. doi:10.1111/jnc.13861.
Zhang H, Yang B, Mu X, Ahmed SS, Su Q, He R, et al. Several rAAV vectors efficiently cross the blood-brain barrier and transduce neurons and astrocytes in the neonatal mouse central nervous system. Mol Ther. 2011;19(8):1440–8. doi:10.1038/mt.2011.98.
Pillay S, Meyer NL, Puschnik AS, Davulcu O, Diep J, Ishikawa Y, et al. An essential receptor for adeno-associated virus infection. Nature. 2016;530(7588):108–12. doi:10.1038/nature16465.
Kattenhorn LM, Tipper CH, Stoica L, Geraghty DS, Wright TL, Clark KR, et al. Adeno-associated virus gene therapy for liver disease. Hum Gene Ther. 2016;27(12):947–61. doi:10.1089/hum.2016.160.
Wang D, Zhong L, Nahid MA, Gao G. The potential of adeno-associated viral vectors for gene delivery to muscle tissue. Expert Opin Drug Deliv. 2014;11(3):345–64. doi:10.1517/17425247.2014.871258.
Su H, Yeghiazarians Y, Lee A, Huang Y, Arakawa-Hoyt J, Ye J, et al. AAV serotype 1 mediates more efficient gene transfer to pig myocardium than AAV serotype 2 and plasmid. J Gene Med. 2008;10(1):33–41. doi:10.1002/jgm.1129.
Greenberg B, Yaroshinsky A, Zsebo KM, Butler J, Felker GM, Voors AA, et al. Design of a phase 2b trial of intracoronary administration of AAV1/SERCA2a in patients with advanced heart failure: the CUPID 2 trial (calcium up-regulation by percutaneous administration of gene therapy in cardiac disease phase 2b). JACC Heart Fail. 2014;2(1):84–92. doi:10.1016/j.jchf.2013.09.008.
Hadri L, Kratlian RG, Benard L, Maron BA, Dorfmuller P, Ladage D, et al. Therapeutic efficacy of AAV1.SERCA2a in monocrotaline-induced pulmonary arterial hypertension. Circulation. 2013;128(5):512–23. doi:10.1161/circulationaha.113.001585.
Zincarelli C, Soltys S, Rengo G, Rabinowitz JE. Analysis of AAV serotypes 1–9 mediated gene expression and tropism in mice after systemic injection. Mol Ther. 2008;16(6):1073–80. doi:10.1038/mt.2008.76.
Kotchey NM, Adachi K, Zahid M, Inagaki K, Charan R, Parker RS, et al. A potential role of distinctively delayed blood clearance of recombinant adeno-associated virus serotype 9 in robust cardiac transduction. Mol Ther. 2011;19(6):1079–89. doi:10.1038/mt.2011.3.
Tarantal AF, Lee CC, Martinez ML, Aravind A, Samulski RJ. Systemic and persistent muscle gene expression in rhesus monkeys with a liver de-targeted adeno-associated virus (AAV) vector. Hum Gene Ther. 2017;. doi:10.1089/hum.2016.130.
Bish LT, Morine K, Sleeper MM, Sanmiguel J, Wu D, Gao G, et al. Adeno-associated virus (AAV) serotype 9 provides global cardiac gene transfer superior to AAV1, AAV6, AAV7, and AAV8 in the mouse and rat. Hum Gene Ther. 2008;19(12):1359–68. doi:10.1089/hum.2008.123.
Scott LJ. Alipogene tiparvovec: a review of its use in adults with familial lipoprotein lipase deficiency. Drugs. 2015;75(2):175–82. doi:10.1007/s40265-014-0339-9.
Limberis MP, Vandenberghe LH, Zhang L, Pickles RJ, Wilson JM. Transduction efficiencies of novel AAV vectors in mouse airway epithelium in vivo and human ciliated airway epithelium in vitro. Mol Ther. 2009;17(2):294–301. doi:10.1038/mt.2008.261.
