
Abstract
Hypertrophic cardiomyopathy (HCM) affects as many as 1 in 200 people in the adult population globally. Patients may present with exertional dyspnea, presyncope or syncope, atrial and ventricular arrhythmias, heart failure, and even sudden cardiac death. Current guideline-based therapy involves medical therapy for treatment of symptoms in milder forms of the disease and surgical or catheter-based septal reduction therapies in obstructive HCM. Until recently, there has existed a gap between these two approaches that is now being filled by a new class of drugs, cardiac myosin inhibitors, which directly target the underlying disease process in HCM. Current investigations examine the effects of two cardiac myosin inhibitors on reported symptoms, echocardiographic evidence of disease, and the associated need for septal reduction. This paper reviews the contemporary evidence for the use of cardiac myosin inhibitors in HCM in adults and highlights future directions for this exciting field of cardiovascular medicine.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Cardiac myosin inhibitor therapy is an exciting new class of pharmacological therapy for management of obstructive hypertrophic cardiomyopathy. |
Ongoing clinical trials are investigating the efficacy of cardiac myosin inhibitor therapy in nonobstructive hypertrophic cardiomyopathy. |
This class of pharmacological therapy is poised to potentially revolutionize the management of hypertrophic cardiomyopathy. |
1 Introduction
Hypertrophic cardiomyopathy (HCM) is a complex cardiac condition with potential for life-threatening complications, characterized by abnormal thickening of the heart muscle, primarily of the left ventricle [1]. It represents one of the most prevalent inherited cardiovascular diseases, affecting as many as 1 in 200 individuals worldwide [1, 2]. Significant hypertrophy coupled with dynamic left ventricular outflow tract (LVOT) obstruction due to systolic anterior motion (SAM) of the mitral valve result in dyspnea, presyncope/syncope, and angina [3]. Both obstructive HCM (oHCM) and the nonobstructive form (nHCM) pose a substantial burden on affected individuals and their families, as they can lead to heart failure, arrhythmias, and sudden cardiac death [1]. While various therapeutic strategies have been employed to manage HCM, there remains an unmet need for more effective and targeted treatments. Myosin inhibitors have emerged as a promising class of drugs for the treatment of HCM, offering a novel approach to address the underlying pathophysiological mechanisms of this complex disorder.
2 Genetics of HCM
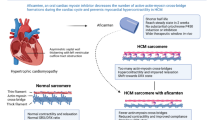
HCM has been associated with any of 1400 reported genetic variants encoding both sarcomeric and nonsarcomeric proteins [4]. Approximately 60% of HCM cases can be attributed to currently identified variants, with the most commonly involved genes being MYH7 and MYBPC3, which encode β-myosin heavy chain 7 and myosin-binding protein C3, respectively [5]. Missense variants in MYH7 and frameshift variants in MYBPC3 account for approximately half of familial HCM cases [3]. Altered expression of these proteins can produce excessive crossbridge formation (Fig. 1) between actin and myosin during cardiac muscle contraction [4]. Confirming this relationship, selective silencing of a β-myosin heavy chain variant prevented development of the disease in an HCM mouse model [6].

Graphic illustration of cardiac myosin inhibition in hypertrophic cardiomyopathy. LV left ventricle, LVOT left ventricular outflow tract
While variants in eight main genes encoding sarcomeric proteins account for as much as 90% of known familial HCM, pathogenic variants of genes encoding Z-disk proteins and other nonsarcomeric proteins are increasingly being shown to play a role in the etiology of HCM [7, 8]. Recent genome-wide association studies have shown strong associations between nonsarcomeric variants and HCM in a polygenic fashion, as well as a strong association between nonsarcomeric variants and increased diastolic blood pressure identified using Mendelian randomization [9]. These findings, along with the variable penetrance of well-established disease-driving genes as well as the apparent non-Mendelian patterns of inheritance in families without a well-studied gene variant, suggest a complex genetic etiology that is still far from being fully understood [8, 9].
Many pathogenic variants in HCM are associated with an increased rate of actin–myosin crossbridge formation. Specifically, variants of myosin and myosin binding protein C may weaken or destabilize the “off state” of cardiac myosin heads that is responsible for relaxation, causing more myosin heads to be available for cross-bridge formation [10]. This increased tendency for myosin to interact with actin results in hypercontractility and impaired relaxation of the heart muscle [10], leading to hypertrophy, fibrosis, and diastolic dysfunction [4, 11]. This dysregulation of actin–myosin cross-bridging makes myosin a key target for intervention.
3 Mechanism of Action of Mavacamten
Mavacamten was synthesized as an inhibitor of cardiac myosin adenosine triphosphatase (ATPase), allowing cardiac myosin to more easily occupy the super-relaxed “off state,” thus decreasing the hypercontractile state that is the sarcomeric hallmark of hypertrophic cardiomyopathy. While only a small number of animal or ex vivo studies assessed the mechanism of action of mavacamten, they are remarkable in the wide range of methods used and bring important insight into how to think about the way mavacamten impacts cardiac physiology.
The overriding consensus is that by stabilizing the super-relaxed state of cardiac myosin, mavacamten acutely decreases hypercontractility and improves relaxation in a dose-dependent manner. These effects are thought to be the drivers of long-term improvement in left ventricular (LV) mass, myocardial fibrosis, and myocyte disarray, which are the key pathologic components that define the HCM phenotype.
