
Abstract
Lupus erythematosus comprises a spectrum of autoimmune diseases that may affect various organs (systemic lupus erythematosus [SLE]) or the skin only (cutaneous lupus erythematosus [CLE]). Typical combinations of clinical, histological and serological findings define clinical subtypes of CLE, yet there is high interindividual variation. Skin lesions arise in the course of triggers such as ultraviolet (UV) light exposure, smoking or drugs; keratinocytes, cytotoxic T cells and plasmacytoid dendritic cells (pDCs) establish a self-perpetuating interplay between the innate and adaptive immune system that is pivotal for the pathogenesis of CLE. Therefore, treatment relies on avoidance of triggers and UV protection, topical therapies (glucocorticosteroids, calcineurin inhibitors) and rather unspecific immunosuppressive or immunomodulatory drugs. Yet, the advent of licensed targeted therapies for SLE might also open new perspectives in the management of CLE. The heterogeneity of CLE might be attributable to individual variables and we speculate that the prevailing inflammatory signature defined by either T cells, B cells, pDCs, a strong lesional type I interferon (IFN) response, or combinations of the above might be suitable to predict therapeutic response to targeted treatment. Therefore, pretherapeutic histological assessment of the inflammatory infiltrate could stratify patients with refractory CLE for T-cell-directed therapies (e.g. dapirolizumab pegol), B-cell-directed therapies (e.g. belimumab), pDC-directed therapies (e.g. litifilimab) or IFN-directed therapies (e.g. anifrolumab). Moreover, Janus kinase (JAK) and spleen tyrosine kinase (SYK) inhibitors might broaden the therapeutic armamentarium in the near future. A close interdisciplinary exchange with rheumatologists and nephrologists is mandatory for optimal treatment of lupus patients to define the best therapeutic strategy.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Cutaneous lupus erythematosus comprises a variety of clinical manifestations and displays interindividual heterogeneity. |
Keratinocytes, cytotoxic T lymphocytes and plasmacytoid dendritic cells establish a self-perpetuating interplay between the innate and adaptive immune system in lesional skin, which is orchestrated by type I interferons (IFNs). |
Lesional cutaneous B cells are emerging disease modulators with potential functions in fine-tuning autoreactive T cells and antigen-presenting cells. |
Targeted therapeutic advances could be most effective if chosen based on individual inflammatory phenotypes with regard to abundance of B cells, plasmacytoid dendritic cells, a strong type I IFN signature, or combinations of the above. |
1 Introduction
Lupus erythematosus comprises a spectrum of autoimmune diseases that may affect various organs (systemic lupus erythematosus [SLE]) or the skin only (cutaneous lupus erythematosus [CLE]). The latter has traditionally been grouped from a clinical perspective with regard to the onset and course of symptoms, into acute, subacute, intermittent and chronic CLE (Table 1) [1]. Typical combinations of clinical, histological and serological findings define these subtypes (Fig. 1), yet there is high interindividual variation. Disease manifestations with more acute onset (malar rash, exanthematic CLE, and bullous LE) segregate from chronic cutaneous manifestations (e.g. discoid CLE), with a declining tendency for systemic disease and detectable circulating antibodies. Excluding organ involvement is important in all newly diagnosed CLE patients [2] and skin symptoms are a commonly expected finding in SLE patients. Unspecific skin findings and vasculopathic reactions such as livedo racemosa, Raynaud phenomenon, and urticarial vasculitis may accompany any type of CLE but are more frequently seen in patients with SLE [3]. Alopecia is also a common finding and discoid scarring lesions must be separated from unspecific diffuse non-scarring alopecia [4]. Overlap between CLE and other cutaneous inflammatory diseases is possible. For example, an overlap between CLE and lichen planus (‘lichenoid CLE) harboring clinical and histological features of both diseases with or without serologic markers is mentioned repeatedly in the literature, which may cause diagnostic confusion [5]. Progression from CLE to SLE may occur but the reported numbers vary greatly in the literature between 5 and 25%, which largely depends on the patient characteristics and clinical risk factors in the study populations [6]. Furthermore, different clinical subtypes of CLE are accompanied with varying risk for development of systemic disease. In a Swedish association study for example, progression from initially isolated skin disease to systemic disease occurred in up to 18% of patients, most commonly in patients with subacute CLE [7]. In summary, risk for development of systemic disease is highest in patients presenting with acute CLE (ACLE), and lowest in chronic discoid LE (CDLE) [1]. Noteworthy, sociocultural factors (ethnicity, sex, social income, education level) are increasingly recognized to have a major impact on the severity of disease, which is comparable with other chronic inflammatory dermatoses such as psoriasis and hidradenitis suppurativa [8]. The overall incidence of SLE and CLE is comparable and female patients far outnumber males in both groups.

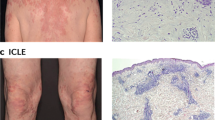
Overview of cutaneous lupus erythematosus subtypes and schematic illustration of pathogenic mechanisms in CLE. (a) Clinical classification of CLE based on chronicity of lesions. The most common subgroups represent ACLE, SCLE, ICLE, and CCLE, of which CDLE is the most common form. (b) Concept of inflammatory signature-oriented, therapy-directed histological evaluation. Pathophysiologically, CLE is understood to be an IFN-driven autoimmune skin disease characterized by cytotoxic lesional inflammation with activation of primarily TLR-independent innate immune pathways as depicted. With a corresponding genetic background, trigger factors lead to cell stress and subsequently apoptosis. Due to several defective mechanisms (reduced phagocytosis, DNAse/TREX1 defects, IFN hyperactivation), necroptosis occurs, leading to an inflammatory response with release of DAMPs (e.g. endogenous nucleic acids, HMGB1), autoantigens such as Ro52, and cytokines such as CXCL chemokines, ILs, and IFNs. DCs sense potential autoantigens and present them to lymphocytes in nearby lymph nodes. This leads to lymphocytic differentiation with subsequent cytotoxic effector functions against keratinocytes, as well as production of autoantibodies. Recruited pDCs are stimulated to express type I and type III IFN after recognition of released nucleic acids, thus amplifying lesional inflammation. Histologically, different inflammatory signatures can be assessed in the lesional tissue of CLE patients, e.g. a B-cell- or pDC-rich infiltrate or a strong IFN signature. Targeted therapeutic options are directed against different inflammatory cytokines and their receptors or intracellular targets. T lymphocytes: (A) S1P receptor 1 antagonists (amiselimod); (B) CD40L antagonists (dapirolizumab pegol, frexalimab). B lymphocytes: (C) BAFF receptor antagonists such as the monoclonal antibody belimumab; (D) CD20 antibodies, e.g. rituximab; (E) fusion proteins that bind to BAFF and TACI (telitacicept); (F) the cereblon E3 ligation modulator iberdomide. pDCs: (G) BDCA2 inhibition (litifilimab); (H) anti-LILRA4 antibody daxdilimab. IFN-associated pathways: (I) IFNAR1 inhibition (anifrolumab); (K) JAK inhibition (e.g. filgotinib, tofacitinib, delgocitinib), TYK2 inhibition (deucravacitinib). Other intracellular pathways: (L) SYK inhibition (lanraplenib). CLE cutaneous lupus erythematosus, ACLE acute CLE, SCLE subacute CLE, ICLE intermittent CLE, CCLE chronic CLE, CDLE chronic discoid LE, IFN interferon, TLR toll-like receptor, DAMPs danger-associated molecular patterns, ILs interleukins, pDCs plasmacytoid dendritic cells, SIP spingosine-1-phosphate, BAFF B-cell-activating factor, TACI transmembrane activator and CAML interactor, BDCA2 blood dendritic cell antigen 2, LILRA4 leukocyte immunoglobulin-like receptor subfamily A member 4, IFNAR1 interferon-α/β receptor α chain, JAK Janus kinase, TYK tyrosine kinase, SYK spleen tyrosine kinase, HMGB1 high mobility group box 1, DCs dendritic cells, CD40L CD40 ligand, pDCs plasmacytoid dendritic cells
With emerging understanding of the pathophysiology of CLE and the underlying interplay between the innate and adaptive immune systems, the advent and licensing of targeted therapies for the treatment of SLE raises questions about a potential use of these drugs in patients suffering from disease limited to the skin [9]. This review provides an update on the most recent pathophysiological findings with a focus on immunology in CLE. We also address diagnostic and therapeutic challenges due to the complexity of the disease and speculate on future therapeutic directions.
2 Pathogenesis
The pathogenesis of CLE is complex and has been widely studied [10]. Briefly summarized, in all clinical subtypes of CLE, a self-amplifying inflammatory loop is established between cells of both the innate and adaptive immune system (Fig. 1) [11]. Recruitment of these cells occurs in the course of keratinocytic cell death due to environmental triggers such as ultraviolet (UV) radiation and drugs. Several studies suggest smoking is a risk factor [12, 13]. Release of cytosolic and nuclear debris into the extracellular space typically leads to activation of danger-associated receptors, which then sparks the recruitment of specific inflammatory cells. Central key to the pathogenesis of LE is overexpression of interferons (IFNs), which leads to an inflammatory loop mimicking an antiviral response [14]. Yet, only susceptible individuals develop disease manifestations, largely depending on genetics, epigenetics and other variables such as hormones, skin, and gut microbiome [15]. Thus far, it is not clear why given patients develop a specific clinical phenotype.
Over the last years, there has been emerging evidence that apart from autoreactive T cells and plasmacytoid dendritic cells (pDCs), B cells could also play a major role in the orchestration of the inflammatory response. In this review we focus on immunology while omitting environmental pathogenetic aspects such as (passive) smoking and UV exposure, as these aspects have been extensively reviewed elsewhere [1, 16]. Of note, new onset and exacerbation of autoimmune disease, including CLE, was described in the context of coronavirus disease 2019 (COVID-19) infection and vaccination, which is still of importance in light of the ongoing pandemic [17]. Histopathology is also out of the scope of this article and detailed information can be found in another review by our group [18].
2.1 Genetics and Epigenetics
Only a small portion of lupus patients harbor a genetic variant of CLE. Namely, a monogenetic mutation in the TREX1 gene, which encodes an enzyme responsible for cytosolic degradation of deoxyribonucleic acid (DNA) [19], may be detected in these patients. Because of DNase deficiency, high levels of cytosolic DNA accumulate and are sensed by specific receptors, which ultimately activates the type I IFN system. Typical symptoms include recurrent swelling and pernio-like nodules in acral areas. This disease manifestation is known as familial chilblain LE. Over the last years, a growing list of gene mutations capable of triggering lupus-like skin lesions was identified [19]. In particular, gene mutations in SAMHD1 and complement factor C2 raise the individual risk to develop CLE lesions [20]. The vast majority of CLE patients bear no specific genetic mutations; however, there is an abundance of associated gene polymorphisms associated with a higher risk of CLE. The involved genes encode for proteins involved in cell death cascades (apoptosis, ubiquitination), clearance of cell debris (e.g. immune complexes), cellular adhesion and activation or regulation of the immune system (innate immune system activation, B-cell/T-cell function) [20].
2.2 Pathophysiology and Immunology
As non-inflammatory cells, keratinocytes contribute to lesional inflammation in CLE. An initial trigger, such as UV radiation, smoking or drugs causes keratinocyte apoptosis. UV radiation leads to an upregulation of autoantigens, such as Ro52, in keratinocytes, inducing and activating the proinflammatory pathways [15, 21]. Apoptotic keratinocytes present antigens, which can be recognized by autoantibodies in autoantibody-positive patients [22]. Autoantibodies against ribonucleoproteins could also have an independent pathophysiological role, as they trigger the development of lupus lesions in mice [23]. Interestingly, UV radiation or other damaging triggers initially lead to keratinocytic cell death and chemokine production in the whole epidermal layer [24], however, later in established CLE lesions, keratinocytic apoptosis and proinflammatory chemokine production is limited to the dermoepidermal junction, resulting in interface dermatitis [25]. Even keratinocytes from uninvolved (non-lesional) skin of CLE patients are more sensitive to UV radiation-induced cytotoxicity compared with keratinocytes from healthy donors, which leads to the assumption of disease predisposition [26, 27]. Following the initial keratinocyte damage, secondary necroptosis of keratinocytes further sparks lesional release of nucleic acids and danger-associated molecular patterns (DAMPs). The latter include high mobility group box 1 protein (HMGB1), a proinflammatory cytokine, which can also function as autoantibody in CLE [28]. UV radiation also leads to DNA damage, generating immune-stimulatory DNA motifs, such as 8-hydroxyguanosine [29]. Phagocytic clearance of apoptotic cells and nucleic acids can be impaired in CLE [27]. Nucleic acids are recognized via pattern recognition receptors (PRRs), including MDA5, RIG-I and cGAS–STING, expressed by keratinocytes leading to production of IFN-regulated genes [29]. The response in keratinocytes is toll-like receptor (TLR)-independent [1]. Keratinocytes produce IFNκ and IFNλ (type I and type III IFNs), which, by autocrine secretion, further induce the keratinocytic production of IFN-regulated proinflammatory cytokines, including interleukin (IL)-6, and chemokines, including CXCL9, CXCL10 and CXCL11, which are CXCR3 ligands [29,30,31]. The IFN response is, among others, mediated via Janus kinase (JAK)-signal transducer and activator of transcription (STAT) signaling [30]. The aforementioned chemokines interact with (autoreactive) cytotoxic T cells via CXCR3-binding, also promoting keratinocyte cell death by recruitment of cytotoxic T cells [32, 33]. Nucleic acid motifs also activate the inflammasome via melanoma 2 (AIM2) [34]. Interestingly, IFN-k is upregulated and basal phospho-STAT (pSTAT) activity is higher even in healthy-appearing skin of CLE patients compared with skin of patients with other chronic inflammatory skin disease (psoriasis) [30]. As outlined, the inflammatory interplay is complex and numerous inflammatory cells contribute to CLE pathology. Therefore, in the following sections, we outline recent findings considering specific types of cells in the orchestration of inflammation in SLE and CLE.