Buning H, Huber A, Zhang L, Meumann N, Hacker U. Engineering the AAV capsid to optimize vector-host-interactions. Curr Opin Pharmacol. 2015;24:94–104. doi:10.1016/j.coph.2015.08.002.
Bartel MA, Weinstein JR, Schaffer DV. Directed evolution of novel adeno-associated viruses for therapeutic gene delivery. Gene Ther. 2012;19(6):694–700. doi:10.1038/gt.2012.20.
Kotterman MA, Schaffer DV. Engineering adeno-associated viruses for clinical gene therapy. Nat Rev Genet. 2014;15(7):445–51. doi:10.1038/nrg3742.
Dalkara D, Byrne LC, Klimczak RR, Visel M, Yin L, Merigan WH, et al. In vivo-directed evolution of a new adeno-associated virus for therapeutic outer retinal gene delivery from the vitreous. Sci Transl Med. 2013;5(189):189ra76. doi:10.1126/scitranslmed.3005708.
Castle MJ, Turunen HT, Vandenberghe LH, Wolfe JH. Controlling AAV tropism in the nervous system with natural and engineered capsids. Methods Mol Biol. 2016;1382:133–49. doi:10.1007/978-1-4939-3271-9_10.
Tseng YS, Agbandje-McKenna M. Mapping the AAV capsid host antibody response toward the development of second generation gene delivery vectors. Front Immunol. 2014;5:9. doi:10.3389/fimmu.2014.00009.
Li C, Diprimio N, Bowles DE, Hirsch ML, Monahan PE, Asokan A, et al. Single amino acid modification of adeno-associated virus capsid changes transduction and humoral immune profiles. J Virol. 2012;86(15):7752–9. doi:10.1128/jvi.00675-12.
Bartel M, Schaffer D, Buning H. Enhancing the clinical potential of AAV vectors by capsid engineering to evade pre-existing immunity. Front Microbiol. 2011;2:204. doi:10.3389/fmicb.2011.00204.
Maersch S, Huber A, Buning H, Hallek M, Perabo L. Optimization of stealth adeno-associated virus vectors by randomization of immunogenic epitopes. Virology. 2010;397(1):167–75. doi:10.1016/j.virol.2009.10.021.
Warrington KH Jr, Gorbatyuk OS, Harrison JK, Opie SR, Zolotukhin S, Muzyczka N. Adeno-associated virus type 2 VP2 capsid protein is nonessential and can tolerate large peptide insertions at its N terminus. J Virol. 2004;78(12):6595–609. doi:10.1128/JVI.78.12.6595-6609.2004.
Ried MU, Girod A, Leike K, Buning H, Hallek M. Adeno-associated virus capsids displaying immunoglobulin-binding domains permit antibody-mediated vector retargeting to specific cell surface receptors. J Virol. 2002;76(9):4559–66. doi:10.1128/jvi.76.9.4559-4566.2002.
Munch RC, Muth A, Muik A, Friedel T, Schmatz J, Dreier B, et al. Off-target-free gene delivery by affinity-purified receptor-targeted viral vectors. Nat Commun. 2015;6:6246. doi:10.1038/ncomms7246.
Munch RC, Janicki H, Volker I, Rasbach A, Hallek M, Buning H, et al. Displaying high-affinity ligands on adeno-associated viral vectors enables tumor cell-specific and safe gene transfer. Mol Ther. 2013;21(1):109–18. doi:10.1038/mt.2012.186.
Aslanidi GV, Rivers AE, Ortiz L, Song L, Ling C, Govindasamy L, et al. Optimization of the capsid of recombinant adeno-associated virus 2 (AAV2) vectors: the final threshold? PLoS One. 2013;8(3):e59142. doi:10.1371/journal.pone.0059142.
Zhong L, Li B, Mah CS, Govindasamy L, Agbandje-McKenna M, Cooper M, et al. Next generation of adeno-associated virus 2 vectors: point mutations in tyrosines lead to high-efficiency transduction at lower doses. Proc Natl Acad Sci USA. 2008;105(22):7827–32. doi:10.1073/pnas.0802866105.