Sewanan et al. developed a mechanical testing setup that produced force-velocity curves and work loops of human engineered heart tissues [12]. They used this system to assess response of human heart tissue to mavacamten. They have shown, in an isometric setting, that while contractility decreased, relaxation (time from peak to 50% relaxation) occurred faster [12]. When shortening was allowed, mavacamten decreased the slope of myocyte end-systolic force/length relationship (a surrogate for end systolic pressure/volume relationship), indicating that mavacamten decreases contractility of healthy myocardium while speeding up relaxation [12].
Awinda et al. studied muscle mechanics in isolated papillary muscle strips obtained from either wild type RLC mice or HCM mice that expressed N47K RLC variant [13]. Mavacamten reduced calcium sensitivity to contraction while it increased the MgADP release rates for both genotypes, thereby contributing to faster cross-bridge detachment, which speeds up myocardial relaxation during diastole [13].
In a series of experiments, Green et al. used their mouse model of HCM to assess whether a small-molecule inhibitor of sarcomere power might ameliorate the disease at its source and abolish histologic features of HCM [14]. They have shown that in vivo, mavacamten decreased fractional shortening (a surrogate of ejection fraction) [14]. In addition, mavacamten slowed down development of LV hypertrophy during aging but only in mutant mice; finally, mavacamten led to a decrease of existing LV hypertrophy again in mutant mice, compared with no regression in wild type mice [14]. Most importantly, it prevented the development of fibrosis and myocardial disarray, thus ameliorating all three pathognomonic cellular markers of HCM [14].
These studies address the cellular mechanisms of mavacamten but not the impact of mavacamten on dynamic LVOT obstruction. This is understandable, as no small animal model of LVOT obstruction exists and it would be difficult to image LVOT obstruction in small animal models. However, it is well known that cats often exhibit an HCM phenotype and that isoproterenol delivery can induce LVOT obstruction [15]. Stern et al. showed that mavacamten efficiently decreased provoked LVOT gradients, thus giving mechanistic support for the use of mavacamten clinically [15].
4 Current Guideline-Based Medical Therapy and Associated Challenges
Currently available drug therapy for patients with dynamic LVOT obstruction seeks to achieve improvement in patient symptomatology. For symptomatic patients, nonvasodilating β-blockers are used for their negative chronotropic and inotropic effects [16, 17]. Until recently, β-blockers were the only medical therapy in obstructive HCM (oHCM) supported by evidence from a randomized controlled trial, rather than small cohort studies [17, 18]. Negative inotropes such as nondihydropyridine calcium channel blockers (CCBs) and disopyramide have also been used for pharmacologic LVOT obstruction treatment [16]. The utility of disopyramide is limited by the side effect profile of the drug, which includes anticholinergic effects and enhanced conduction through the atrioventricular (AV) node, particularly in atrial fibrillation. For this reason, disopyramide is not used as a monotherapy, as its side effect profile must be mitigated by β-blockers and nondihydropyridine calcium channel blockers [19].
There is a clinical dilemma in treating LVOT obstruction with comorbid conditions such as hypertension, atrial fibrillation, and heart failure, as current guidelines recommend discontinuing the use of vasodilators such as dihydropyridine calcium channel blockers, angiotensin-converting enzyme (ACE) inhibitors, and angiotensin receptor blockers (ARBs), which are cornerstones of hypertension and heart failure treatment, because preload and afterload-reduction can exacerbate LVOT obstruction and associated symptoms [16].
Management of advanced symptomatic oHCM often necessitates septal reduction therapy (SRT), which is achieved surgically via septal myectomy or by alcohol septal ablation [16]. SRT candidates usually have a dynamic peak LVOT gradient of at least 50 mmHg [16], and SRT is effective in reducing resting and poststress LVOT gradients in patients who do not respond to β-blockade and CCBs/disopyramide [20, 21]. It is clear that between the current guideline-recommended medical therapies and surgical options, there exists an opportunity to more directly address the disease process in HCM. Recognition of this gap has led to the development of novel myosin inhibitors for HCM [10] (Table 1).
5 Mavacamten
Mavacamten is an allosteric, selective, and reversible inhibitor of cardiac myosin ATPase. It reduces the rate at which myosin interacts with actin to form the cross-bridges that are responsible for muscle contraction (Fig. 1). This reduction in excess cross-bridge formation in turn leads to a super-relaxed state and associated reduction in LVOT obstruction, improvement of left ventricular filling pressures, and decreased myocardial wall stress [22]. Mavacamten is metabolized via CYP2C19 (74%), CYP3A4 (18%), and CYP2C9 (8%) with a half-life of 6–9 days in CYP2C19 normal metabolizers and up to 23 days in poor metabolizers [23]. To date, three major human studies have shown the efficacy of mavacamten as a novel therapy in HCM.
The PIONEER-HCM trial was a nonrandomized, open-label phase 2 trial that served as a proof-of-concept study for mavacamten. It included 21 patients with oHCM with New York Heart Association (NYHA) class II–III symptoms who also had LV hypertrophy demonstrated by LV wall thickness of ≥ 15 mm or ≥ 13 mm in patients with family history of HCM [22]. The primary endpoint of the study was to evaluate postexercise LVOT gradient reduction at 12 weeks, with secondary endpoints of changes in peak oxygen consumption (pVO2), resting and Valsalva LVOT gradients, resting left ventricular ejection fraction (LVEF), and numerically rated dyspnea score [22]. Cohort A (n = 11) received a weight-based starting dose of mavacamten 10 mg for weight ≤ 60 kg and 15 mg daily for weight > 60 kg after discontinuing existing therapies including β-blockers, CCBs, and disopyramide at least 2 weeks prior to start [22]. Doses were adjusted based on percentage decrease in LVEF from baseline at week 4, and the median dose in cohort A over the course of the study was 15 mg/day (range 10–20 mg/day) [22]. Cohort B (n = 10) continued β-blockers, if applicable, and started a lower dose of 2 mg/day that was increased to 5 mg/day at the end of week 4 for participants whose resting LVOT gradients decreased less than 50% from baseline [22]. Exclusion criteria included exertional syncope in the previous 6 months, sustained ventricular tachycardia, LV systolic dysfunction (LVEF < 45%), and history of persistent or paroxysmal atrial fibrillation in the past year [22].