2.2.1 Dendritic Cells
After initial keratinocyte cell death, antigen-presenting cells (APCs) sense accumulating nucleic acids, among them dendritic cells (DCs) and pDCs. pDCs are recruited to the skin lesions via CXCL-chemokine interaction with CXCR3 [24]. PDCs sense nucleic acids mostly via TLRs, especially TLR7 and TLR9 [35]. Uptake of nucleic acids and immunecomplexes may be achieved by endocytosis via TLR9 and cluster of differentiation (CD) 32, at least in SLE [36]. Upon PRR activation, pDCs produce large amounts of type I and type III IFNs, cytokines and ILs, further orchestrating the autoimmune circle [37, 38]. The presence of type I IFN is necessary for pDC maturation and migration [39].
PDC infiltrates are observed in a great proportion of skin biopsies and can form clusters in CLE skin lesions [38, 40]; however, not all skin lesions harbor a pDC infiltrate [41]. Recently, single-cell ribonucleic acid (RNA) and spatial RNA sequencing has shown that even healthy-appearing skin of CLE patients contains a type I IFN-rich environment and that CD16+ DCs undergo IFN priming in the skin, leading to proinflammatory subtypes [42]. Because of their central role in CLE pathophysiology, pDCs are an attractive therapeutic target. One potential targeted pDC therapy is the blood DC antigen 2 (BDCA2) receptor, which is exclusively expressed on pDCs [43]. BDCA2 suppresses IFN induction [44].
2.2.2 T cells
Lesional inflammatory infiltrates mainly consist of T cells, B cells, DCs, natural killer (NK) cells, and, infrequently, neutrophils [25, 45]. CXCR3-expressing T cells are recruited into skin lesions via CXCL10. Physiologically, T cells recognize antigens presented by APCs via T-cell receptor (TCR)/major histocompatibility complex (MHC) interaction. Upon TCR engagement, downstream signaling pathways are activated, leading to various T-cell functions. T cells have a lower threshold of activation in lupus patients [46]. Due to defective CD3 chains, the spleen tyrosine kinase (SYK) and Fc receptor γ-chain (FcRγ) association results in higher phosphorylation of signaling molecules and an enhanced calcium influx, leading to enhanced TCR downstream signaling [47]. Additionally, transcription factors lead to differential expression of numerous genes, including the CD40 ligand (CD40L) [48], a co-stimulatory molecule engaged in B-cell interaction, promoting B-cell functions such as proliferation, differentiation, antibody production, and class switching [46]. Increased CD40L does not only have an impact on B cells interacting with T cells but also on APCs. It leads to increased expression of co-stimulatory receptors on APCs, further intensifying the TCR signal [49]. Several different pathways have been described as defective (such as cyclic adenosine monophosphate-dependent phosphorylation, protein kinase C) or increased (such as phosphatidylinositol-3 kinase (PI3K) [46]. Apart from altered pathway signaling, lupus T cells display differential DNA methylation of several genes, leading to differential gene expression [46]. Furthermore, SLE patients show an IL-2 deficiency [50]. IL-2 is important for T-cell polarization, and decreased IL-2 expression enhances inflammatory T-helper 17 (Th17) cell formation [51].
Upon activation, cytotoxic T cells target keratinocytes of the basal epidermal layer, histologically resulting in interface dermatitis [52]. However, this applies for CLE subtypes with superficial involvement and plays a minor part in dermal or subcutaneous CLE subtypes such as LE profundus. Cytotoxic markers such as granzyme B expressed by CD8+ T cells are present in CLE skin lesions and are likely to be induced by IFN [53, 54]. Interestingly, granzyme B expression is higher in scarring lesions of CDLE compared with non-scarring lesions of subacute CLE, suggesting a pathophysiologic role in scarring lesions in CLE [54].
Initiation of cutaneous inflammation is likely to be triggered by Th2 cells, but fully established lesions shift to a Th1-dominated inflammation [52, 55]). Th1 cells stimulate type I IFN production of cytotoxic T cells and macrophages [52, 56]; not only cytotoxic T cells are responsible for keratinocytic apoptosis. Lesional CD4+ T cells can directly induce keratinocytic apoptosis via FAS/FAS ligand (FAS-L) interaction [57]. T-helper cells produce IL-21, inducing the expression of granzyme B in pDCs, promoting NK cells to attack keratinocytes [58, 59]. On the other hand, type I IFNs negatively regulate granzyme B production by pDCs [58]. Th cells can react to nucleosomes released from dying cells and induce (anti-DNA-)antibody production of B cells in SLE [60,61,62]. Th clones in lupus produce IL-2, IFNγ, and IL-4 [63], and CD4+ T cells overexpress perforin, which is epigenetically regulated via DNA methylation [64].
The number of CD4+, CD8+ regulatory, and γδ-T cells is significantly reduced in CLE compared with other inflammatory skin diseases or healthy individuals, and impairment of regulatory immunosuppressive function contributes to the autoimmune circle [46, 65, 66]. Furthermore, emerging evidence points out that composition of the inflammatory infiltrate differs among CLE subtypes. CD4+ T cells and FOXP3+ T cells are significantly reduced in skin lesions of patients with subacute CLE compared with CDLE, as is the CD4/CD8 ratio [67].
2.2.3 B Cells and Plasma cells
B cells harbor a central role in LE pathogenesis by the production of autoantibodies against nuclear components and their complex interplay with T cells [18, 68,69,70]. The capacity of B cells to produce antibodies is enhanced by different IFNs, however, prolonged type I IFN exposure drives autoantibody production [71]. A new mouse model established the role of IL-21 and TLR7/9 in the context of B-cell recruitment to inflammation sites in CLE lesions and localized antibody production [72]. IL-17 recruits immune cells and augments antibody production of B cells in SLE [73]. SLE patients frequently present with antinuclear antibodies (ANAs) but only a minority of CLE patients display detectable autoantibody levels in the serum [74]. Similarly to SLE, autoantibodies against ribonucleoproteins (anti-Ro antibodies) and La are frequently found in SCLE but fewer in CDLE [75]. Different studies reported a specificity of autoantibody presence and CLE subtype [76]. The presence of antibodies are in accordance with the HLA-DR3 phenotype in SLE [74], and the presence of different antibodies (e.g. Ro or LA) are associated with disease severity in SLE [77]. IL-17 recruits immune cells and augments antibody production of B cells in SLE [73]. Besides autoantibody production, B-cell migration, receptor engagement, antigen presentation, cytokine responsiveness and production, survival, differentiation and class-switching are IFN-dependent [71].
Recently, the understanding of the pathophysiological role of B cells in LE has shifted, since strong B-cell signatures and lesional B-cell infiltrates have been described in patients with autoantibody-negative CLE [41, 78]. Beside antibody production, B cells can contribute to the autoimmune reaction by different mechanisms. For example, emerging evidence points towards an antigen-presenting, T-cell activating function of B cells [41]. Lesional B-cell infiltration varies among LE subtypes [41, 79]. B cells can form clusters and arrange in lymphoid-like structures in the skin, called tertiary lymphoid organs/structures (TLO). In different subtypes of CLE, the formation of dense B-cell clusters or TLOs has been described, e.g. in LE profundus or CDLE [41, 80]. TLOs are highly organized structures containing T and B cells, contributing to autoimmunity [81], and have been described in detail in lupus nephritis [82, 83]. B cells can harvest a regulatory function, as is described for SLE [84], and can interact with keratinocytes via B-cell-activating factor (BAFF/Blys) and its receptor in both SLE and CLE, whereby BAFF is expressed by lesional keratinocytes and the associated receptors (BAFF-receptor [BAFF-r], transmembrane activator and CAML interactor [TACI], B-cell maturation antigen [BCMA]) by B cells [41, 85,86,87]. BAFF is a membrane-bound or soluble factor necessary for B-cell maturation [88]. BAFF expression in keratinocytes can be induced by immunostimulatory DNA motifs, highlighting its significance in CLE [86]. B cells produce high levels of cytokines such as IL-6, which in turn is important for B-cell survival [18, 89].
During the maturation process, B cells exhibit immunoglobulin class switching and somatic hypermutation to differentiate into antibody-secreting plasma cells; those processes can occur either in germinal centers or extrafollicular locations, and both have been described in SLE [90]. Somatic hypermutation and isotype switching are dependent on CD40 and IL-21 [18]. Plasma cell differentiation is supported by Th cells [90]. After activation of naïve B cells, plasma cells are generated and persistently produce antibodies while receiving survival signals, mediated by the BAFF axis and IL-6, originating from adjacent cells [81]. Plasma cells can reside and accumulate at the site of inflammation [91]. Even in the absence of antigens, plasma cells can produce antibodies, as they receive survival signals via BAFF or IL-6 [18]. Similar to naïve B cells, plasma cells are responsive to IFN [92]. Different plasma cell subsets have been described to secrete autoantibodies against different structures in SLE [93, 94].
2.2.4 Natural Killer Cells
In SLE, peripheral NK cell levels show an inverse correlation with disease activity [76]. Lupus NK cells secrete higher IFN levels compared with healthy controls, and cytotoxic functions are impaired [95, 96]. NK cells are enriched at lesional inflammation and are able to proliferate in CLE skin lesions [97]. The definitive role of NK cells remains unclear regarding CLE pathophysiology.
2.2.5 Neutrophil Granulocytes
Neutrophil granulocytes are early responders in the course of tissue damage. Neutrophils produce antimicrobial peptides (AMPs; e.g. LL-37) and reactive oxygen species (ROS) [98], and establish neutrophil extracellular traps (NETS), which are nets consisting of chromatin, histones and other intracellular content [99, 100]. The process of NET formation, ‘NETosis’, can be a source of immunogenic materials, and along with release of AMPs, has been linked to autoimmunity. A recent study showed a high molecular heterogeneity in pathogenic neutrophil subsets in SLE (so-called low-density granulocytes [LGS]) with, among others, differences in NET formation and response to type I IFNs, displaying a high number of IFN-induced genes [101]. LGS have been associated with increased vascular inflammation and arterial dysfunction in SLE [102]. Not only are neutrophils prone to NETosis, but also impaired degradation of NETs due to enzymatic blockade or antibody formation can promote SLE activity [103].
Upon NET formation and by release of other immune cells, complexes formed by double-stranded DNA (dsDNA) and LL-37 are taken up by pDCs via endocytosis and are recognized via TLR9, leading to activation and type I IFN production in SLE [104]. LL-37/dsDNA complexes can serve as autoantigens [98]. Higher levels of LL-37 and other AMPs have also been observed in CLE skin lesions, as well as skin lesions from SLE patients compared with healthy controls [105, 106]. Furthermore, NETs are present in several CLE subtypes (panniculitis, ACLE, DLE) [107]; however, it has yet to be elucidated if, and to what extent, those subsets of neutrophils and AMPs play a pathophysiologic role in CLE.