Zhong L, Li B, Jayandharan G, Mah CS, Govindasamy L, Agbandje-McKenna M, et al. Tyrosine-phosphorylation of AAV2 vectors and its consequences on viral intracellular trafficking and transgene expression. Virology. 2008;381(2):194–202. doi:10.1016/j.virol.2008.08.027.
Mays LE, Wang L, Lin J, Bell P, Crawford A, Wherry EJ, et al. AAV8 induces tolerance in murine muscle as a result of poor APC transduction, T cell exhaustion, and minimal MHCI upregulation on target cells. Mol Ther. 2014;22(1):28–41. doi:10.1038/mt.2013.134.
Basner-Tschakarjan E, Mingozzi F. Cell-mediated immunity to AAV vectors, evolving concepts and potential solutions. Front Immunol. 2014;5:350. doi:10.3389/fimmu.2014.00350.
Louis Jeune V, Joergensen JA, Hajjar RJ, Weber T. Pre-existing anti-adeno-associated virus antibodies as a challenge in AAV gene therapy. Hum Gene Ther Methods. 2013;24(2):59–67. doi:10.1089/hgtb.2012.243.
Sudres M, Cire S, Vasseur V, Brault L, Da Rocha S, Boisgerault F, et al. MyD88 signaling in B cells regulates the production of Th1-dependent antibodies to AAV. Mol Ther. 2012;20(8):1571–81. doi:10.1038/mt.2012.101.
Ferrand M, Da Rocha S, Corre G, Galy A, Boisgerault F. Serotype-specific binding properties and nanoparticle characteristics contribute to the immunogenicity of rAAV1 vectors. Mol Ther. 2015;23(6):1022–33. doi:10.1038/mt.2015.59.
Mays LE, Wilson JM. The complex and evolving story of T cell activation to AAV vector-encoded transgene products. Mol Ther. 2011;19(1):16–27. doi:10.1038/mt.2010.250.
Uhrig S, Coutelle O, Wiehe T, Perabo L, Hallek M, Buning H. Successful target cell transduction of capsid-engineered rAAV vectors requires clathrin-dependent endocytosis. Gene Ther. 2012;19(2):210–8. doi:10.1038/gt.2011.78.
Balakrishnan B, Jayandharan GR. Basic biology of adeno-associated virus (AAV) vectors used in gene therapy. Curr Gene Ther. 2014;14(2):86–100.
Duan D, Yue Y, Yan Z, Yang J, Engelhardt JF. Endosomal processing limits gene transfer to polarized airway epithelia by adeno-associated virus. J Clin Investig. 2000;105(11):1573–87. doi:10.1172/jci8317.
Pien GC, Basner-Tschakarjan E, Hui DJ, Mentlik AN, Finn JD, Hasbrouck NC, et al. Capsid antigen presentation flags human hepatocytes for destruction after transduction by adeno-associated viral vectors. J Clin Investig. 2009;119(6):1688–95. doi:10.1172/jci36891.
Finn JD, Hui D, Downey HD, Dunn D, Pien GC, Mingozzi F, et al. Proteasome inhibitors decrease AAV2 capsid derived peptide epitope presentation on MHC class I following transduction. Mol Ther. 2010;18(1):135–42. doi:10.1038/mt.2009.257.
Nonnenmacher M, Weber T. Intracellular transport of recombinant adeno-associated virus vectors. Gene Ther. 2012;19(6):649–58. doi:10.1038/gt.2012.6.
Rogers GL, Suzuki M, Zolotukhin I, Markusic DM, Morel LM, Lee B, et al. Unique roles of TLR9- and MyD88-dependent and -independent pathways in adaptive immune responses to AAV-mediated gene transfer. J Innate Immun. 2015;7(3):302–14. doi:10.1159/000369273.
Faust SM, Bell P, Cutler BJ, Ashley SN, Zhu Y, Rabinowitz JE, et al. CpG-depleted adeno-associated virus vectors evade immune detection. J Clin Investig. 2013;123(7):2994–3001. doi:10.1172/jci68205.