The authors of PIONEER-HCM reported improvements in the primary and secondary outcomes for both cohort A and B [22]. For the primary outcome in cohort A, there was a decrease in the postexercise LVOT gradient from 103 ± 50 mmHg at baseline to 19 ± 13 mmHg at 12 weeks for a mean change of −89.5 mmHg [95% confidence intervals (CI) −138 mmHg to −40.7 mmHg, p = 0.008] [22]. The secondary outcomes in cohort A included a mean increase in pVO2 by 3.5 mL/kg/min ± 3.3, improvements in resting LVOT gradient with mean change −48 mmHg (p = 0.006) and Valsalva LVOT gradient with mean change −85 mmHg (p = 0.002) [22]. For cohort A, the median dose of mavacamten at 12 weeks was 15 mg daily with final dose ranging from 10 mg to 20 mg daily [22].
For the primary outcome in cohort B, the mean postexercise LVOT gradient decreased by a mean change of 25 mmHg (95% CI −138.3 to −40.7 mmHg, p = 0.02) [22]. Secondary outcomes included resting LVOT gradient reduction of −25 mmHg (p = 0.020), resting LVOT gradient mean change of −49 mmHg (p = 0.004) [22]. There was an increase in pVO2 by a mean of 1.7 mL/kg/min, and resting LVEF showed a mean change of −6% [22]. All ten patients in cohort B were titrated to mavacamten 5 mg daily by week 12 of therapy [22].
There was a large proportion of patients who showed improved NYHA scores in both groups (7/11 patients in cohort A and 8/10 patients in cohort B) [22]. Average Kansas City Cardiomyopathy Questionnaire (KCCQ) overall summary scores improved by 10.5 and 6.1 points in cohorts A and B, respectively [22]. Dyspnea scores also improved in both groups, and mavacamten was overall well tolerated, with 80% reporting only mild symptoms [22]. Four patients experienced atrial fibrillation, with one withdrawing from the study, which may have been related to initiation of mavacamten or discontinuation of metoprolol and disopyramide [22]. Notably, the focus of PIONEER-HCM on both LVOT gradients and patient-centric measures of functional status (pVO2) and quality of life (KCCQ, NYHA status) marks an approach to drug trials that is sure to be emulated.
The EXPLORER-HCM trial (n = 251) was a phase 3, multicenter, randomized, double-blind, placebo-controlled, parallel-group trial of adult patients with a diagnosis of oHCM who were randomized to either placebo (n = 128) or oral mavacamten (n = 123) with starting dose of 5 mg daily increasing to 15 mg daily over 14 weeks [24]. Total treatment duration was 30 weeks, with a subsequent wash-out period of 8 weeks [24]. The primary endpoint was a 1.5 mL/kg per min or greater increase in pVO2 and at least one NYHA class reduction or a 3.0 mL/kg per min or greater pVO2 increase without NYHA class worsening [24]. Secondary endpoints assessed changes in postexercise LVOT gradient, pVO2, NYHA class, KCCQ clinical summary score (KCCQ-CSS), and Hypertrophic Cardiomyopathy Symptom Questionnaire Shortness-of-Breath subscore (HCMSQ-SoB). A total of 92% of patients were allowed to continue background β-blocker or calcium channel blocker therapy, and 19 patients, 11 mavacamten and 8 placebo, included in the study had previous septal reduction therapy.
At the end of treatment, 45 (37%) of the patients in the mavacamten group met the primary endpoint compared to 22 (17%) of the placebo group (p = 0.0005). The authors reported that there was an improvement in all secondary endpoints for the mavacamten group with greater reductions in postexercise LVOT gradient (−47 mmHg) compared with placebo (−10 mmHg) (p = 0.0001), as well as an average pVO2 increase of 1.4 mL/kg per min compared with a decrease of 0.1 mL/kg per min in the placebo group (p = 0.0006), as well as improvement in KCCQ scores and in NYHA class for 65% of patients in the mavacamten group versus 35% of patients in the placebo group (p = 0.0001). It is worth mentioning the considerable percentage of improved NYHA and KCCQ scores in the placebo group. This fact may be both a result of the dynamic nature of HCM symptoms, and an inevitable consequence of using “soft” endpoints. The relatively high proportion of nonresponders may be attributed again to the use of “soft” endpoints, in which patients’ symptoms may be affected by other cardiac and noncardiac comorbidities and also possibly to the presence of HCM phenotypes that respond differently to treatment. A sustained reduction in all LVOT gradients to < 30 mmHg was achieved by 27 % of patients versus 1% in the placebo group, which is considered a complete response to therapy. Additionally, mavacamten reduced LVOT gradient to < 50 mmHg in 74% of patients versus 21% of patients in the placebo group. An analysis of patients who were not on background β-blocker therapy shows improvements in the primary outcome for patients who were in the mavacamten group versus the placebo group. A 5-year extension (MAVA-LTE) is ongoing, with 36-week interim results showing continued LVOT gradient decrease from baseline. Mean resting LVOT gradient change for 25 patients was −27.4 ± 33.33 mmHg, and the mean Valsalva LVOT gradient change was −45.7 ± 39.9 mmHg.