2.2.6 Macrophages
Monocytes and macrophages hold different biological functions, such as phagocytosis or cytokine production [108]. Monocytes exert antigen-presenting properties in SLE [109]. Contrary results have been published on whether differing monocyte and macrophage numbers in lupus patients versus healthy controls are found. Several studies describe an impairment in uptake of apoptotic material and prolonged phagocytosis, leading to accumulation of potential autoantigens and further immune stimulation lupus [110, 111]. One study reported an impaired phagocytosis capacity of macrophages from SLE patients only in the presence of patients’ serum [111]. Macrophages from SLE patients have an impaired adhesion capacity [110]. Furthermore, macrophages can be classified into M1 macrophages, which harbor inflammatory and destructive properties and are induced by IFN, while M2 macrophages, which harbor regulatory properties, are involved in tissue repair and are induced by IL-4 or IL-13 [112]. In SLE, polarization tends towards M1 macrophages, as M1 genes (e.g. STAT1 and SOCS3) were among differentially expressed genes in monocytes from SLE patients [113, 114]. In a mouse model, adoptive M2 macrophage transfer led to decreased SLE severity [115].
One study found FAS-L-expressing macrophages enriched around hair follicles in CLE patients, potentially being responsible for lupus-associated scarring alopecia via a direct FAS/FAS-L interaction with keratinocytes of hair follicles [57]. More detail about the potential functions of macrophages in SLE is outlined elsewhere [116]. Of note, a recent study suggested a pathophysiologic, inflammatory role for the microRNA (miRNA) miR-4512 in monocytes and macrophages in SLE via the TLR4-CXCL2 axis [117].
3 Treatment and Future Directions
Based on the aforementioned pathophysiology of CLE, treatment mainly relies on the avoidance of typical triggers and dampening of the effects of key immunologic reactions. We briefly recapitulate commonly used substances and reflect on developments that are more recent.
3.1 Conventional Treatments
A variety of immunosuppressive and immunomodulatory drugs are in use for CLE (see Table 2 for an overview). There is a striking lack of licensed drugs both in Europe and the United States (US). Being a chronic-relapsing inflammatory skin disease, long-term remissions are rare in CLE and many patients require continued treatment. In a longitudinal cohort study, factors associated with a lower chance of long-term remission were smoking and discoid CLE [118].
3.1.1 Topical Treatment
Optimal broad-spectrum sunscreen is mandatory for all patients. Topical corticosteroids (TCS), especially more potent agents such as fluocinonide, are the first-line options for circumscribed CLE [119]. Intralesional application with triamcinolone suspension may also be suitable in specific localizations such as the scalp. However, use is limited by typical adverse effects (skin atrophy) and lack of efficiency in widespread disease. Topical calcineurin inhibitors (TCI; pimecrolimus, tacrolimus) may be used (off-label) for sustaining remissions, especially in facial lesions, but they often fail to control flares of the disease. The response varies among clinical subtypes. Some authors argue for off-label use of topical retinoids (tazarotene, tretinoin) in hypertrophic lesions [119]. Topical treatment is also a mainstay as adjunct to systemic treatments [120].
3.1.2 Systemic Treatment
In widespread CLE or circumscribed CLE irresponsive to topical treatment, a variety of systemic agents are in use, mostly with a low level of evidence. Recommended first-line treatments for active or widespread lesions are systemic glucocorticosteroids (e.g. prednisolone, dexamethasone) that typically exert a broad immunosuppressive effect, and antimalarials with an immunomodulatory effect. Both groups of drugs have been available for decades and are licensed for this indication. Among the antimalarials, hydroxychloroquine (HCQ) is generally favored above chloroquine (CQ) due to a more favorable adverse effect profile. Mepacrine may be used if HCQ and CQ are not well tolerated or if monotherapy fails to achieve disease control. The exact mode of action of antimalarials in CLE is still not exactly defined, yet dampening of TLRs and inhibition of the production of type I IFNs have been described [1]. A recent study found variability in the therapeutic response to antimalarials defined by alternating immune profiles of responders and non-responders [121]. Predictive biomarkers for therapeutic response are missing. To prevent osteoporosis in the course of sun protection and glucocorticosteroid use, vitamin D supplementation should be incorporated in the treatment plan [122]. Second- and third-line treatments according to the German [119, 123] and British [124] national guidelines are summarized in Table 2. Drugs most commonly used include methotrexate (MTX), dapsone, and mycophenolate mofetil (MMF) [125], whereas immunosuppressive agents in use for SLE [126], such as azathioprine (AZA), are not recommended for the management of CLE. In a recent retrospective analysis of CLE patients irresponsive to antimalarials, MTX and MMF showed similar therapeutic efficiency in different clinical subgroups [127]. Dermatologic peculiarities include the use of acitretin for hypertrophic CLE lesions and the use of thalidomide and lenalidomide for severe or refractory cases. Acitretin was found to be an effective treatment option in a recent prospective, open-label, uncontrolled study with an acceptable safety profile [128]. The use of lenalidomide is supported by retrospective observational studies [129] and case series [130]; however, strict contraceptive measures must be taken into account, and neurotoxicity limits its use. All of the aforementioned immunosuppressive or immunomodulatory drugs have various effects on the pathophysiology of CLE but none of them act in a targeted manner. The current state of CLE treatment is concisely summarized in previous works that deserve mentioning [131, 132].
3.2 T-Cell-Directed Treatments
As stated previously, the inflammatory reaction in CLE may be skewed towards a prevailing of different inflammatory cell types. We discuss targeted treatments based on our aforementioned approach, with a focus on T cells, B cells, pDCs, or IFN preponderance. A more general review about recent developments in targeted therapy of autoimmune skin diseases has been reported elsewhere [133]. We therefore omitted some targeted approaches using cytokine blockade that is well-established in psoriasis (e.g. IL-12/23 blockade, ustekinumab) [134].
Various drugs are in development either to reduce the effects of cytotoxic T cells or to promote the anti-inflammatory properties of regulatory T cells (Table 3). Amiselimod (MT-1303) is a functional antagonist of sphingosine 1-phosphate (S1P) receptor 1 that showed good tolerability in a multicenter, open-label, phase Ib clinical trial for SLE patients [135]. S1P is involved in the egress of T cells from secondary lymphoid organs to sites of inflammation. A reduction of skin symptoms was described in patients finishing the 24-week trial period according to the Systemic Lupus Erythematosus Disease Activity Index 2000 (SLEDAI-2K), without mentioning further details. Another potential target is the CD40 ligand; antagonistic drugs may interfere with antigen presentation to T cells. Use of dapirolizumab pegol was assessed in a randomized, placebo-controlled, phase II study in patients with moderate to severe active SLE. Improvements in various clinical measures, including in the Cutaneous Lupus Erythematosus Disease Area and Severity Index (CLASI), compared with placebo, were observed although the primary objective was not met [136]. Another CD40 ligand antagonist is frexalimab (SAR441344). Recruiting clinical trials in the indications of SLE and primary Sjögren’s syndrome are currently ongoing, however no results are available yet. Another T-cell-directed approach is the selective expansion of regulatory T cells, and an agent of interest is efavaleukin alfa (AMG 592). Results from a phase Ib study in 35 subjects with SLE are available and demonstrated a favorable safety profile [137]. Clinical efficacy will be investigated in phase II studies.
3.3 B-Cell-Directed Treatments
Modulation of B-cell activity has been within the scope of therapeutic research in SLE and CLE for a while. Similar to antimalarials, the individual therapeutic response is hard to predict, which may be attributable to the variety of functions of B cells in fine tuning the inflammatory response as outlined earlier. However, a B-cell-directed therapy was the first to achieve US FDA and European Medicines Agency (EMA) approval for SLE in decades, which highlights the potential of B-cell modulation.
3.3.1 B-Cell-Activating Factor Receptor Inhibition
BAFF is a cytokine crucial for the development, survival and differentiation of B cells, and is also known as a B-lymphocyte stimulator (BLyS). A closely related protein with similar functions is A PRoliferation-Inducing Ligand (APRIL), also known as CD256. Both proteins may bind to three receptors, i.e. BAFF-r, BCMA, and TACI. Monoclonal antibodies and fusion proteins capable of interfering with the ligand–receptor interaction are potential candidates for the treatment of CLE. Notably, belimumab, a monoclonal antibody binding to BAFF, was licensed for the treatment of refractory SLE in 2011 [138], although the pivotal study did not precisely assess clinical outcome regarding skin lesions. A first case series regarding successful use of belimumab in five patients with CLE was published 2017 [139], followed by case reports [140] and a monocentric case series with seven patients [141]. In 2020, a multicenter, retrospective, observational trial was published that included 16 CLE patients [142]. After 6 months of treatment, half of the patients showed at least a 50% reduction in CLASI scores. Finally, in 2021, a prospective, observational study with five patients with CLE was published and showed favorable results [143]. Currently, the efficacy of belimumab for therapy-resistant skin manifestations in LE patients is being investigated in a phase III, multicenter, randomized, double-blind, placebo-controlled, 24-week trial (BELI-SKIN, EUDRA-CT: 2017-003051-35) and the results are eagerly anticipated. Another monoclonal antibody binding to BAFF (tabalumab) did not meet key clinical endpoints in a phase III study in SLE and there is limited information on the efficacy on cutaneous lesions [144]. Fusion proteins binding to BAFF and TACI represent a similar mode of action. Atacicept showed the capacity to reduce flares in patients with SLE in a phase IIb clinical trial, but data on CLE are limited [145]. Telitacicept is currently under investigation for SLE and received fast-track designation status by the FDA in light of positive results. The Chinese National Medical Products Administration (NMPA) granted telitacicept conditional marketing approval for the treatment of adult patients with active, autoantibody-positive SLE [146]. Thus far, there is no detailed information regarding the effects of cutaneous manifestations.
3.3.2 B-Cell-Depleting Therapies
CD20-based depletion of B cells was a milestone in the therapy of hematologic malignancies and numerous autoimmune diseases. To date, rituximab, a first-in-class CD20 monoclonal antibody, failed to achieve licensing in SLE [147]. In a prospective study with 82 SLE patients, 32 had severe mucocutaneous involvement before or after treatment. Only patients with ACLE showed a favorable response to rituximab, while chronic CLE patients failed to show improvement [148]. In a single-center, retrospective cohort study in Great Britain, 38 of 50 (76%) CLE patients receiving rituximab improved regarding mucocutaneous symptoms, with somewhat lower numbers for patients with subacute CLE and chronic CLE [149]. Notably, some authors describe ongoing complete remissions following only two infusions of rituximab in CLE [150]. Other B-cell-depleting agents are under clinical investigation for SLE, including obinutuzumab [151], obexelimab [152], and ocrelizumab [153], and might show alternating efficacy on mucocutaneous lesions in LE patients when compared with rituximab. Thus far, there are very limited clinical data on CLE with these agents.
3.3.3 Inhibition of Differentiation of B Cells to Plasma Cells
Iberdomide, a cereblon E3 ligase modulator that promotes degradation of transcription factors involved in autoimmunity, is under investigation for SLE. The results of a multicenter, phase II study including 288 patients were recently published. The outcome after 24 weeks was favorable in the iberdomide group when compared with placebo [154]. The reduction of CLASI was analyzed as a secondary endpoint. Sixty-four patients had a CLASI-A score of at least 10 at baseline, and the differences between the iberdomide and placebo groups with respect to a CLASI reduction of 50% ranged between 5.3% and 24.0% in different dosing groups. In light of this therapeutic range, patient selection seems to be crucial, which should be determined in further studies.
3.3.4 Plasma-Cell-Directed Therapies
In order to reduce circulating autoantibodies, one therapeutic strategy is the reduction of plasma-cell activity. Use of daratumumab to deplete long-lived plasma cells was described to be successful in SLE, as published in a small case series [155]. Proteasome inhibitors are in use for myeloma therapy and are under investigation for certain autoimmune diseases; however, clinical trials investigating the use of bortezomib and ixazomib for SLE were terminated. At this point, the use of these agents in CLE appears unlikely in the near future, although in certain combinatorial settings, proteasome inhibitors might be of use for severe cases of SLE [156].
3.4 Plasmacytoid Dendritic Cell-Directed Treatments
pDCs are major stakeholders of the innate immune system and are actively involved in the early inflammatory response. Their role in CLE is well-established as they are producers of large amounts of lesional type I IFNs, which then amplifies inflammatory loops. BDCA2 is an important surface antigen exclusively expressed on the cell membrane of pDCs and was identified as a therapeutic target [157]. A humanized monoclonal antibody binding to BDCA2 (litifilimab) is under clinical investigation for CLE with or without systemic disease. The results of a phase II trial with dosing between 50 and 450 mg subcutaneously biweekly over the course of 16 weeks are available [158]. One hundred and thirty-two patients were enrolled in the trial, with the difference from baseline CLASI in the treatment arms ranging between 24.3 and 33.4%, which was statistically significant compared with the placebo arm. Tolerability was acceptable and the authors concluded that larger and longer trials are needed to determine the effects of litifilimab in CLE [158].