Greenberg B, Butler J, Felker GM, Ponikowski P, Voors AA, Pogoda JM, et al. Prevalence of AAV1 neutralizing antibodies and consequences for a clinical trial of gene transfer for advanced heart failure. Gene Ther. 2016;23(3):313–9. doi:10.1038/gt.2015.109.
Mingozzi F, High KA. Immune responses to AAV vectors: overcoming barriers to successful gene therapy. Blood. 2013;122(1):23–36. doi:10.1182/blood-2013-01-306647.
Kotin RM. Large-scale recombinant adeno-associated virus production. Hum Mol Genet. 2011;20(R1):R2–6. doi:10.1093/hmg/ddr141.
Clement N, Grieger JC. Manufacturing of recombinant adeno-associated viral vectors for clinical trials. Mol Ther Methods Clin Dev. 2016;3:16002. doi:10.1038/mtm.2016.2.
Wright JF. Manufacturing and characterizing AAV-based vectors for use in clinical studies. Gene Ther. 2008;15(11):840–8. doi:10.1038/gt.2008.65.
Batchu RB, Shammas MA, Wang JY, Munshi NC. Interaction of adeno-associated virus Rep78 with p53: implications in growth inhibition. Can Res. 1999;59(15):3592–5.
Schmidt M, Afione S, Kotin RM. Adeno-associated virus type 2 Rep78 induces apoptosis through caspase activation independently of p53. J Virol. 2000;74(20):9441–50.
Mietzsch M, Hering H, Hammer EM, Agbandje-McKenna M, Zolotukhin S, Heilbronn R. OneBac 2.0: Sf9 cell lines for production of AAV1, AAV2 and AAV8 vectors with minimal encapsidation of foreign DNA. Hum Gene Ther Methods. 2017;28(1):15–22. doi:10.1089/hgtb.2016.164.
Mietzsch M, Grasse S, Zurawski C, Weger S, Bennett A, Agbandje-McKenna M, et al. OneBac: platform for scalable and high-titer production of adeno-associated virus serotype 1–12 vectors for gene therapy. Hum Gene Ther. 2014;25(3):212–22. doi:10.1089/hum.2013.184.
Arden E, Metzger JM. Inexpensive, serotype-independent protocol for native and bioengineered recombinant adeno-associated virus purification. J Biol Methods. 2016;3(2):1–7. doi:10.14440/jbm.2016.102.
Smith RH, Levy JR, Kotin RM. A simplified baculovirus-AAV expression vector system coupled with one-step affinity purification yields high-titer rAAV stocks from insect cells. Mol Ther. 2009;17(11):1888–96. doi:10.1038/mt.2009.128.
Potter M, Lins B, Mietzsch M, Heilbronn R, Van Vliet K, Chipman P, et al. A simplified purification protocol for recombinant adeno-associated virus vectors. Mol Ther Methods Clin Dev. 2014;1:14034. doi:10.1038/mtm.2014.34.
Ayuso E, Mingozzi F, Bosch F. Production, purification and characterization of adeno-associated vectors. Curr Gene Ther. 2010;10(6):423–36.
Tomono T, Hirai Y, Okada H, Adachi K, Ishii A, Shimada T, et al. Ultracentrifugation-free chromatography-mediated large-scale purification of recombinant adeno-associated virus serotype 1 (rAAV1). Mol Ther Methods Clin Dev. 2016;3:15058. doi:10.1038/mtm.2015.58.
Rayaprolu V, Kruse S, Kant R, Venkatakrishnan B, Movahed N, Brooke D, et al. Comparative analysis of adeno-associated virus capsid stability and dynamics. J Virol. 2013;87(24):13150–60. doi:10.1128/jvi.01415-13.
Bennett J, Wellman J, Marshall KA, McCague S, Ashtari M, DiStefano-Pappas J, et al. Safety and durability of effect of contralateral-eye administration of AAV2 gene therapy in patients with childhood-onset blindness caused by RPE65 mutations: a follow-on phase 1 trial. Lancet (Lond, Engl). 2016;388(10045):661–72. doi:10.1016/s0140-6736(16)30371-3.