Treatment-emergent serious adverse events leading to decrease in LVEF less than 50% occurred in one patient following a serious cardiac event [24]. During the 30-week treatment period, there were an additional three patients who experienced a transient decrease in LVEF of less than 50%. Subsequently at week 30 end of treatment visits, four more patients experienced LVEF less than 50%. The authors of the study reported that three of the four patients who experienced LVEF reduction to less than 50% recovered to baseline after the 8-week washout. The fourth patient recovered to an LVEF of 50% [24]. Of note, no significant difference was found in the incidence of atrial fibrillation between the mavacamten and placebo group (2% versus 3%) [24].
Two important substudies of EXPLORER-HCM were conducted to examine the effects of mavacamten on cardiac structure and function. First, the cardiac magnetic resonance (CMR) imaging substudy examined changes in left ventricular mass index from baseline to week 30 as a primary endpoint and included additional exploratory endpoints of left atrial volume index, LV function, changes in cellular hypertrophy, and myocardial fibrosis among others. At 30 weeks, the mean LV mass index was 15.8 g/m2 (95% CI, −22.6 to −9.0) lower in the mavacamten group than placebo. There were also greater reductions in LV wall thickness and maximum left atrial volume index in the mavacamten group compared with placebo. Thus, this substudy demonstrated that treatment with mavacamten can produce reverse remodeling effects in addition to the previously reported improvements in NYHA and KCCQ scores, LVOT gradients, and exercise capacity [25].
A second substudy examined the effect of mavacamten on echocardiographic features in HCM. Analysis of echocardiography for all patients in the mavacamten (n = 123) and placebo (n = 128) was conducted. For patients in the mavacamten group, who at baseline displayed SAM of the mitral valve (n = 94 of 123), treatment for 30 weeks showed complete resolution of SAM in 80.9% (n = 76 of 94) compared with 34% of patients in the placebo group (n = 33 of 97). Mean decrease from baseline left atrial volume index was –7.5 mL/m2 with mavacamten compared with −0.1 mL/m2 with placebo. Mean septal e′ increased by 0.7 cm/s in the mavacamten group compared with a decrease of 0.02 cm/s in the placebo group. Mean septal E/e′ decreased by 3.5 with mavacamten compared with 0.3 with placebo. Mean lateral E/e′ decreased by 3.8 with mavacamten compared with an increase of 0.04 in the placebo group [26].
VALOR-HCM was a multicenter, randomized, placebo-controlled trial of 112 patients on maximally tolerated medical therapy with oHCM and NYHA class III–IV symptoms (or class II with exertional syncope/presyncope) who at baseline were eligible for septal reduction therapy (SRT) [27]. The primary endpoint of the study was whether patients underwent SRT, were continually eligible for SRT, or became ineligible on the basis of NYHA class symptoms and LVOT gradients after a treatment period of 16 weeks. Secondary endpoints include reductions of NYHA class, resting LVOT gradient, Valsalva LVOT gradient, and KCCQ-23 score. The mean age of the group was 60 ±12 years, and 46% were on β-blockers, 15% on calcium channel blockers, 32% on combination therapy and 20% on disopyramide during the study [27, 28]. Patients in the mavacamten group were initiated on 5 mg daily and titrated up to 15 mg daily. At 4 weeks if the LVOT gradient was < 30 mmHg during Valsalva maneuver the dose would be reduced to 2.5 mg daily and reevaluated for continued titration at week 8 for continued dose titration [16, 17].
At 16 weeks, 43 of 56 placebo patients (76.8%) and 10 of 56 mavacamten patients (17.9%) met guideline criteria or underwent SRT [27]. Two patients had a drop in LVEF below 50% and treatment was paused temporarily, but otherwise, there were no severe adverse events resulting in withdrawal from the trial [27].
A long-term extension of VALOR-HCM is ongoing, with the placebo group crossing over to receive mavacamten 5 mg daily at week 16, then subsequently undergo dose titration at weeks 20, 24, and 28 [29]. The most recent interim results at week 56 showed that 5 of 56 patients (8.9%) in the original mavacamten group and 10 of 52 patients (19.2%) in the placebo-then-mavacamten group met composite SRT criteria; 3 patients in each group chose to undergo SRT. At week 56, 96, of 108 patients (89%) continue taking mavacamten long term [29].
Mavacamten received Federal Drug Administration approval in the USA in April 2022 and European Commission approval in June 2023 after the publication of the results of the EXPLORER-HCM and VALOR-HCM trials. Dosing titration starts at 2.5–5 mg daily up to a maximum recommended dose of 15 mg daily [23]. Availability of mavacamten is restricted to specialty pharmacies due to the CAMZYOS Risk Evaluation and Mitigation Strategies (REMS) program requirement for all patients who are initiating therapy [30]. The CAMZYOS REMS program requires that patients undergo LVOT gradient after Valsalva assessment at initiation, weeks 4, 8, and 12 of therapy. Following this, the program requires echocardiographic evaluation starting at week 12 of therapy and repeated every 12 weeks to assess systolic function, with 4-week serial echocardiograms required after any dose adjustment. This precaution has been put in place due to the number of patients who developed an EF of < 50% in the EXPLORE-HCM and VALOR-HCM studies.