Another mode of pDC inhibition is the use of a monoclonal antibody directed at leukocyte immunoglobulin-like receptor subfamily A member 4 (LILRA4) [daxdilimab]. An open-label extension study (phase II) is ongoing and the estimated enrollment is 156 patients with SLE. The primary outcome measure is safety, however no results are available as yet. It will be very interesting to follow this therapeutic approach and to determine which CLE patients could benefit the most.
3.5 Interferon-I-Directed Therapies
When considering the major role of type 1 IFNs in CLE, it is self-evident to evaluate therapeutic interventions regarding these cytokines. Disappointing results of therapeutic trials with monoclonal antibodies directed at IFNs led to shifting the focus on the corresponding receptor (IFNAR1), which turned out to be more effective. Indeed, only recently, an IFN-targeted treatment was licensed for SLE, which might point towards therapeutic potential for (subgroups) of CLE patients. Downstream signaling of IFNAR1 mainly relies on JAK1–3 and tyrosine kinase 2 (TYK2), which is another promising approach for both topical and systemic treatment modalities.
3.5.1 Interferon Receptor Inhibition
The concept of IFNAR inhibition was found to be well tolerated and effective across multiple clinical endpoints in a phase IIb trial including 305 patients with moderate to severe SLE [159], which led to the initiation of further development of the drug. In a placebo-controlled, phase III study (TULIP-2), 362 patients with active SLE were randomized to receive 300 mg of intravenous anifrolumab or placebo every 4 weeks over a course of 48 weeks [160]. The primary outcome measure of the British Isles Lupus Assessment Group (BILAG)-based Composite Lupus Assessment (BICLA) reduction was statistically higher in the verum group. Interestingly, in this trial, skin symptoms were assessed as a secondary endpoint defined by improvement of CLASI by at least 50% (CLASI50). Of the patients with a baseline CLASI of >10, within the anifrolumab group twice as many patients reached CLASI50 compared with the placebo group. A post hoc analysis of the twin phase III studies TULIP-1 and TULIP-2 established comparable findings for cutaneous disease manifestation [161]. Adverse effects most commonly observed included upper respiratory infections, zoster, and influenza, which might be attributable to interference with the antiviral cellular immune response. A recently published placebo-controlled, phase III extension trial underlined the favorable safety profile of anifrolumab, even in light of the ongoing COVID-19 pandemic [162]. A pooled safety analysis of the phase II and III trials also underlined the good tolerability [163]. The possibility of reducing the daily dose of glucocorticoids might even be favorable considering the risk of infection in subjects with LE. In light of these interesting results in SLE patients, first reports regarding the use of anifrolumab in CLE patients are available. Only recently, successful use in a case series of three patients was published by an American group [164]. Interestingly, the patients reported were pretreated heavily with second- and third-line treatments, even belimumab. Similarly, another case report described a reduction in CLASI from 17 to 7 within 8 weeks of treatment initiation in an SLE patient with predominant skin involvement [165]. After all, IFNAR1 inhibition might be a breakthrough innovation for the therapy of certain CLE patients, however more data are necessary for treatment allocation considering the high cost of the drug at this point in time.
3.5.2 Janus Kinase-Signal Transducer and Activator of Transcription (JAK-STAT) Pathway
Given their substantial role in signal transduction of various proinflammatory cytokines, including IFN signaling, a multitude of JAK inhibitors, SYK inhibitors, and TYK2 inhibitors are under clinical development for different inflammatory skin diseases, including CLE. As they are small molecules, administration may be oral or even topical, potentially limiting adverse effects, which are dose-dependent. Treatment approaches include selective inhibition of singular JAKs (potentially more appropriate for systemic treatment) or more broad inhibition of various JAKs (potentially more appropriate for topical treatment), depending on the specificity of the drugs.
In vitro data support the role of JAK1 and SYK in the pathophysiology of CLE [166, 167]. Use of orally available filgotinib (a JAK1 inhibitor) or lanraplenib (an SYK inhibitor) was assessed in a phase IIb study in 47 subjects with moderate to severe CLE [168]. The drugs were generally well tolerated, however the primary endpoint of CLASI50 at week 12 was not met. Efficacy was somewhat higher in the filgotinib group compared with the lanraplenib group. In 2022, French authors reported an impressive clinical response in a heavily pretreated patient with an absolute CLASI of 58 upon therapeutic challenge with oral upadacitinib 15 mg, a selective JAK1 inhibitor [169]. HCQ was continued during therapy and the patient showed a reduction in CLASI50 within 3 months. Similar reports are available for oral administration of baricitinib, a JAK1 and JAK2 inhibitor, following the encouraging results of a phase IIb trial for SLE [170, 171]. The results from two randomized phase III studies in subjects with SLE were recently posted and showed somewhat disencouraging results [172]. Tofacitinib, a JAK1 and JAK3 inhibitor, is currently under clinical investigation in an actively recruiting, open-label, phase II study for young subjects with CLE and SLE. A case series reported improvement of at least 50% skin involvement in two of three heavily pretreated CLE patients receiving tofacitinib 5 mg twice daily [173]. However, it is noteworthy that JAK inhibitors might be associated with an increased risk of major cardiovascular events. Furthermore, thromboembolic events as a safety signal were observed for tofacitinib in a surveillance study in patients with rheumatoid arthritis [174]. SLE and CLE patients are at increased risk for these events, therefore further study is needed to evaluate safety in this cohort of patients.
Selective TYK2 inhibition might be less prone to off-target adverse effects, including disruption of hematopoiesis and cardiovascular adverse effects. Results from a phase IIb trial of deucravacitinib, an allosteric selective TYK2 inhibitor, in subjects with active SLE are encouraging [175]. Three hundred and sixty-three patients were randomized to receive either placebo or 3 mg/6 mg deucravacitinib twice daily, or 12 mg once daily, and while the adverse effects were comparable between the groups, a statistically significant larger number of patients achieved CLASI50 reduction in the 3 mg group at week 48 compared with placebo. In light of these results, larger studies are planned to evaluate the potential therapeutic use in SLE and CLE patients.
Even though the aforementioned clinical trial with systemic SYK inhibition did not reach its primary endpoint, topical SYK inhibition might be of value as overexpression of SYK is frequently found in cutaneous LE lesions. A phase Ib study established a favorable safety profile for GSK2646264, however clinical efficacy did not statistically differ from placebo [176]. Another candidate that was in clinical development was R932333, a dual JAK3 and SYK inhibitor. A phase IIb study enrolled 54 patients to receive 6% verum twice daily versus placebo (vehicle). There were no serious adverse events but efficacy was not superior compared with placebo. The topical pan-JAK inhibitor delgocitinib is already available in Japan. A case report of successful treatment of facial lesions of subacute CLE with delgocitinib 0.5% ointment was recently published [177]. In light of the advent of licensing of topical ruxolitinib for atopic dermatitis and vitiligo in the US, first reports about use in CLE patients are now available [178]. More study is needed regarding this topical approach, yet potent topical JAK inhibitors might expand the therapeutic armamentarium for CLE patients, especially for specific sites such as the face and the scalp.
Apart from our focused approach in this review, even more drugs are under investigation for CLE or SLE. For further reading, we refer to other reviews dealing with emerging therapies in CLE or other connective tissue diseases [179,180,181].
4 Summary and Outlook
CLE is a complex disease with many facets, which might be reflected partly by a varying dominance of specific immune cell subsets and cytokine profiles. Still, a given clinical or histological pattern fails to reliably predict cytokine or gene expression. A better comprehension of molecular pathways and individual disease-perpetuating factors will render the way forward to personalized treatment options. The advent of targeted treatment options licensed for SLE might also pave the way to more precise therapeutic interventions in CLE. Entering an era of precision medicine in CLE will be of benefit for both patients and treating physicians, and upcoming data of active clinical trials and real-world data will help to further delineate the underlying multidimensional interrelations. A close interdisciplinary exchange with rheumatologists and nephrologists is mandatory for optimal treatment of lupus patients to define the best therapeutic strategy.
References
Wenzel J. Cutaneous lupus erythematosus: new insights into pathogenesis and therapeutic strategies. Nat Rev Rheumatol. 2019;15:519–32. https://doi.org/10.1038/s41584-019-0272-0.
Patel J, Borucki R, Werth VP. An update on the pathogenesis of cutaneous lupus erythematosus and its role in clinical practice. Curr Rheumatol Rep. 2020;22:69. https://doi.org/10.1007/s11926-020-00946-z.
Kuhn A, Wenzel J, Bijl M. Lupus erythematosus revisited. Semin Immunopathol. 2016;38:97–112. https://doi.org/10.1007/s00281-015-0550-0.
Udompanich S, Chanprapaph K, Suchonwanit P. Hair and scalp changes in cutaneous and systemic lupus erythematosus. Am J Clin Dermatol. 2018;19:679–94. https://doi.org/10.1007/s40257-018-0363-8.
Jicha KI, Wang DM, Miedema JR, Diaz LA. Cutaneous lupus erythematosus/lichen planus overlap syndrome. JAAD Case Rep. 2021;17:130–51. https://doi.org/10.1016/j.jdcr.2021.09.031.
Zhou W, Wu H, Zhao M, Lu Q. New insights into the progression from cutaneous lupus to systemic lupus erythematosus. Expert Rev Clin Immunol. 2020;16:829–37. https://doi.org/10.1080/1744666X.2020.1805316.
Grönhagen CM, Fored CM, Granath F, Nyberg F. Cutaneous lupus erythematosus and the association with systemic lupus erythematosus: a population-based cohort of 1088 patients in Sweden. Br J Dermatol. 2011;164:1335–41. https://doi.org/10.1111/j.1365-2133.2011.10272.x.
Walker AM, Lu G, Clifton SC, Ogunsanya ME, Chong BF. Influence of socio-demographic factors in patients with cutaneous lupus erythematosus. Front Med (Lausanne). 2022;9:916134. https://doi.org/10.3389/fmed.2022.916134.
Kuhn A, Landmann A, Wenzel J. Advances in the treatment of cutaneous lupus erythematosus. Lupus. 2016;25:830–7. https://doi.org/10.1177/0961203316641771.
Li Q, Wu H, Zhou S, Zhao M, Lu Q. An Update on the Pathogenesis of Skin Damage in Lupus. Curr Rheumatol Rep. 2020;22:16. https://doi.org/10.1007/s11926-020-00893-9.
Fetter T, Wenzel J. Cutaneous lupus erythematosus: The impact of self-amplifying innate and adaptive immune responses and future prospects of targeted therapies. Exp Dermatol. 2020;29:1123–32. https://doi.org/10.1111/exd.14146.
Ker KJ, Teske NM, Feng R, Chong BF, Werth VP. Natural history of disease activity and damage in patients with cutaneous lupus erythematosus. J Am Acad Dermatol. 2018;79:1053-1060.e3. https://doi.org/10.1016/j.jaad.2018.06.040.
Kuhn A, Sigges J, Biazar C, Ruland V, Patsinakidis N, Landmann A, et al. Influence of smoking on disease severity and antimalarial therapy in cutaneous lupus erythematosus: analysis of 1002 patients from the EUSCLE database. Br J Dermatol. 2014;171:571–9. https://doi.org/10.1111/bjd.13006.
Crow MK. Type I interferon in the pathogenesis of lupus. J Immunol. 2014;192:5459–68. https://doi.org/10.4049/jimmunol.1002795.
Stannard JN, Kahlenberg JM. Cutaneous lupus erythematosus: updates on pathogenesis and associations with systemic lupus. Curr Opin Rheumatol. 2016;28:453–9. https://doi.org/10.1097/BOR.0000000000000308.
Kuhn A, Wenzel J, Weyd H. Photosensitivity, apoptosis, and cytokines in the pathogenesis of lupus erythematosus: a critical review. Clin Rev Allergy Immunol. 2014;47:148–62. https://doi.org/10.1007/s12016-013-8403-x.
Niebel D, Novak N, Wilhelmi J, Ziob J, Wilsmann-Theis D, Bieber T, et al. Cutaneous adverse reactions to COVID-19 vaccines: insights from an immuno-dermatological perspective. Vaccines (Basel). 2021. https://doi.org/10.3390/vaccines9090944.
Fetter T, Braegelmann C, de Vos L, Wenzel J. Current concepts on pathogenic mechanisms and histopathology in cutaneous lupus erythematosus. Front Med (Lausanne). 2022;9:915828. https://doi.org/10.3389/fmed.2022.915828.
Lo MS. Monogenic Lupus. Curr Rheumatol Rep. 2016;18:71. https://doi.org/10.1007/s11926-016-0621-9.