Testa F, Maguire AM, Rossi S, Pierce EA, Melillo P, Marshall K, et al. Three-year follow-up after unilateral subretinal delivery of adeno-associated virus in patients with Leber congenital Amaurosis type 2. Ophthalmology. 2013;120(6):1283–91. doi:10.1016/j.ophtha.2012.11.048.
Duan D. Systemic delivery of adeno-associated viral vectors. Curr Opin Virol. 2016;21:16–25. doi:10.1016/j.coviro.2016.07.006.
Mingozzi F, High KA. Therapeutic in vivo gene transfer for genetic disease using AAV: progress and challenges. Nat Rev Genet. 2011;12(5):341–55. doi:10.1038/nrg2988.
Nakai H, Montini E, Fuess S, Storm TA, Grompe M, Kay MA. AAV serotype 2 vectors preferentially integrate into active genes in mice. Nat Genet. 2003;34(3):297–302. doi:10.1038/ng1179.
Chandler RJ, Sands MS, Venditti CP. Recombinant adeno-associated viral integration and genotoxicity: insights from animal models. Hum Gene Ther. 2017;28(4):314–22. doi:10.1089/hum.2017.009.
Nault JC, Datta S, Imbeaud S, Franconi A, Mallet M, Couchy G, et al. Recurrent AAV2-related insertional mutagenesis in human hepatocellular carcinomas. Nat Genet. 2015;47(10):1187–93. doi:10.1038/ng.3389.
Salmon F, Grosios K, Petry H. Safety profile of recombinant adeno-associated viral vectors: focus on alipogene tiparvovec (Glybera®). Expert Rev Clin Pharmacol. 2014;7(1):53–65. doi:10.1586/17512433.2014.852065.
Hocquemiller M, Giersch L, Audrain M, Parker S, Cartier N. Adeno-associated virus-based gene therapy for CNS diseases. Hum Gene Ther. 2016;27(7):478–96. doi:10.1089/hum.2016.087.
Ojala DS, Amara DP, Schaffer DV. Adeno-associated virus vectors and neurological gene therapy. Neuroscientist. 2015;21(1):84–98. doi:10.1177/1073858414521870.
Yang B, Li S, Wang H, Guo Y, Gessler DJ, Cao C, et al. Global CNS transduction of adult mice by intravenously delivered rAAVrh.8 and rAAVrh.10 and nonhuman primates by rAAVrh.10. Mol Ther. 2014;22(7):1299–309. doi:10.1038/mt.2014.68.
Gray SJ, Nagabhushan Kalburgi S, McCown TJ, Jude Samulski R. Global CNS gene delivery and evasion of anti-AAV-neutralizing antibodies by intrathecal AAV administration in non-human primates. Gene Ther. 2013;20(4):450–9. doi:10.1038/gt.2012.101.
Wang H, Yang B, Qiu L, Yang C, Kramer J, Su Q, et al. Widespread spinal cord transduction by intrathecal injection of rAAV delivers efficacious RNAi therapy for amyotrophic lateral sclerosis. Hum Mol Genet. 2014;23(3):668–81. doi:10.1093/hmg/ddt454.
Saraiva J, Nobre RJ, Pereira de Almeida L. Gene therapy for the CNS using AAVs: the impact of systemic delivery by AAV9. J Control Release. 2016;241:94–109. doi:10.1016/j.jconrel.2016.09.011.
Bourdenx M, Dutheil N, Bezard E, Dehay B. Systemic gene delivery to the central nervous system using Adeno-associated virus. Front Mol Neurosci. 2014;7:50. doi:10.3389/fnmol.2014.00050.
Selkoe DJ, Hardy J. The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol Med. 2016;8(6):595–608. doi:10.15252/emmm.201606210.
Anand R, Kaushal A, Wani WY, Gill KD. Road to Alzheimer’s disease: the pathomechanism underlying. Pathobiology. 2012;79(2):55–71. doi:10.1159/000332218.