Following FDA approval, real-world patient data is starting to accumulate under the REMS program. Data made available by one tertiary care center illustrated the clinical experience of the first 150 patients with oHCM treated with mavacamten at this center [31]. The mean age of patients was 65 years, 83% were on β-blockers, and 61% had NYHA class III symptoms [31]. All patients started mavacamten 5 mg and doses were titrated on the basis of symptom assessment and echocardiographic assessment, both of which were performed at baseline and every 4 weeks thereafter for the first 12 weeks with titration occurring at the end of this period [31]. Results were reported after 261±143 days of treatment, at which time 69 (46%) patients had ≥ 1 NYHA class and 27 (18%) additional patients had ≥ 2 NYHA class improvement [31]. The mean Valsalva LVOT gradient decreased from 72±43 mmHg at baseline to 29±31 mmHg at 4 weeks, 29±28 mmHg at 8 weeks, and 30±29 mmHg at 12 weeks [31]. All patients had Valsalva LVOT gradients ≥ 30 mmHg at baseline, which reduced to 29% at 4 weeks, 28% at 8 weeks, and 30% at 12 weeks. A total of 40 patients reported no improvement in symptoms, though in this group Valsalva LVOT gradient decreased from 73±39 mmHg to 34±27 mmHg at 4 weeks, 35±28 mmHg at 8 weeks, and 30±24 mmHg at 12 weeks (p < 0.001) [31]. No patients in the cohort underwent SRT or developed heart failure requiring admission, and three patients paused mavacamten treatment due to LVEF decreased below 50% [31]. Notably, the rate of drug interruption due to LVEF reduction (2%) was lower than in EXPLORER-HCM (5%), which the authors attribute to a slower titration approach than was used in clinical trials [31].
The major drug interactions with mavacamten are driven by negative inotropic effect and cytochrome P450 (CYP) system interactions. An increase in negative inotropic effect occurs when combining mavacamten with nondihydropyridine calcium channel blockers or with β-blockers. When β-blockers are necessary, there is no recommended reduction in mavacamten dose but rather a monitoring of LVEF when initiating the combination. It is not recommended to combine both nondihydropyridine CCB and β-blockers together with mavacamten [23]. Mavacamten is metabolized mainly through CYP2C19 and CYP3A4; therefore, inhibitors and inducers of these enzymes may affect the drug’s plasma concentrations, increasing the risk of systolic dysfunction [23]. Additionally, mavacamten may cause fetal toxicity based on findings in animal studies, and confirmation of absence of pregnancy is needed when applicable [23]. Furthermore, mavacamten may decrease the efficacy of certain combined hormonal contraceptives [23]. As such, a mavacamten pregnancy surveillance program is underway to monitor maternal, fetal, and infant outcomes as a result of mavacamten exposure during pregnancy or breastfeeding [32].
6 Aficamten
A second-in-class cardiac myosin inhibitor, aficamten has a shorter half-life than mavacamten (3.4 days compared with 7–9 days), allowing for more frequent dosage adjustment [33]. Preclinical data has shown a prolonged half-life of 4.6 days in CYP2D6 poor metabolizers, though poor metabolizers did not appear to have reduced clearance of the drug, as no increase was observed in the area under the plasma drug concentration-time curve between CYP2D6 poor and normal metabolizers [34].
The REDWOOD-HCM trial was a phase 2 dose-finding study with primary endpoints of reported drug-related adverse events, serious adverse events, and decreases in LVEF below 50% [35]. Secondary endpoints of the study were changes in resting and Valsalva LVOT gradients. The experimental group was divided into cohort 1 (n = 14), which received a starting dose of 5 mg daily that was titrated up to a maximum of 15 mg over 10 weeks, and cohort 2 (n = 14), which received a starting dose of 10 mg daily titrated up to a maximum of 30 mg at 10 weeks. Exclusion criteria included paroxysmal atrial fibrillation during screening, planned or prior septal reduction therapy, history of LVEF < 45%, and use of disopyramide in the prior 4 weeks. As such, patients taking disopyramide were subsequently included as a third cohort to further aficamten’s safety.
In cohort 1, resting LVOT gradients decreased at week 10 from 54±25 mmHg to 13±4 mmHg (p = 0.0003 versus placebo) and gradients with Valsalva decreased from 74±25 mmHg to 38 ± 14 mmHg (p = 0.001 versus placebo) [35]. In cohort 2, resting at week 10 gradients decreased from 58±36 mmHg to 15±22 mmHg (p = 0.0004 versus placebo) and Valsalva gradients decreased from 82±37 mmHg to 30±30 mm Hg (p = 0.0001 versus placebo) [35]. Valsalva gradients in both groups returned to baseline after a 2-week washout period. In cohort 1, LVEF decreased from 73%±6% to 67%±9% (p = 0.007) and in cohort 2 from 75%±6% to 64%±8% (p = 0.0001) with no change in the placebo group [35]. Analysis of the relationship between aficamten dose and LVEF revealed a dose-dependent decrease with a mean reduction in LVEF of 0.6% per mg of aficamten administered [35]. The average final dose in cohort 1 was aficamten 10 mg and in cohort 2 the average final dose was aficamten 14 mg [35].