Chen HW, Barber G, Chong BF. The genetic landscape of cutaneous lupus erythematosus. Front Med (Lausanne). 2022;9:916011. https://doi.org/10.3389/fmed.2022.916011.
Liu Y, Xu M, Min X, Wu K, Zhang T, Li K, et al. TWEAK/Fn14 activation participates in Ro52-mediated photosensitization in cutaneous lupus erythematosus. Front Immunol. 2017;8:651. https://doi.org/10.3389/fimmu.2017.00651.
Reich A, Meurer M, Viehweg A, Muller DJ. Narrow-band UVB-induced externalization of selected nuclear antigens in keratinocytes: implications for lupus erythematosus pathogenesis. Photochem Photobiol. 2009;85:1–7. https://doi.org/10.1111/j.1751-1097.2008.00480.x.
Greiling TM, Dehner C, Chen X, Hughes K, Iñiguez AJ, Boccitto M, et al. Commensal orthologs of the human autoantigen Ro60 as triggers of autoimmunity in lupus. Sci Transl Med. 2018. https://doi.org/10.1126/scitranslmed.aan2306.
Meller S, Winterberg F, Gilliet M, Müller A, Lauceviciute I, Rieker J, et al. Ultraviolet radiation-induced injury, chemokines, and leukocyte recruitment: an amplification cycle triggering cutaneous lupus erythematosus. Arthritis Rheum. 2005;52:1504–16. https://doi.org/10.1002/art.21034.
Wenzel J, Zahn S, Mikus S, Wiechert A, Bieber T, Tüting T. The expression pattern of interferon-inducible proteins reflects the characteristic histological distribution of infiltrating immune cells in different cutaneous lupus erythematosus subsets. Br J Dermatol. 2007;157:752–7. https://doi.org/10.1111/j.1365-2133.2007.08137.x.
Furukawa F, Itoh T, Wakita H, Yagi H, Tokura Y, Norris DA, Takigawa M. Keratinocytes from patients with lupus erythematosus show enhanced cytotoxicity to ultraviolet radiation and to antibody-mediated cytotoxicity. Clin Exp Immunol. 1999;118:164–70. https://doi.org/10.1046/j.1365-2249.1999.01026.x.
Kuhn A, Herrmann M, Kleber S, Beckmann-Welle M, Fehsel K, Martin-Villalba A, et al. Accumulation of apoptotic cells in the epidermis of patients with cutaneous lupus erythematosus after ultraviolet irradiation. Arthritis Rheum. 2006;54:939–50. https://doi.org/10.1002/art.21658.
Hayashi A, Nagafuchi H, Ito I, Hirota K, Yoshida M, Ozaki S. Lupus antibodies to the HMGB1 chromosomal protein: epitope mapping and association with disease activity. Mod Rheumatol. 2009;19:283–92. https://doi.org/10.1007/s10165-009-0151-7.
Scholtissek B, Zahn S, Maier J, Klaeschen S, Braegelmann C, Hoelzel M, et al. Immunostimulatory endogenous nucleic acids drive the lesional inflammation in cutaneous lupus erythematosus. J Invest Dermatol. 2017;137:1484–92. https://doi.org/10.1016/j.jid.2017.03.018.
Sarkar MK, Hile GA, Tsoi LC, Xing X, Liu J, Liang Y, et al. Photosensitivity and type I IFN responses in cutaneous lupus are driven by epidermal-derived interferon kappa. Ann Rheum Dis. 2018;77:1653–64. https://doi.org/10.1136/annrheumdis-2018-213197.
Zahn S, Rehkämper C, Kümmerer BM, Ferring-Schmidt S, Bieber T, Tüting T, Wenzel J. Evidence for a pathophysiological role of keratinocyte-derived type III interferon (IFNλ) in cutaneous lupus erythematosus. J Invest Dermatol. 2011;131:133–40. https://doi.org/10.1038/jid.2010.244.
Wenzel J, Tüting T. Identification of type I interferon-associated inflammation in the pathogenesis of cutaneous lupus erythematosus opens up options for novel therapeutic approaches. Exp Dermatol. 2007;16:454–63. https://doi.org/10.1111/j.1600-0625.2007.00556.x.
Braegelmann C, Fetter T, Niebel D, Dietz L, Bieber T, Wenzel J. Immunostimulatory endogenous nucleic acids perpetuate interface dermatitis-translation of pathogenic fundamentals into an in vitro model. Front Immunol. 2020;11:622511. https://doi.org/10.3389/fimmu.2020.622511.
Kopfnagel V, Wittmann M, Werfel T. Human keratinocytes express AIM2 and respond to dsDNA with IL-1β secretion. Exp Dermatol. 2011;20:1027–9. https://doi.org/10.1111/j.1600-0625.2011.01382.x.
Guiducci C, Tripodo C, Gong M, Sangaletti S, Colombo MP, Coffman RL, Barrat FJ. Autoimmune skin inflammation is dependent on plasmacytoid dendritic cell activation by nucleic acids via TLR7 and TLR9. J Exp Med. 2010;207:2931–42. https://doi.org/10.1084/jem.20101048.
Means TK, Latz E, Hayashi F, Murali MR, Golenbock DT, Luster AD. Human lupus autoantibody-DNA complexes activate DCs through cooperation of CD32 and TLR9. J Clin Invest. 2005;115:407–17. https://doi.org/10.1172/JCI23025.
Kraman M, Bambrough PJ, Arnold JN, Roberts EW, Magiera L, Jones JO, et al. Suppression of antitumor immunity by stromal cells expressing fibroblast activation protein-alpha. Science. 2010;330:827–30. https://doi.org/10.1126/science.1195300.
Farkas L, Beiske K, Lund-Johansen F, Brandtzaeg P, Jahnsen FL. Plasmacytoid dendritic cells (natural interferon- alpha/beta-producing cells) accumulate in cutaneous lupus erythematosus lesions. Am J Pathol. 2001;159:237–43. https://doi.org/10.1016/s0002-9440(10)61689-6.
Asselin-Paturel C, Brizard G, Chemin K, Boonstra A, O’Garra A, Vicari A, Trinchieri G. Type I interferon dependence of plasmacytoid dendritic cell activation and migration. J Exp Med. 2005;201:1157–67. https://doi.org/10.1084/jem.20041930.
Vermi W, Lonardi S, Morassi M, Rossini C, Tardanico R, Venturini M, et al. Cutaneous distribution of plasmacytoid dendritic cells in lupus erythematosus. Selective tropism at the site of epithelial apoptotic damage. Immunobiology. 2009;214:877–86. https://doi.org/10.1016/j.imbio.2009.06.013.
de Vos L, Guel T, Niebel D, Bald S, ter Steege A, Bieber T, Wenzel J. Characterization of B cells in lupus erythematosus skin biopsies in the context of different immune cell infiltration patterns. Front Med (Lausanne). 2022;9:1037408. https://doi.org/10.3389/fmed.2022.1037408.
Billi AC, Ma F, Plazyo O, Gharaee-Kermani M, Wasikowski R, Hile GA, et al. Nonlesional lupus skin contributes to inflammatory education of myeloid cells and primes for cutaneous inflammation. Sci Transl Med. 2022;14:eabn2263. https://doi.org/10.1126/scitranslmed.abn2263.
Furie R, Werth VP, Merola JF, Stevenson L, Reynolds TL, Naik H, et al. Monoclonal antibody targeting BDCA2 ameliorates skin lesions in systemic lupus erythematosus. J Clin Investig. 2019;129:1359–71. https://doi.org/10.1172/JCI124466.
Dzionek A, Sohma Y, Nagafune J, Cella M, Colonna M, Facchetti F, et al. BDCA-2, a novel plasmacytoid dendritic cell-specific type II C-type lectin, mediates antigen capture and is a potent inhibitor of interferon alpha/beta induction. J Exp Med. 2001;194:1823–34. https://doi.org/10.1084/jem.194.12.1823.
Lipsker D, Saurat J-H. Neutrophilic cutaneous lupus erythematosus. At the edge between innate and acquired immunity? Dermatology. 2008;216:283–6. https://doi.org/10.1159/000113940.
Mak A, Kow NY. The pathology of T cells in systemic lupus erythematosus. J Immunol Res. 2014. https://doi.org/10.1155/2014/419029.
Ghosh D, Tsokos GC, Kyttaris VC. c-Jun and Ets2 proteins regulate expression of spleen tyrosine kinase in T cells. J Biol Chem. 2012;287:11833–41. https://doi.org/10.1074/jbc.M111.333997.
Kyttaris VC, Wang Y, Juang Y-T, Weinstein A, Tsokos GC. Increased levels of NF-ATc2 differentially regulate CD154 and IL-2 genes in T cells from patients with systemic lupus erythematosus. J Immunol. 2007;178:1960–6. https://doi.org/10.4049/jimmunol.178.3.1960.
Kow NY, Mak A. Costimulatory pathways: physiology and potential therapeutic manipulation in systemic lupus erythematosus. Clin Dev Immunol. 2013. https://doi.org/10.1155/2013/245928.
Solomou EE, Juang YT, Gourley MF, Kammer GM, Tsokos GC. Molecular basis of deficient IL-2 production in T cells from patients with systemic lupus erythematosus. J Immunol. 2001;166:4216–22. https://doi.org/10.4049/jimmunol.166.6.4216.
Crispín JC, Oukka M, Bayliss G, Cohen RA, van Beek CA, Stillman IE, et al. Expanded double negative T cells in patients with systemic lupus erythematosus produce IL-17 and infiltrate the kidneys. J Immunol. 2008;181:8761–6. https://doi.org/10.4049/jimmunol.181.12.8761.
Wenzel J, Wörenkämper E, Freutel S, Henze S, Haller O, Bieber T, Tüting T. Enhanced type I interferon signalling promotes Th1-biased inflammation in cutaneous lupus erythematosus. J Pathol. 2005;205:435–42. https://doi.org/10.1002/path.1721.
Grassi M, Capello F, Bertolino L, Seia Z, Pippione M. Identification of granzyme B-expressing CD-8-positive T cells in lymphocytic inflammatory infiltrate in cutaneous lupus erythematosus and in dermatomyositis. Clin Exp Dermatol. 2009;34:910–4. https://doi.org/10.1111/j.1365-2230.2009.03297.x.
Wenzel J, Uerlich M, Wörrenkämper E, Freutel S, Bieber T, Tüting T. Scarring skin lesions of discoid lupus erythematosus are characterized by high numbers of skin-homing cytotoxic lymphocytes associated with strong expression of the type I interferon-induced protein MxA. Br J Dermatol. 2005;153:1011–5. https://doi.org/10.1111/j.1365-2133.2005.06784.x.
Haddadi N-S, Mande P, Brodeur TY, Hao K, Ryan GE, Moses S, et al. Th2 to Th1 transition is required for induction of skin lesions in an inducible and recurrent murine model of cutaneous lupus-like inflammation. Front Immunol. 2022;13:883375. https://doi.org/10.3389/fimmu.2022.883375.
Yang J, Murphy TL, Ouyang W, Murphy KM. Induction of interferon-γ production in Th1 CD4+ T cells: evidence for two distinct pathways for promoter activation. Eur J Immunol. 1999;29:548–55. https://doi.org/10.1002/(SICI)1521-4141(199902)29:02%3c548::AID-IMMU548%3e3.0.CO;2-Z.
Nakajima M, Nakajima A, Kayagaki N, Honda M, Yagita H, Okumura K. Expression of Fas ligand and its receptor in cutaneous lupus: implication in tissue injury. Clin Immunol Immunopathol. 1997;83:223–9. https://doi.org/10.1006/clin.1997.4352.
Salvi V, Vermi W, Cavani A, Lonardi S, Carbone T, Facchetti F, et al. IL-21 May promote granzyme B-dependent NK/plasmacytoid dendritic cell functional interaction in cutaneous lupus erythematosus. J Invest Dermatol. 2017;137:1493–500. https://doi.org/10.1016/j.jid.2017.03.016.
Karrich JJ, Jachimowski LCM, Nagasawa M, Kamp A, Balzarolo M, Wolkers MC, et al. IL-21-stimulated human plasmacytoid dendritic cells secrete granzyme B, which impairs their capacity to induce T-cell proliferation. Blood. 2013;121:3103–11. https://doi.org/10.1182/blood-2012-08-452995.
Shivakumar S, Tsokos GC, Datta SK. T cell receptor alpha/beta expressing double-negative (CD4-/CD8-) and CD4+ T helper cells in humans augment the production of pathogenic anti-DNA autoantibodies associated with lupus nephritis. J Immunol. 1989;143:103–12.