St-Amour I, Cicchetti F, Calon F. Immunotherapies in Alzheimer’s disease: too much, too little, too late or off-target? Acta Neuropathol. 2016;131(4):481–504. doi:10.1007/s00401-015-1518-9.
Shimada M, Abe S, Takahashi T, Shiozaki K, Okuda M, Mizukami H, et al. Prophylaxis and treatment of Alzheimer’s disease by delivery of an adeno-associated virus encoding a monoclonal antibody targeting the amyloid Beta protein. PLoS One. 2013;8(3):e57606. doi:10.1371/journal.pone.0057606.
Fish KM, Ishikawa K. Advances in gene therapy for heart failure. Discov Med. 2015;19(105):285–91.
Greenberg B. Gene therapy for heart failure. J Cardiol. 2015;66(3):195–200. doi:10.1016/j.jjcc.2015.02.006.
Greenberg B, Butler J, Felker GM, Ponikowski P, Voors AA, Desai AS, et al. Calcium upregulation by percutaneous administration of gene therapy in patients with cardiac disease (CUPID 2): a randomised, multinational, double-blind, placebo-controlled, phase 2b trial. Lancet (Lond, Engl). 2016;387(10024):1178–86. doi:10.1016/s0140-6736(16)00082-9.
Griesenbach U, Pytel KM, Alton EW. Cystic fibrosis gene therapy in the UK and elsewhere. Hum Gene Ther. 2015;26(5):266–75. doi:10.1089/hum.2015.027.
Burney TJ, Davies JC. Gene therapy for the treatment of cystic fibrosis. Appl Clin Genet. 2012;5:29–36. doi:10.2147/TACG.S8873.
Schuster BS, Kim AJ, Kays JC, Kanzawa MM, Guggino WB, Boyle MP, et al. Overcoming the cystic fibrosis sputum barrier to leading adeno-associated virus gene therapy vectors. Mol Ther. 2014;22(8):1484–93. doi:10.1038/mt.2014.89.
Limberis MP, Adam VS, Wong G, Gren J, Kobasa D, Ross TM, et al. Intranasal antibody gene transfer in mice and ferrets elicits broad protection against pandemic influenza. Sci Transl Med. 2013;5(187):187ra72. doi:10.1126/scitranslmed.3006299.
Munsell EV, Ross NL, Sullivan MO. Journey to the center of the cell: current nanocarrier design strategies targeting biopharmaceuticals to the cytoplasm and nucleus. Curr Pharm Des. 2016;22(9):1227–44.
Lesher-Perez SC, Segura T, Moraes C. Getting there is half the battle: recent advances in delivering therapeutics. Integr Biol. 2016;8(1):8–11. doi:10.1039/c5ib90052b.
Wu Z, Miller E, Agbandje-McKenna M, Samulski RJ. Alpha 2,3 and alpha2,6 N-linked sialic acids facilitate efficient binding and transduction by adeno-associated virus types 1 and 6. J Virol. 2006;80(18):9093–103. doi:10.1128/jvi.00895-06.
Burger C, Gorbatyuk OS, Velardo MJ, Peden CS, Williams P, Zolotukhin S, et al. Recombinant AAV viral vectors pseudotyped with viral capsids from serotypes 1, 2, and 5 display differential efficiency and cell tropism after delivery to different regions of the central nervous system. Mol Ther. 2004;10(2):302–17. doi:10.1016/j.ymthe.2004.05.024.
Wang C, Wang CM, Clark KR, Sferra TJ. Recombinant AAV serotype 1 transduction efficiency and tropism in the murine brain. Gene Ther. 2003;10(17):1528–34. doi:10.1038/sj.gt.3302011.
Xiao W, Chirmule N, Berta SC, McCullough B, Gao G, Wilson JM. Gene therapy vectors based on adeno-associated virus type 1. J Virol. 1999;73(5):3994–4003.
Summerford C, Samulski RJ. Membrane-associated heparan sulfate proteoglycan is a receptor for adeno-associated virus type 2 virions. J Virol. 1998;72(2):1438–45.