The third cohort taking concurrent disopyramide (n = 13) received between 5–15 mg aficamten daily based on on-site echocardiogram findings and were on an average disopyramide dose of 346 mg/day [35]. Mean resting LVOT gradient was decreased by 27±22 mmHg (p < 0.0001), and mean Valsalva LVOT gradient was decreased by 28±32. 11 (85%) (p = 0.0002) [35]. A total of 11 patients (85%) reported NYHA class improvement of at least one level. Of note, all eight patients with baseline NYHA class III experienced NYHA class improvement. Two of five patients who were NYHA class II did not experience an improvement in NYHA class but did experience improvement in LVOT-G. [24] The authors reported there were no significant adverse events in cohort 3 [35].
7 Future Directions
Further research opportunities exist to evaluate the utility of cardiac myosin inhibitors in HCM. In particular, it remains to be seen if patients with nonobstructive HCM (nHCM) will derive similar benefits. To this end, ODYSSEY-HCM [36], a multicenter double-blind clinical trial has recently completed patient enrolment to assess the effects of mavacamten in nHCM. Inclusion criteria include a diagnosis of HCM with NYHA class II/III symptoms as well as peak LVOT gradients below 30 mmHg at rest and below 50 mmHg with Valsalva or exercise. Primary endpoints of this trial will be change in KCCQ-23 clinical summary score and change in baseline pVO2 at 48 weeks. Secondary endpoints will include changes in ventilatory efficiency slope (VE/VCO2), proportion of participants with at least 1 NYHA class improvement from baseline, changes in N-terminal pro B-type natriuretic peptide (NT-proBNP), and changes in HCM symptom questionnaire shortness of breath (HCMSQ-SoB) scores [36]. Given the reverse remodeling effects seen in the EXPLORER-HCM subsets discussed earlier, ODYSSEY-HCM holds much promise in elucidating the effects of mavacamten on cellular markers, echocardiographic indicators of disease, and symptoms in patients who do not stand to benefit from SRT (Table 2).
Similarly, the efficacy and safety of aficamten in nHCM will be assessed in the ACACIA-HCM trial, which is currently enrolling participants [37]. Participants will receive either placebo or between 5 and 20 mg of aficamten per day (guided by echocardiographic assessment) for up to 72 weeks. The primary endpoint of the study will be change in KCCQ-CSS scores, and secondary endpoints will include changes in a composite of Z-scores of pVO2 and VE/VCO2 slope, proportion of patients with at least 1 class improvement in NYHA functional class, and change in NT-proBNP.
A number of patient populations were not included in existing literature. Almost all patients in PIONEER-HCM, EXPLORER-HCM, AND VALOR-HCM were white, and several comorbidities were excluded, including atrial fibrillation at the time of initiating therapy. Specific data are not available regarding whether mavacamten is effective in patients with anomalous papillary muscles and/or mitral valve subvalvular apparatus.
It is important to note the current lack of long-term data regarding the use of cardiac myosin inhibitors. The completion of the EXPLORER-HCM and VALOR-HCM long-term extension studies will provide important guidance for decision-making regarding the optimal use of mavacamten, particularly with regards to its role in the algorithm for HCM management alongside or possibly in place of other available therapies (Fig. 2). Furthermore, cost effectiveness will remain a concern when weighing cardiac myosin inhibitor therapy against septal reduction therapy. One estimate puts the mean cost of septal myectomy in the USA at $42,000 [38], while the list price of mavacamten is $89,500 annually [39]. At present, decisions in favor of one over the other will hinge on careful discussion of patient-specific risks and benefits, such as access to centers of surgical excellence, preferences regarding long-term medication use, and surgical eligibility.

Proposed and possible future roles of CMI in HCM treatment in accordance with the 2024 AHA/ACC hypertrophic cardiomyopathy guidelines [40]. ACC American College of Cardiology, AHA American Heart Association, CMI cardiac myosin inhibitor, HCM hypertrophic cardiomyopathy, SRT septal reduction therapy
8 Conclusions
Cardiac myosin inhibitors are a revolutionary new class of medical therapy for HCM with a disease-specific mechanism of action. With approved clinical indications for the treatment of oHCM, myosin inhibition is already benefiting many symptomatic patients clinically, and exciting opportunities exist to assess whether it is equally efficacious in nonobstructive HCM. As clinical data continues to accumulate, it is becoming increasingly clear that cardiac myosin inhibitor therapy will play a key role in addressing the burden of HCM.
References
Maron BJ, Maron MS. Hypertrophic cardiomyopathy. Lancet. 2013;381(9862):242–55.
Semsarian C, Ingles J, Maron MS, Maron BJ. New perspectives on the prevalence of hypertrophic cardiomyopathy. J Am Coll Cardiol. 2015;65(12):1249–54.
Marian AJ, Braunwald E. Hypertrophic cardiomyopathy: genetics, pathogenesis, clinical manifestations, diagnosis, and therapy. Circ Res. 2017;121(7):749–70.
Cheng Z, Fang T, Huang J, Guo Y, Alam M, Qian H. Hypertrophic cardiomyopathy: from phenotype and pathogenesis to treatment. Front Cardiovasc Med. 2021;8: 722340.
Ingles J, Burns C, Bagnall RD, Lam L, Yeates L, Sarina T, et al. Nonfamilial hypertrophic cardiomyopathy: prevalence, natural history, and clinical implications. Circ Cardiovasc Genet. 2017. https://doi.org/10.1161/CIRCGENETICS.116.001620.