Rajagopalan S, Zordan T, Tsokos GC, Datta SK. Pathogenic anti-DNA autoantibody-inducing T helper cell lines from patients with active lupus nephritis: isolation of CD4-8- T helper cell lines that express the gamma delta T-cell antigen receptor. Proc Natl Acad Sci U S A. 1990;87:7020–4. https://doi.org/10.1073/pnas.87.18.7020.
Lu L, Kaliyaperumal A, Boumpas DT, Datta SK. Major peptide autoepitopes for nucleosome-specific T cells of human lupus. J Clin Invest. 1999;104:345–55. https://doi.org/10.1172/JCI6801.
Voll RE, Roth EA, Girkontaite I, Fehr H, Herrmann M, Lorenz HM, Kalden JR. Histone-specific Th0 and Th1 clones derived from systemic lupus erythematosus patients induce double-stranded DNA antibody production. Arthritis Rheum. 1997;40:2162–71. https://doi.org/10.1002/art.1780401210.
Luo Y, Zhang X, Zhao M, Lu Q. DNA demethylation of the perforin promoter in CD4(+) T cells from patients with subacute cutaneous lupus erythematosus. J Dermatol Sci. 2009;56:33–6. https://doi.org/10.1016/j.jdermsci.2009.06.010.
Franz B, Fritzsching B, Riehl A, Oberle N, Klemke C-D, Sykora J, et al. Low number of regulatory T cells in skin lesions of patients with cutaneous lupus erythematosus. Arthritis Rheum. 2007;56:1910–20. https://doi.org/10.1002/art.22699.
Filaci G, Bacilieri S, Fravega M, Monetti M, Contini P, Ghio M, et al. Impairment of CD8+ T suppressor cell function in patients with active systemic lupus erythematosus. J Immunol. 2001;166:6452–7. https://doi.org/10.4049/jimmunol.166.10.6452.
Gambichler T, Pätzholz J, Schmitz L, Lahner N, Kreuter A. FOXP3+ and CD39+ regulatory T cells in subtypes of cutaneous lupus erythematosus. J Eur Acad Dermatol Venereol. 2015;29:1972–7. https://doi.org/10.1111/jdv.13123.
Kil LP, Hendriks RW. Aberrant B cell selection and activation in systemic lupus erythematosus. Int Rev Immunol. 2013;32:445–70. https://doi.org/10.3109/08830185.2013.786712.
Lipsky PE. Systemic lupus erythematosus: an autoimmune disease of B cell hyperactivity. Nat Immunol. 2001;2:764–6. https://doi.org/10.1038/ni0901-764.
Lerman I, Mitchell DC, Richardson CT. Human cutaneous B cells: what do we really know? Ann Transl Med. 2021;9:440. https://doi.org/10.21037/atm-20-5185.
Kiefer K, Oropallo MA, Cancro MP, Marshak-Rothstein A. Role of type I interferons in the activation of autoreactive B cells. Immunol Cell Biol. 2012;90:498–504. https://doi.org/10.1038/icb.2012.10.
Zhou S, Li Q, Zhou S, Zhao M, Lu L, Wu H, Lu Q. A novel humanized cutaneous lupus erythematosus mouse model mediated by IL-21-induced age-associated B cells. J Autoimmun. 2021;123:102686. https://doi.org/10.1016/j.jaut.2021.102686.
Crispín JC, Tsokos GC. IL-17 in systemic lupus erythematosus. J Biomed Biotechnol. 2010. https://doi.org/10.1155/2010/943254.
Sontheimer RD, Maddison PJ, Reichlin M, Jordon RE, Stastny P, Gilliam JN. Serologic and HLA associations in subacute cutaneous lupus erythematosus, a clinical subset of lupus erythematosus. Ann Intern Med. 1982;97:664–71. https://doi.org/10.7326/0003-4819-97-5-664.
Patsinakidis N, Gambichler T, Lahner N, Moellenhoff K, Kreuter A. Cutaneous characteristics and association with antinuclear antibodies in 402 patients with different subtypes of lupus erythematosus. J Eur Acad Dermatol Venereol. 2016;30:2097–104. https://doi.org/10.1111/jdv.13769.
Garelli CJ, Refat MA, Nanaware PP, Ramirez-Ortiz ZG, Rashighi M, Richmond JM. Current insights in cutaneous lupus erythematosus immunopathogenesis. Front Immunol. 2020;11:1353. https://doi.org/10.3389/fimmu.2020.01353.
Wasicek CA, Reichlin M. Clinical and serological differences between systemic lupus erythematosus patients with antibodies to Ro versus patients with antibodies to Ro and La. J Clin Invest. 1982;69:835–43. https://doi.org/10.1172/jci110523.
Abernathy-Close L, Lazar S, Stannard J, Tsoi LC, Eddy S, Rizvi SM, et al. B cell signatures distinguish cutaneous lupus erythematosus subtypes and the presence of systemic disease activity. Front Immunol. 2021;12:775353. https://doi.org/10.3389/fimmu.2021.775353.
Jenks SA, Wei C, Bugrovsky R, Hill A, Wang X, Rossi FM, et al. B cell subset composition segments clinically and serologically distinct groups in chronic cutaneous lupus erythematosus. Ann Rheum Dis. 2021;80:1190–200. https://doi.org/10.1136/annrheumdis-2021-220349.
Kogame T, Yamashita R, Hirata M, Kataoka TR, Kamido H, Ueshima C, et al. Analysis of possible structures of inducible skin-associated lymphoid tissue in lupus erythematosus profundus. J Dermatol. 2018;45:1117–21. https://doi.org/10.1111/1346-8138.14498.
Fetter T, Niebel D, Braegelmann C, Wenzel J. Skin-associated B cells in the pathogenesis of cutaneous autoimmune diseases-implications for therapeutic approaches. Cells. 2020. https://doi.org/10.3390/cells9122627.
Steinmetz OM, Velden J, Kneissler U, Marx M, Klein A, Helmchen U, et al. Analysis and classification of B-cell infiltrates in lupus and ANCA-associated nephritis. Kidney Int. 2008;74:448–57. https://doi.org/10.1038/ki.2008.191.
Chang A, Henderson SG, Brandt D, Liu N, Guttikonda R, Hsieh C, et al. In situ B cell-mediated immune responses and tubulointerstitial inflammation in human lupus nephritis. J Immunol. 2011;186:1849–60. https://doi.org/10.4049/jimmunol.1001983.
Yang X, Yang J, Chu Y, Xue Y, Xuan D, Zheng S, Zou H. T follicular helper cells and regulatory B cells dynamics in systemic lupus erythematosus. PLoS ONE. 2014;9:e88441. https://doi.org/10.1371/journal.pone.0088441.
Chen Y, Yang M, Di Long LQ, Zhao M, Wu H, Lu Q. Abnormal expression of BAFF and its receptors in peripheral blood and skin lesions from systemic lupus erythematosus patients. Autoimmunity. 2020;53:192–200. https://doi.org/10.1080/08916934.2020.1736049.
Wenzel J, Landmann A, Vorwerk G, Kuhn A. High expression of B lymphocyte stimulator in lesional keratinocytes of patients with cutaneous lupus erythematosus. Exp Dermatol. 2018;27:95–7. https://doi.org/10.1111/exd.13419.
Chong BF, Tseng L-C, Kim A, Miller RT, Yancey KB, Hosler GA. Differential expression of BAFF and its receptors in discoid lupus erythematosus patients. J Dermatol Sci. 2014;73:216–24. https://doi.org/10.1016/j.jdermsci.2013.11.007.
Mackay F, Browning JL. BAFF: a fundamental survival factor for B cells. Nat Rev Immunol. 2002;2:465–75. https://doi.org/10.1038/nri844.
Liu M, Guo Q, Wu C, Sterlin D, Goswami S, Zhang Y, et al. Type I interferons promote the survival and proinflammatory properties of transitional B cells in systemic lupus erythematosus patients. Cell Mol Immunol. 2019;16:367–79. https://doi.org/10.1038/s41423-018-0010-6.
Malkiel S, Barlev AN, Atisha-Fregoso Y, Suurmond J, Diamond B. Plasma cell differentiation pathways in systemic lupus erythematosus. Front Immunol. 2018;9:427. https://doi.org/10.3389/fimmu.2018.00427.
Karaaslan S, Tomayko MM. A niche for plasma cells: the skin. J Invest Dermatol. 2019;139:2411–4. https://doi.org/10.1016/j.jid.2019.06.133.
Keller EJ, Patel NB, Patt M, Nguyen JK, Jørgensen TN. Partial protection from lupus-like disease by B-cell specific type I interferon receptor deficiency. Front Immunol. 2020;11:616064. https://doi.org/10.3389/fimmu.2020.616064.
Wang S, Wang J, Kumar V, Karnell JL, Naiman B, Gross PS, et al. IL-21 drives expansion and plasma cell differentiation of autoreactive CD11chiT-bet+ B cells in SLE. Nat Commun. 2018;9:1758. https://doi.org/10.1038/s41467-018-03750-7.
Ma K, Li J, Wang X, Lin X, Du W, Yang X, et al. TLR4+CXCR4+ plasma cells drive nephritis development in systemic lupus erythematosus. Ann Rheum Dis. 2018;77:1498–506. https://doi.org/10.1136/annrheumdis-2018-213615.
Henriques A, Teixeira L, Inês L, Carvalheiro T, Gonçalves A, Martinho A, et al. NK cells dysfunction in systemic lupus erythematosus: relation to disease activity. Clin Rheumatol. 2013;32:805–13. https://doi.org/10.1007/s10067-013-2176-8.
Schepis D, Gunnarsson I, Eloranta M-L, Lampa J, Jacobson SH, Kärre K, Berg L. Increased proportion of CD56bright natural killer cells in active and inactive systemic lupus erythematosus. Immunology. 2009;126:140–6. https://doi.org/10.1111/j.1365-2567.2008.02887.x.
Hofmann SC, Bosma A, Bruckner-Tuderman L, Vukmanovic-Stejic M, Jury EC, Isenberg DA, Mauri C. Invariant natural killer T cells are enriched at the site of cutaneous inflammation in lupus erythematosus. J Dermatol Sci. 2013;71:22–8. https://doi.org/10.1016/j.jdermsci.2013.04.012.
Moreno-Angarita A, Aragón CC, Tobón GJ. Cathelicidin LL-37: a new important molecule in the pathophysiology of systemic lupus erythematosus. J Transl Autoimmun. 2020;3:100029. https://doi.org/10.1016/j.jtauto.2019.100029.
Papayannopoulos V. Neutrophil extracellular traps in immunity and disease. Nat Rev Immunol. 2018;18:134–47. https://doi.org/10.1038/nri.2017.105.
Yu Y, Su K. Neutrophil extracellular traps and systemic lupus erythematosus. J Clin Cell Immunol. 2013. https://doi.org/10.4172/2155-9899.1000139.
Mistry P, Nakabo S, O’Neil L, Goel RR, Jiang K, Carmona-Rivera C, et al. Transcriptomic, epigenetic, and functional analyses implicate neutrophil diversity in the pathogenesis of systemic lupus erythematosus. Proc Natl Acad Sci. 2019;116:25222–8. https://doi.org/10.1073/pnas.1908576116.
Carlucci PM, Purmalek MM, Dey AK, Temesgen-Oyelakin Y, Sakhardande S, Joshi AA, et al. Neutrophil subsets and their gene signature associate with vascular inflammation and coronary atherosclerosis in lupus. JCI Insight. 2018. https://doi.org/10.1172/jci.insight.99276.
Hakkim A, Fürnrohr BG, Amann K, Laube B, Abed UA, Brinkmann V, et al. Impairment of neutrophil extracellular trap degradation is associated with lupus nephritis. Proc Natl Acad Sci. 2010;107:9813–8. https://doi.org/10.1073/pnas.0909927107.
Lande R, Ganguly D, Facchinetti V, Frasca L, Conrad C, Gregorio J, et al. Neutrophils activate plasmacytoid dendritic cells by releasing self-DNA-peptide complexes in systemic lupus erythematosus. Sci Transl Med. 2011;3:73ra19. https://doi.org/10.1126/scitranslmed.3001180.
Kreuter A, Jaouhar M, Skrygan M, Tigges C, Stücker M, Altmeyer P, et al. Expression of antimicrobial peptides in different subtypes of cutaneous lupus erythematosus. J Am Acad Dermatol. 2011;65:125–33. https://doi.org/10.1016/j.jaad.2010.12.012.
Sun C-L, Zhang F-Z, Li P, Bi L-Q. LL-37 expression in the skin in systemic lupus erythematosus. Lupus. 2011;20:904–11. https://doi.org/10.1177/0961203311398515.