Qing K, Mah C, Hansen J, Zhou S, Dwarki V, Srivastava A. Human fibroblast growth factor receptor 1 is a co-receptor for infection by adeno-associated virus 2. Nat Med. 1999;5(1):71–7. doi:10.1038/4758.
Akache B, Grimm D, Pandey K, Yant SR, Xu H, Kay MA. The 37/67-kilodalton laminin receptor is a receptor for adeno-associated virus serotypes 8, 2, 3, and 9. J Virol. 2006;80(19):9831–6. doi:10.1128/jvi.00878-06.
Kashiwakura Y, Tamayose K, Iwabuchi K, Hirai Y, Shimada T, Matsumoto K, et al. Hepatocyte growth factor receptor is a coreceptor for adeno-associated virus type 2 infection. J Virol. 2005;79(1):609–14. doi:10.1128/jvi.79.1.609-614.2005.
Asokan A, Hamra JB, Govindasamy L, Agbandje-McKenna M, Samulski RJ. Adeno-associated virus type 2 contains an integrin alpha5beta1 binding domain essential for viral cell entry. J Virol. 2006;80(18):8961–9. doi:10.1128/jvi.00843-06.
Walters RW, Yi SM, Keshavjee S, Brown KE, Welsh MJ, Chiorini JA, et al. Binding of adeno-associated virus type 5 to 2,3-linked sialic acid is required for gene transfer. J Biol Chem. 2001;276(23):20610–6. doi:10.1074/jbc.M101559200.
Walters RW, Agbandje-McKenna M, Bowman VD, Moninger TO, Olson NH, Seiler M, et al. Structure of adeno-associated virus serotype 5. J Virol. 2004;78(7):3361–71.
Di Pasquale G, Davidson BL, Stein CS, Martins I, Scudiero D, Monks A, et al. Identification of PDGFR as a receptor for AAV-5 transduction. Nat Med. 2003;9(10):1306–12. doi:10.1038/nm929.
Davidson BL, Stein CS, Heth JA, Martins I, Kotin RM, Derksen TA, et al. Recombinant adeno-associated virus type 2, 4, and 5 vectors: transduction of variant cell types and regions in the mammalian central nervous system. Proc Natl Acad Sci USA. 2000;97(7):3428–32. doi:10.1073/pnas.050581197.
Weller ML, Amornphimoltham P, Schmidt M, Wilson PA, Gutkind JS, Chiorini JA. Epidermal growth factor receptor is a co-receptor for adeno-associated virus serotype 6. Nat Med. 2010;16(6):662–4. doi:10.1038/nm.2145.
Ng R, Govindasamy L, Gurda BL, McKenna R, Kozyreva OG, Samulski RJ, et al. Structural characterization of the dual glycan binding adeno-associated virus serotype 6. J Virol. 2010;84(24):12945–57. doi:10.1128/jvi.01235-10.
Blankinship MJ, Gregorevic P, Allen JM, Harper SQ, Harper H, Halbert CL, et al. Efficient transduction of skeletal muscle using vectors based on adeno-associated virus serotype 6. Mol Ther. 2004;10(4):671–8. doi:10.1016/j.ymthe.2004.07.016.
Nam HJ, Lane MD, Padron E, Gurda B, McKenna R, Kohlbrenner E, et al. Structure of adeno-associated virus serotype 8, a gene therapy vector. J Virol. 2007;81(22):12260–71. doi:10.1128/jvi.01304-07.
Nakai H, Fuess S, Storm TA, Muramatsu S, Nara Y, Kay MA. Unrestricted hepatocyte transduction with adeno-associated virus serotype 8 vectors in mice. J Virol. 2005;79(1):214–24. doi:10.1128/jvi.79.1.214-224.2005.
Gao G, Vandenberghe LH, Alvira MR, Lu Y, Calcedo R, Zhou X, et al. Clades of Adeno-associated viruses are widely disseminated in human tissues. J Virol. 2004;78(12):6381–8. doi:10.1128/JVI.78.12.6381-6388.2004.