Jiang J, Wakimoto H, Seidman JG, Seidman CE. Allele-specific silencing of mutant Myh6 transcripts in mice suppresses hypertrophic cardiomyopathy. Science. 2013;342(6154):111–4.
Walsh R, Thomson KL, Ware JS, Funke BH, Woodley J, McGuire KJ, et al. Reassessment of Mendelian gene pathogenicity using 7,855 cardiomyopathy cases and 60,706 reference samples. Genet Med. 2017;19(2):192–203.
Walsh R, Offerhaus JA, Tadros R, Bezzina CR. Minor hypertrophic cardiomyopathy genes, major insights into the genetics of cardiomyopathies. Nat Rev Cardiol. 2022;19(3):151–67.
Harper AR, Goel A, Grace C, Thomson KL, Petersen SE, Xu X, et al. Common genetic variants and modifiable risk factors underpin hypertrophic cardiomyopathy susceptibility and expressivity. Nat Genet. 2021;53(2):135–42.
Trivedi DV, Adhikari AS, Sarkar SS, Ruppel KM, Spudich JA. Hypertrophic cardiomyopathy and the myosin mesa: viewing an old disease in a new light. Biophys Rev. 2018;10(1):27–48.
Choudhury L, Mahrholdt H, Wagner A, Choi KM, Elliott MD, Klocke FJ, et al. Myocardial scarring in asymptomatic or mildly symptomatic patients with hypertrophic cardiomyopathy. J Am Coll Cardiol. 2002;40(12):2156–64.
Sewanan LR, Shen S, Campbell SG. Mavacamten preserves length-dependent contractility and improves diastolic function in human engineered heart tissue. Am J Physiol Heart Circ Physiol. 2021;320(3):H1112–23.
Awinda PO, Watanabe M, Bishaw Y, Huckabee AM, Agonias KB, Kazmierczak K, et al. Mavacamten decreases maximal force and Ca(2+) sensitivity in the N47K-myosin regulatory light chain mouse model of hypertrophic cardiomyopathy. Am J Physiol Heart Circ Physiol. 2021;320(2):H881–90.
Green EM, Wakimoto H, Anderson RL, Evanchik MJ, Gorham JM, Harrison BC, et al. A small-molecule inhibitor of sarcomere contractility suppresses hypertrophic cardiomyopathy in mice. Science. 2016;351(6273):617–21.
Stern JA, Markova S, Ueda Y, Kim JB, Pascoe PJ, Evanchik MJ, et al. A small molecule inhibitor of sarcomere contractility acutely relieves left ventricular outflow tract obstruction in feline hypertrophic cardiomyopathy. PLoS One. 2016;11(12): e0168407.
Ommen SR, Mital S, Burke MA, Day SM, Deswal A, Elliott P, et al. 2020 AHA/ACC Guideline for the diagnosis and treatment of patients with hypertrophic cardiomyopathy: a report of the American College of Cardiology/American Heart Association Joint Committee on Clinical Practice Guidelines. Circulation. 2020;142(25):e558–631.
Dybro AM, Rasmussen TB, Nielsen RR, Andersen MJ, Jensen MK, Poulsen SH. Randomized trial of metoprolol in patients with obstructive hypertrophic cardiomyopathy. J Am Coll Cardiol. 2021;78(25):2505–17.
Olivotto I, Tomberli B, Spoladore R, Mugelli A, Cecchi F, Camici PG. Hypertrophic cardiomyopathy: the need for randomized trials. Glob Cardiol Sci Pract. 2013;2013(3):243–8.
Adler A, Fourey D, Weissler-Snir A, Hindieh W, Chan RH, Gollob MH, et al. Safety of outpatient initiation of disopyramide for obstructive hypertrophic cardiomyopathy patients. J Am Heart Assoc. 2017. https://doi.org/10.1161/JAHA.116.005152.
Smedira NG, Lytle BW, Lever HM, Rajeswaran J, Krishnaswamy G, Kaple RK, et al. Current effectiveness and risks of isolated septal myectomy for hypertrophic obstructive cardiomyopathy. Ann Thorac Surg. 2008;85(1):127–33.
Ball W, Ivanov J, Rakowski H, Wigle ED, Linghorne M, Ralph-Edwards A, et al. Long-term survival in patients with resting obstructive hypertrophic cardiomyopathy comparison of conservative versus invasive treatment. J Am Coll Cardiol. 2011;58(22):2313–21.
Heitner SB, Jacoby D, Lester SJ, Owens A, Wang A, Zhang D, et al. Mavacamten treatment for obstructive hypertrophic cardiomyopathy: a clinical trial. Ann Intern Med. 2019;170(11):741–8.
Bristol-Myers Squibb. CAMZYOS (mavacamten) [package insert]. U.S. Food and Drug Administration. https://www.accessdata.fda.gov/drugsatfda_docs/label/2022/214998s000lbl.pdf.
Olivotto I, Oreziak A, Barriales-Villa R, Abraham TP, Masri A, Garcia-Pavia P, et al. Mavacamten for treatment of symptomatic obstructive hypertrophic cardiomyopathy (EXPLORER-HCM): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet. 2020;396(10253):759–69.
Saberi S, Cardim N, Yamani M, Schulz-Menger J, Li W, Florea V, et al. Mavacamten favorably impacts cardiac structure in obstructive hypertrophic cardiomyopathy: EXPLORER-HCM cardiac magnetic resonance substudy analysis. Circulation. 2021;143(6):606–8.