Safi R, Al-Hage J, Abbas O, Kibbi A-G, Nassar D. Investigating the presence of neutrophil extracellular traps in cutaneous lesions of different subtypes of lupus erythematosus. Exp Dermatol. 2019;28:1348–52. https://doi.org/10.1111/exd.14040.
Li Y, Lee PY, Reeves WH. Monocyte and macrophage abnormalities in systemic lupus erythematosus. Arch Immunol Ther Exp (Warsz). 2010;58:355–64. https://doi.org/10.1007/s00005-010-0093-y.
Blanco P, Palucka AK, Gill M, Pascual V, Banchereau J. Induction of dendritic cell differentiation by IFN-alpha in systemic lupus erythematosus. Science. 2001;294:1540–3. https://doi.org/10.1126/science.1064890.
Tas SW, Quartier P, Botto M, Fossati-Jimack L. Macrophages from patients with SLE and rheumatoid arthritis have defective adhesion in vitro, while only SLE macrophages have impaired uptake of apoptotic cells. Ann Rheum Dis. 2006;65:216–21. https://doi.org/10.1136/ard.2005.037143.
Bijl M, Reefman E, Horst G, Limburg PC, Kallenberg CGM. Reduced uptake of apoptotic cells by macrophages in systemic lupus erythematosus: correlates with decreased serum levels of complement. Ann Rheum Dis. 2006;65:57–63. https://doi.org/10.1136/ard.2005.035733.
Mantovani A, Sica A, Sozzani S, Allavena P, Vecchi A, Locati M. The chemokine system in diverse forms of macrophage activation and polarization. Trends Immunol. 2004;25:677–86. https://doi.org/10.1016/j.it.2004.09.015.
Labonte AC, Kegerreis B, Geraci NS, Bachali P, Madamanchi S, Robl R, et al. Identification of alterations in macrophage activation associated with disease activity in systemic lupus erythematosus. PLoS ONE. 2018;13:e0208132. https://doi.org/10.1371/journal.pone.0208132.
Mohammadi S, Saghaeian-Jazi M, Sedighi S, Memarian A. Immunomodulation in systemic lupus erythematosus: induction of M2 population in monocyte-derived macrophages by pioglitazone. Lupus. 2017;26:1318–27. https://doi.org/10.1177/0961203317701842.
Li F, Yang Y, Zhu X, Huang L, Xu J. Macrophage polarization modulates development of systemic lupus erythematosus. Cell Physiol Biochem. 2015;37:1279–88. https://doi.org/10.1159/000430251.
Ma C, Xia Y, Yang Q, Zhao Y. The contribution of macrophages to systemic lupus erythematosus. Clin Immunol. 2019;207:1–9. https://doi.org/10.1016/j.clim.2019.06.009.
Yang B, Huang X, Xu S, Li L, Wu W, Dai Y, et al. Decreased miR-4512 Levels in monocytes and macrophages of individuals with systemic lupus erythematosus contribute to innate immune activation and Neutrsophil NETosis by targeting TLR4 and CXCL2. Front Immunol. 2021;12:756825. https://doi.org/10.3389/fimmu.2021.756825.
Fayard D, Francès C, Amoura Z, Breillat P, Mathian A, Senet P, et al. Prevalence and factors associated with long-term remission in cutaneous lupus: a longitudinal cohort study of 141 cases. J Am Acad Dermatol. 2022;87:323–32. https://doi.org/10.1016/j.jaad.2022.03.056.
Worm M, Zidane M, Eisert L, Fischer-Betz R, Foeldvari I, Günther C, et al. S2k guideline: Diagnosis and management of cutaneous lupus erythematosus - Part 2: Therapy, risk factors and other special topics. J Dtsch Dermatol Ges. 2021;19:1371–95. https://doi.org/10.1111/ddg.14491.
Barua DP, Chowdhury MIH, Mowla MR, Reich A, Murrell D, Ruzicka T. Comparison of effectiveness of topical tacrolimus 0.1% vs topical halobetasol propionate 0.05% as an add-on to oral hydroxychloroquine in discoid lupus erythematosus. Dermatol Ther. 2021;34:e14675. https://doi.org/10.1111/dth.14675.
Patel J, Vazquez T, Chin F, Keyes E, Yan D, Diaz D, et al. Multidimensional immune profiling of cutaneous lupus erythematosus in vivo stratified by patient responses to antimalarials. Arthritis Rheumatol. 2022. https://doi.org/10.1002/art.42235.
Kuhn A, Aberer E, Bata-Csörgő Z, Caproni M, Dreher A, Frances C, et al. S2k guideline for treatment of cutaneous lupus erythematosus - guided by the European Dermatology Forum (EDF) in cooperation with the European Academy of Dermatology and Venereology (EADV). J Eur Acad Dermatol Venereol. 2017;31:389–404. https://doi.org/10.1111/jdv.14053.
Worm M, Zidane M, Eisert L, Fischer-Betz R, Foeldvari I, Günther C, et al. S2k guideline: Diagnosis and management of cutaneous lupus erythematosus—part 1: classification, diagnosis, prevention, activity scores. J Dtsch Dermatol Ges. 2021;19:1236–47. https://doi.org/10.1111/ddg.14492.
O’Kane D, McCourt C, Meggitt S, D’Cruz DP, Orteu CH, Benton E, et al. British Association of Dermatologists guidelines for the management of people with cutaneous lupus erythematosus 2021. Br J Dermatol. 2021;185:1112–23. https://doi.org/10.1111/bjd.20597.
Kreuter A, Tomi NS, Weiner SM, Huger M, Altmeyer P, Gambichler T. Mycophenolate sodium for subacute cutaneous lupus erythematosus resistant to standard therapy. Br J Dermatol. 2007;156:1321–7. https://doi.org/10.1111/j.1365-2133.2007.07826.x.
Fanouriakis A, Kostopoulou M, Alunno A, Aringer M, Bajema I, Boletis JN, et al. 2019 update of the EULAR recommendations for the management of systemic lupus erythematosus. Ann Rheum Dis. 2019;78:736–45. https://doi.org/10.1136/annrheumdis-2019-215089.
Keyes E, Jobanputra A, Feng R, Grinnell M, Vazquez T, Diaz D, Werth VP. Comparative responsiveness of cutaneous lupus erythematosus patients to methotrexate and mycophenolate mofetil: A cohort study. J Am Acad Dermatol. 2022;87:447–8. https://doi.org/10.1016/j.jaad.2021.09.017.
Laosakul K, Chiewchanvit S, Chuamanochan M, Tovanabutra N. Acitretin treatment in antimalarial-refractory/intolerant discoid lupus erythematosus: a prospective, open-label, uncontrolled study. Lupus. 2022;31:575–81. https://doi.org/10.1177/09612033221086878.
Aitmehdi R, Arnaud L, Francès C, Senet P, Monfort J-B, de Risi-Pugliese T, et al. Long-term efficacy and safety outcomes of lenalidomide for cutaneous lupus erythematosus: a multicenter retrospective observational study of 40 patients. J Am Acad Dermatol. 2021;84:1171–4. https://doi.org/10.1016/j.jaad.2020.11.014.
Reymann V, Bessis D, Bergeret B, Lipsker D, Du-Thanh A, Terrail N, et al. Efficacy and safety of low-dose oral lenalidomide in refractory cutaneous lupus erythematosus: an open series of 19 cases. J Eur Acad Dermatol Venereol. 2021;35:e113–5. https://doi.org/10.1111/jdv.16839.
Okon LG, Werth VP. Cutaneous lupus erythematosus: diagnosis and treatment. Best Pract Res Clin Rheumatol. 2013;27:391–404. https://doi.org/10.1016/j.berh.2013.07.008.
Shi H, Gudjonsson JE, Kahlenberg JM. Treatment of cutaneous lupus erythematosus: current approaches and future strategies. Curr Opin Rheumatol. 2020;32:208–14. https://doi.org/10.1097/BOR.0000000000000704.
Braegelmann C, Niebel D, Wenzel J. Targeted therapies in autoimmune skin diseases. J Invest Dermatol. 2022;142:969-975.e7. https://doi.org/10.1016/j.jid.2021.08.439.
Petty AJ, Floyd L, Henderson C, Nicholas MW. Cutaneous lupus erythematosus: progress and challenges. Curr Allergy Asthma Rep. 2020;20:12. https://doi.org/10.1007/s11882-020-00906-8.
Tanaka Y, Kondo K, Ichibori A, Yanai Y, Susuta Y, Inoue S, Takeuchi T. Amiselimod, a sphingosine 1-phosphate receptor-1 modulator, for systemic lupus erythematosus: a multicenter, open-label exploratory study. Lupus. 2020;29:1902–13. https://doi.org/10.1177/0961203320966385.
Furie RA, Bruce IN, Dörner T, Leon MG, Leszczyński P, Urowitz M, et al. Phase 2, randomized, placebo-controlled trial of dapirolizumab pegol in patients with moderate-to-severe active systemic lupus erythematosus. Rheumatology (Oxford). 2021;60:5397–407. https://doi.org/10.1093/rheumatology/keab381.
Tchao N, Sarkar N, Hu X, Zhang R, Milmont C, Shi Jin Y, et al. AB0432 efavaleukin alfa, a novel IL-2 mutein, selectively expands regulatory T cells in patients with SLE: final results of a phase 1b multiple ascending dose study. Ann Rheum Dis. 2022;81:1343–4. https://doi.org/10.1136/annrheumdis-2022-eular.2244.
Furie R, Petri M, Zamani O, Cervera R, Wallace DJ, Tegzová D, et al. A phase III, randomized, placebo-controlled study of belimumab, a monoclonal antibody that inhibits B lymphocyte stimulator, in patients with systemic lupus erythematosus. Arthritis Rheum. 2011;63:3918–30. https://doi.org/10.1002/art.30613.
Vashisht P, Borghoff K, O’Dell JR, Hearth-Holmes M. Belimumab for the treatment of recalcitrant cutaneous lupus. Lupus. 2017;26:857–64. https://doi.org/10.1177/0961203316682097.
Bosch-Amate X, Morgado-Carrasco D, Combalia A, Giavedoni P, Espinosa G, Mascaró JM. Successful treatment of two cases of refractory cutaneous lupus erythematosus with belimumab. Indian J Dermatol Venereol Leprol. 2021;87:421–4. https://doi.org/10.25259/IJDVL_1032_18.
Dresco F, Puzenat E, Delobeau M, Salard D, Lihoreau T, Pelletier F, Aubin F. Lupus érythémateux cutanés réfractaires traités par bélimumab : étude descriptive monocentrique [Resistant and progressive cutaneous lupus erythematosus treated with belimumab: A retrospective monocentric study]. Rev Med Intern. 2020;41:152–9. https://doi.org/10.1016/j.revmed.2019.12.004.
Salle R, Chasset F, Kottler D, Picard-Dahan C, Jannic A, Mekki N, et al. Belimumab for refractory manifestations of cutaneous lupus: A multicenter, retrospective observational study of 16 patients. J Am Acad Dermatol. 2020;83:1816–9. https://doi.org/10.1016/j.jaad.2020.05.058.
Torrente-Segarra V, Peramiquel L, Bonet M. Belimumab in subacute cutaneous lupus erythematosus. Lupus. 2021;30:2017–8. https://doi.org/10.1177/09612033211033989.
Isenberg DA, Petri M, Kalunian K, Tanaka Y, Urowitz MB, Hoffman RW, et al. Efficacy and safety of subcutaneous tabalumab in patients with systemic lupus erythematosus: results from ILLUMINATE-1, a 52-week, phase III, multicentre, randomised, double-blind, placebo-controlled study. Ann Rheum Dis. 2016;75:323–31. https://doi.org/10.1136/annrheumdis-2015-207653.
Merrill JT, Wallace DJ, Wax S, Kao A, Fraser PA, Chang P, Isenberg D. Efficacy and safety of atacicept in patients with systemic lupus erythematosus: results of a twenty-four-week, multicenter, randomized, double-blind, placebo-controlled, parallel-arm. Phase IIb Study Arthritis Rheumatol. 2018;70:266–76. https://doi.org/10.1002/art.40360.
Fan Y, Gao D, Zhang Z. Telitacicept, a novel humanized, recombinant TACI-Fc fusion protein, for the treatment of systemic lupus erythematosus. Drugs Today (Barc). 2022;58:23–32. https://doi.org/10.1358/dot.2022.58.1.3352743.
Wise LM, Stohl W. Belimumab and rituximab in systemic lupus erythematosus: a tale of two B cell-targeting agents. Front Med (Lausanne). 2020;7:303. https://doi.org/10.3389/fmed.2020.00303.