Pan X, Yue Y, Zhang K, Lostal W, Shin JH, Duan D. Long-term robust myocardial transduction of the dog heart from a peripheral vein by adeno-associated virus serotype-8. Hum Gene Ther. 2013;24(6):584–94.
Wang Z, Zhu T, Qiao C, Zhou L, Wang B, Zhang J, et al. Adeno-associated virus serotype 8 efficiently delivers genes to muscle and heart. Nat Biotechnol. 2005;23(3):321–8. doi:10.1038/nbt1073.
Loiler SA, Tang Q, Clarke T, Campbell-Thompson ML, Chiodo V, Hauswirth W, et al. Localized gene expression following administration of adeno-associated viral vectors via pancreatic ducts. Mol Ther. 2005;12(3):519–27. doi:10.1016/j.ymthe.2005.04.017.
Wang AY, Peng PD, Ehrhardt A, Storm TA, Kay MA. Comparison of adenoviral and adeno-associated viral vectors for pancreatic gene delivery in vivo. Hum Gene Ther. 2004;15(4):405–13. doi:10.1089/104303404322959551.
Shen S, Bryant KD, Brown SM, Randell SH, Asokan A. Terminal N-linked galactose is the primary receptor for adeno-associated virus 9. J Biol Chem. 2011;286(15):13532–40. doi:10.1074/jbc.M110.210922.
Wright JF. Transient transfection methods for clinical adeno-associated viral vector production. Hum Gene Ther. 2009;20(7):698–706. doi:10.1089/hum.2009.064.
Lock M, Alvira M, Vandenberghe LH, Samanta A, Toelen J, Debyser Z, et al. Rapid, simple, and versatile manufacturing of recombinant adeno-associated viral vectors at scale. Hum Gene Ther. 2010;21(10):1259–71. doi:10.1089/hum.2010.055.
Grieger JC, Soltys SM, Samulski RJ. Production of recombinant adeno-associated virus vectors using suspension HEK293 cells and continuous harvest of vector from the culture media for GMP FIX and FLT1 clinical vector. Mol Ther. 2016;24(2):287–97. doi:10.1038/mt.2015.187.
Aslanidi G, Lamb K, Zolotukhin S. An inducible system for highly efficient production of recombinant adeno-associated virus (rAAV) vectors in insect Sf9 cells. Proc Natl Acad Sci USA. 2009;106(13):5059–64. doi:10.1073/pnas.0810614106.
Cecchini S, Virag T, Kotin RM. Reproducible high yields of recombinant adeno-associated virus produced using invertebrate cells in 0.02- to 200-liter cultures. Hum Gene Ther. 2011;22(8):1021–30. doi:10.1089/hum.2010.250.
Thomas DL, Wang L, Niamke J, Liu J, Kang W, Scotti MM, et al. Scalable recombinant adeno-associated virus production using recombinant herpes simplex virus type 1 coinfection of suspension-adapted mammalian cells. Hum Gene Ther. 2009;20(8):861–70. doi:10.1089/hum.2009.004.
Clement N, Knop DR, Byrne BJ. Large-scale adeno-associated viral vector production using a herpesvirus-based system enables manufacturing for clinical studies. Hum Gene Ther. 2009;20(8):796–806. doi:10.1089/hum.2009.094.
Thorne BA, Takeya RK, Peluso RW. Manufacturing recombinant adeno-associated viral vectors from producer cell clones. Hum Gene Ther. 2009;20(7):707–14. doi:10.1089/hum.2009.070.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflicts of interest
Michael F. Naso, Brian Tomkowicz, and William L. Perry III are employees of Janssen Research and Development. William R. Strohl has no conflicts of interest to declare.
Funding
No funding was received for the preparation of this review.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution-NonCommercial 4.0 International License (http://creativecommons.org/licenses/by-nc/4.0/), which permits any noncommercial use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Naso, M.F., Tomkowicz, B., Perry, W.L. et al. Adeno-Associated Virus (AAV) as a Vector for Gene Therapy. BioDrugs 31, 317–334 (2017). https://doi.org/10.1007/s40259-017-0234-5
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40259-017-0234-5