Hegde SM, Lester SJ, Solomon SD, Michels M, Elliott PM, Nagueh SF, et al. Effect of mavacamten on echocardiographic features in symptomatic patients with obstructive hypertrophic cardiomyopathy. J Am Coll Cardiol. 2021;78(25):2518–32.
Desai MY, Owens A, Geske JB, Wolski K, Naidu SS, Smedira NG, et al. Myosin inhibition in patients with obstructive hypertrophic cardiomyopathy referred for septal reduction therapy. J Am Coll Cardiol. 2022;80(2):95–108.
Desai MY, Wolski K, Owens A, Naidu SS, Geske JB, Smedira NG, et al. Study design and rationale of VALOR-HCM: evaluation of mavacamten in adults with symptomatic obstructive hypertrophic cardiomyopathy who are eligible for septal reduction therapy. Am Heart J. 2021;239:80–9.
Desai MY, Owens A, Wolski K, Geske JB, Saberi S, Wang A, et al. Mavacamten in patients with hypertrophic cardiomyopathy referred for septal reduction: week 56 results from the VALOR-HCM randomized clinical trial. JAMA Cardiol. 2023;8(10):968–77.
CAMZYOS REMS: MyoKardia; 2023. https://www.camzyosrems.com.
Desai MY, Hajj-Ali A, Rutkowski K, Ospina S, Gaballa A, Emery M, et al. Real-world experience with mavacamten in obstructive hypertrophic cardiomyopathy: observations from a tertiary care center. Prog Cardiovasc Dis. 2024. https://doi.org/10.1016/j.pcad.2024.02.001.
Bristol-Myers Squibb. Mavacamten Pregnancy Surveillance Program 2023. clinicaltrials.gov/study/NCT05939700.
Chuang C, Collibee S, Ashcraft L, Wang W, Vander Wal M, Wang X, et al. Discovery of aficamten (CK-274), a next-generation cardiac myosin inhibitor for the treatment of hypertrophic cardiomyopathy. J Med Chem. 2021;64(19):14142–52.
Malik FI, Robertson LA, Armas DR, Robbie EP, Osmukhina A, Xu D, et al. A Phase 1 dose-escalation study of the cardiac myosin inhibitor aficamten in healthy participants. JACC Basic Transl Sci. 2022;7(8):763–75.
Maron MS, Masri A, Choudhury L, Olivotto I, Saberi S, Wang A, et al. Phase 2 study of aficamten in patients with obstructive hypertrophic cardiomyopathy. J Am Coll Cardiol. 2023;81(1):34–45.
Bristol-Myers Squibb. A study of mavacamten in non-obstructive hypertrophic cardiomyopathy (ODYSSEY-HCM) 2023. https://classic.clinicaltrials.gov/ct2/show/NCT05582395.
Cytokinetics. A phase 3, multi-center, randomized, double-blind trial to evaluate the efficacy and safety of aficamten compared to placebo in adults with symptomatic non-obstructive hypertrophic cardiomyopathy (ACACIA-HCM) ClinicalTrials.gov2023. https://classic.clinicaltrials.gov/ct2/show/NCT06081894.
Panaich SS, Badheka AO, Chothani A, Mehta K, Patel NJ, Deshmukh A, et al. Results of ventricular septal myectomy and hypertrophic cardiomyopathy (from Nationwide Inpatient Sample [1998-2010]). Am J Cardiol. 2014;114(9):1390–5.
Rakowski H, Bataiosu R. can novel myosin inhibitors defer septal reduction therapy in HCM? At what risk and at what cost? Circulation. 2023;147(11):864–6.
Ommen SR, Ho CY, Asif IM, Balaji S, Burke MA, Day SM, et al. 2024 AHA/ACC/AMSSM/HRS/PACES/SCMR Guideline for the management of hypertrophic cardiomyopathy: a report of the American Heart Association/American College of Cardiology Joint Committee on Clinical Practice Guidelines. Circulation. 2024;149(23):e1239–311.
Acknowledgements
M.Y.D. acknowledges the Haslam family endowed chair in cardiovascular medicine.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
The current study was funded by unrestricted philanthropic gifts by the Ratner family, Stinson family and Anderson family for M.Y.D.’s research. S.O. and K.R. have received salary support from unrestricted philanthropic gifts by the Haslam family, Ratner family, Stinson family, and Anderson family.
Disclosures
M.Y.D. is a consultant and has research agreements with Bristol Myers Squibb, Cytokinetics, Tenaya, Viz-AI and Edgewise. N.S. is a consultant for Bristol Myers Squibb. No industry support was utilized in the conduct of this study. J.K.K., B.X., R.B., N.T., K.R., S.O., M.T., and Z.B.P. have no potential conflicts of interest that might be relevant to this manuscript.
Ethics Approval
Not applicable.
Consent to Participate
Not applicable.
Consent for Publication
Not applicable.
Availability of Data and Material (Data Transparency)
Not applicable.
Code Availability (Software Application or Custom Code)
Not applicable.
Author Contributions
All authors contributed to the study conception. The literature search was performed by J.K.K., who also wrote the first draft of the manuscript. B.X., N.T., and R.B. revised subsequent versions of the manuscript, and all authors read and approved the final version of the manuscript.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Kalinski, J.K., Xu, B., Boyd, R. et al. Novel Cardiac Myosin Inhibitor Therapy for Hypertrophic Cardiomyopathy in Adults: A Contemporary Review. Am J Cardiovasc Drugs 24, 591–602 (2024). https://doi.org/10.1007/s40256-024-00667-z
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40256-024-00667-z