Vital EM, Wittmann M, Edward S, Md Yusof MY, MacIver H, Pease CT, et al. Brief report: responses to rituximab suggest B cell-independent inflammation in cutaneous systemic lupus erythematosus. Arthritis Rheumatol. 2015;67:1586–91. https://doi.org/10.1002/art.39085.
Da Quelhas CR, Aguirre-Alastuey ME, Isenberg DA, Saracino AM. Assessment of response to B-cell depletion using rituximab in cutaneous lupus erythematosus. JAMA Dermatol. 2018;154:1432–40. https://doi.org/10.1001/jamadermatol.2018.3793.
Mehta N, Baskaran N, Arava S, Gupta S. B cell depletion therapy using rituximab to induce long-term remission of recalcitrant skin lesions of subacute cutaneous lupus erythematosus. BMJ Case Rep. 2022. https://doi.org/10.1136/bcr-2021-248476.
Arnold J, Dass S, Twigg S, Jones CH, Rhodes B, Hewins P, et al. Efficacy and safety of obinutuzumab in systemic lupus erythematosus patients with secondary non-response to rituximab. Rheumatology (Oxford). 2022;61:4905–9. https://doi.org/10.1093/rheumatology/keac150.
Merrill JT, Guthridge J, Zack D, Foster P, Burington B, Tran L, et al. SAT0187 discrimination of systemic lupus (SLE) patients with clinical response to obexelimab (XMAB\textregistered5871) based on a pattern of immunologic markers. Ann Rheum Dis. 2020;79:1035–6. https://doi.org/10.1136/annrheumdis-2020-eular.2972.
Mysler EF, Spindler AJ, Guzman R, Bijl M, Jayne D, Furie RA, et al. Efficacy and safety of ocrelizumab in active proliferative lupus nephritis: results from a randomized, double-blind, phase III study. Arthritis Rheum. 2013;65:2368–79. https://doi.org/10.1002/art.38037.
Merrill JT, Werth VP, Furie R, van Vollenhoven R, Dörner T, Petronijevic M, et al. Phase 2 trial of iberdomide in systemic lupus erythematosus. N Engl J Med. 2022;386:1034–45. https://doi.org/10.1056/NEJMoa2106535.
Ostendorf L, Burns M, Durek P, Heinz GA, Heinrich F, Garantziotis P, et al. Targeting CD38 with daratumumab in refractory systemic lupus erythematosus. N Engl J Med. 2020;383:1149–55. https://doi.org/10.1056/NEJMoa2023325.
Alexander T, Cheng Q, Klotsche J, Khodadadi L, Waka A, Biesen R, et al. Proteasome inhibition with bortezomib induces a therapeutically relevant depletion of plasma cells in SLE but does not target their precursors. Eur J Immunol. 2018;48:1573–9. https://doi.org/10.1002/eji.201847492.
Karnell JL, Wu Y, Mittereder N, Smith MA, Gunsior M, Yan L, et al. Depleting plasmacytoid dendritic cells reduces local type I interferon responses and disease activity in patients with cutaneous lupus. Sci Transl Med. 2021. https://doi.org/10.1126/scitranslmed.abf8442.
Werth VP, Furie RA, Romero-Diaz J, Navarra S, Kalunian K, van Vollenhoven RF, et al. Trial of anti-BDCA2 antibody litifilimab for cutaneous lupus erythematosus. N Engl J Med. 2022;387:321–31. https://doi.org/10.1056/NEJMoa2118024.
Furie R, Khamashta M, Merrill JT, Werth VP, Kalunian K, Brohawn P, et al. Anifrolumab, an anti-interferon-α receptor monoclonal antibody, in moderate-to-severe systemic lupus erythematosus. Arthritis Rheumatol. 2017;69:376–86. https://doi.org/10.1002/art.39962.
Morand EF, Furie R, Tanaka Y, Bruce IN, Askanase AD, Richez C, et al. Trial of anifrolumab in active systemic lupus erythematosus. N Engl J Med. 2020;382:211–21. https://doi.org/10.1056/NEJMoa1912196.
Morand EF, Furie RA, Bruce IN, Vital EM, Dall’Era M, Maho E, et al. Efficacy of anifrolumab across organ domains in patients with moderate-to-severe systemic lupus erythematosus: a post-hoc analysis of pooled data from the TULIP-1 and TULIP-2 trials. Lancet Rheumatol. 2022;4:e282–92. https://doi.org/10.1016/S2665-9913(21)00317-9.
Kalunian KC, Furie R, Morand EF, Bruce IN, Manzi S, Tanaka Y, et al. A randomized, placebo-controlled phase III extension trial of the long-term safety and tolerability of anifrolumab in active systemic lupus erythematosus. Arthritis Rheumatol. 2022. https://doi.org/10.1002/art.42392.
Tummala R, Abreu G, Pineda L, Michaels MA, Kalyani RN, Furie RA, Morand EF. Safety profile of anifrolumab in patients with active SLE: an integrated analysis of phase II and III trials. Lupus Sci Med. 2021. https://doi.org/10.1136/lupus-2020-000464.
Blum FR, Sampath AJ, Foulke GT. Anifrolumab for treatment of refractory cutaneous lupus erythematosus. Clin Exp Dermatol. 2022. https://doi.org/10.1111/ced.15335.
Plüß M, Piantoni S, Wincup C, Korsten P. rapid response of refractory systemic lupus erythematosus skin manifestations to anifrolumab—a case-based review of clinical trial data suggesting a domain-based therapeutic Approach. J Clin Med. 2022. https://doi.org/10.3390/jcm11123449.
Fetter T, Smith P, Guel T, Braegelmann C, Bieber T, Wenzel J. Selective Janus Kinase 1 Inhibition Is a Promising Therapeutic Approach for Lupus Erythematosus Skin Lesions. Front Immunol. 2020;11:344. https://doi.org/10.3389/fimmu.2020.00344.
Braegelmann C, Hölzel M, Ludbrook V, Dickson M, Turan N, Ferring-Schmitt S, et al. Spleen tyrosine kinase (SYK) is a potential target for the treatment of cutaneous lupus erythematosus patients. Exp Dermatol. 2016;25:375–9. https://doi.org/10.1111/exd.12986.
Werth VP, Fleischmann R, Robern M, Touma Z, Tiamiyu I, Gurtovaya O, et al. Filgotinib or lanraplenib in moderate to severe cutaneous lupus erythematosus: a phase 2, randomized, double-blind, placebo-controlled study. Rheumatology (Oxford). 2022;61:2413–23. https://doi.org/10.1093/rheumatology/keab685.
Calugareanu A, Grolleau C, Le Buanec H, Chasset F, Jachiet M, Battistella M, et al. Clinical efficacy of selective JAK1 inhibition and transcriptome analysis of chronic discoid lupus erythematosus. J Eur Acad Dermatol Venereol. 2022;36:e308–10. https://doi.org/10.1111/jdv.17839.
Joos L, Vetterli F, Jaeger T, Cozzio A, von Kempis J, Rubbert-Roth A. Treatment of refractory subacute cuataneous lupus erythematosus with baricitinib. Clin Exp Dermatol. 2022;47:748–50. https://doi.org/10.1111/ced.15005.
Kreuter A, Licciardi-Fernandez MJ, Burmann S-N, Paschos A, Michalowitz A-L. Baricitinib for recalcitrant subacute cutaneous lupus erythematosus with concomitant frontal fibrosing alopecia. Clin Exp Dermatol. 2022;47:787–8. https://doi.org/10.1111/ced.15044.
Morand EF, Tanaka Y, Furie R, Vital E, van Vollenhoven R, Kalunian K, et al. POS0190 efficacy and safety of baricitinib in patients with systemic lupus erythematosus: results from two randomised, double-blind, placebo-controlled, parallel-group, phase 3 studies. Ann Rheum Dis. 2022;81:327–8. https://doi.org/10.1136/annrheumdis-2022-eular.1968.
Bonnardeaux E, Dutz JP. Oral tofacitinib citrate for recalcitrant cutaneous lupus. JAAD Case Rep. 2022;20:61–4. https://doi.org/10.1016/j.jdcr.2021.09.030.
Ytterberg SR, Bhatt DL, Mikuls TR, Koch GG, Fleischmann R, Rivas JL, et al. Cardiovascular and cancer risk with tofacitinib in rheumatoid arthritis. N Engl J Med. 2022;386:316–26. https://doi.org/10.1056/NEJMoa2109927.
Morand E, Pike M, Merrill JT, van Vollenhoven R, Werth VP, Hobar C, et al. LB0004 efficacy and safety of deucravacitinib, an oral, selective, allosteric tyk2 inhibitor, in patients with active systemic lupus erythematosus: a phase 2, randomized, double-blind, placebo-controlled study. Ann Rheum Dis. 2022;81:209. https://doi.org/10.1136/annrheumdis-2022-eular.5020a.
Walker A, Erwig L, Foster K, Nevin K, Wenzel J, Worm M, et al. Safety, pharmacokinetics and pharmacodynamics of a topical SYK inhibitor in cutaneous lupus erythematosus: a double-blind Phase Ib study. Exp Dermatol. 2021;30:1686–92. https://doi.org/10.1111/exd.14253.
Maruyama A, Katoh N. Subacute cutaneous lupus erythematosus successfully treated with topical delgocitinib. J Dermatol. 2022. https://doi.org/10.1111/1346-8138.16639.
Park JJ, Little AJ, Vesely MD. Treatment of cutaneous lupus with topical ruxolitinib cream. JAAD Case Rep. 2022;28:133–5. https://doi.org/10.1016/j.jdcr.2022.08.038.
Borucki R, Werth VP. Emerging therapies for cutaneous lupus erythematosus. Cutis. 2020;105:276–7.
Skudalski L, Shahriari N, Torre K, Santiago S, Bibb L, Kodomudi V, et al. Emerging therapeutics in the management of connective tissue disease. Part I Lupus erythematosus and Sjögren syndrome. J Am Acad Dermatol. 2022;87:1–18. https://doi.org/10.1016/j.jaad.2021.12.067.
Sprow G, Dan J, Merola JF, Werth VP. Emerging Therapies in cutaneous lupus Erythematosus. Front Med (Lausanne). 2022;9:968323. https://doi.org/10.3389/fmed.2022.968323.
Gordon C, Amissah-Arthur M-B, Gayed M, Brown S, Bruce IN, D’Cruz D, et al. The British Society for Rheumatology guideline for the management of systemic lupus erythematosus in adults. Rheumatology (Oxford). 2018;57:e1–45. https://doi.org/10.1093/rheumatology/kex286.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
Open Access funding enabled and organized by Projekt DEAL. Funding was provided by GlaxoSmithKline for the characterization of autoreactive skin-associated B cells in patients’ skin samples of lupus erythematosus and other inflammatory skin diseases (grant number: #213383 / N-005.0523). LdV is funded by the University Medical Centre Bonn (UKB) BONFOR program (Grant 2022-1A-08).
Conflicts of interest/competing interests
Dennis Niebel has received speakers’ honoraria or travel expense reimbursements from Abbvie, Almirall, BMS, Boehringer Ingelheim, Eli Lilly, GlaxoSmithKline, Janssen-Cilag, Kyowa Kirin, LEO Pharma, L’Oreal, MSD, Novartis, Pfizer, Sanofi-Aventis and UCB. Luke de Vos has received financial support (travel expenses) from Pierre Fabre. Christine Brägelmann has received financial support (travel grants) from L’Oréal, Novartis, ADF and EADV. Joerg Wenzel has received financial support (clinical studies, travel grants, speaker’s fees) from GlaxoSmithKline, Novartis, Medac, Merck/Serono, Roche, Actelion, Pfizer, Spirig, ArrayBio, Biogen, and Incyte. Tanja Fetter declares no competing interests.
Ethical statement and consent
Ethics approval and patient consent to participate were not applicable for this study. Patient’s images displayed in this paper were obtained from patients treated at the University Hospital of Bonn and written informed consent was obtained from all patients to publish the images used.
Author’s contributions
JW conceived the idea for the article, and all authors performed the literature search and discussed the outline of the article. DN and LdV performed the data analysis, and TF drafted the graphic illustration. The first draft of the manuscript was written by DN and LdV. All authors commented on previous versions of the manuscript, and read and approved the final manuscript.
Data and code availability
Non-applicable.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Niebel, D., de Vos, L., Fetter, T. et al. Cutaneous Lupus Erythematosus: An Update on Pathogenesis and Future Therapeutic Directions. Am J Clin Dermatol 24, 521–540 (2023). https://doi.org/10.1007/s40257-023-00774-8
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40257-023-00774-8