
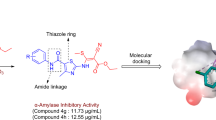
Abstract
Previous studies have shown that 2-arylbenzimidazole derivatives have a strong anti-diabetic effect. To further explore this potential, we develop new analogues of the compound using ligand-based drug design and tested their inhibitory and binding properties through QSAR analyses, molecular docking, dynamic simulations and pharmacokinetic studies. By using quantitative structure activity relationship and ligand-based modification, a highly precise predictive model and design of potent compounds was developed from the derivatives of 2-arylbenzimidazoles. Molecular docking and simulation studies were then conducted to identify the optimal binding poses and pharmacokinetic profiles of the newly generated therapeutic drugs. DFT was employed to optimize the chemical structures of 2-arylbenzimidazole derivatives using B3LYP/6-31G* as the basis set. The model with the highest R2trng set, R2adj, Q2cv, and R2test sets (0.926, 0.912, 0.903, and 0.709 respectively) was chosen to predict the inhibitory activities of the derivatives. Five analogues designed using ligand-based strategy had higher activity than the hit molecule. Additionally, the designed molecules had more favorable MolDock scores than the hit molecule and acarbose and simulation studies confirm on their stability and binding affinities towards the protein. The ADME and druglikeness properties of the analogues indicated that they are safe to consume orally and have a high potential for total clearance. The results of this study showed that the suggested analogues could act as α-amylase inhibitors, which could be used as a basis for the creation of new drugs to treat type 2 diabetes mellitus.



















Similar content being viewed by others


Data availability
All relevant data pertaining to this research are provided within this document or can be obtained upon request.
References
Abdullahi SH, Uzairu A, Ibrahim MT, Umar AB (2021) Chemo-informatics activity prediction, ligand based drug design, Molecular docking and pharmacokinetics studies of some series of 4, 6-diaryl-2-pyrimidinamine derivatives as anti-cancer agents. Bull Natl Res Cent. https://doi.org/10.1186/s42269-021-00631-w
Adegboye AA, Khan KM, Salar U, Aboaba SA, Kanwal CS, Fatima I, Taha M, Wadood A, Mohammad JI, Khan H, Perveen S (2018) 2-Aryl benzimidazoles: synthesis, In vitro α-amylase inhibitory activity, and molecular docking study. Eur J Med Chem 150:248–260. https://doi.org/10.1016/j.ejmech.2018.03.011
Baig MH, Ahmad K, Rabbani G, Danishuddin M, Choi I (2018) Computer aided drug design and its application to the development of potential drugs for neurodegenerative disorders. Current Neuropharmacol 16(6):740–748. https://doi.org/10.2174/1570159X15666171016163510
Bansal Y, Silakari O (2012) The therapeutic journey of benzimidazoles: a review. Bioorg Med Chem 20(21):6208–6236
Belete TM (2020) A recent achievement in the discovery and development of novel targets for the treatment of type-2 diabetes mellitus. J Exp Pharmacol 12:1–15. https://doi.org/10.2147/JEP.S226113
Bickerton GR, Paolini GV, Besnard J, Muresan S, Hopkins AL (2012) Quantifying the chemical beauty of drugs. Nat Chem 4(2):90–98. https://doi.org/10.1038/nchem.1243
Bitencourt-Ferreira G, de Azevedo WF (2019) Molegro virtual docker for docking. Methods Mol Biol 20(53):149–167. https://doi.org/10.1007/978-1-4939-9752-7_10
Bonsembiante L, Targher G, Maffeis C (2021) Type 2 diabetes and dietary carbohydrate intake of adolescents and young adults: what is the impact of different choices. Nutrients 13(10):3344. https://doi.org/10.3390/nu13103344
Camilo LMM, Marfran CDS, Kássio MGL, Francis LM (2019) Improving data splitting for classification applications in spectrochemical analyses employing a random-mutation Kennard-Stone algorithm approach. Bioinformatics 35(24):5257–5263. https://doi.org/10.1093/bioinformatics/btz421
Chandra A, Qamar I, Singh N (2020) Identification of novel and potent small molecule inhibitor of SRPK1: mechanism of dual inhibition of cancer progression. Aging (albany NY) 12:1945–4589. https://doi.org/10.18632/aging.202301
Cherkasov A, Muratov EN, Fourches D, Varnek A, Baskin II, Cronin M, Dearden J, Gramatica P, Martin YC, Todeschini R (2014) QSAR modeling: where have you been? where are you going to? J Med Chem 57:4977–5010
Daina A, Michielin O, Zoete V (2017) SwissADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci Rep 7:42717. https://doi.org/10.1038/srep42717
Docking simulation studies of some novel anticancer compounds on MALME-3M melanoma cell line. Egypt J Med Hum Genet. 22(1).https://doi.org/10.1186/s43042-020-00126
Elshakre ME, Noamaan MA, Moustafa H, Butt H (2020) Density functional theory, chemical reactivity, pharmacological potential and molecular docking of dihydrothiouracil-indenopyridopyrimidines with human-DNA topoisomerase II. Int J Mol Sci 21(4):1253. https://doi.org/10.3390/ijms21041253
Ferreira LT, Borba JVB, Moreira-Filho JT, Rimoldi A, Andrade CH, Costa FTM (2021) QSAR-based virtual screening of natural products database for identification of potent antimalarial hits. Biomolecules 11(3):459. https://doi.org/10.3390/biom11030459
Ibrahim ZY, Uzairu A, Shallangwa GA, Abechi SE (2021) Application of QSAR method in the design of enhanced antimalarial derivatives of azetidine-2-carbonitriles, their molecular docking, drug-likeness, and SwissADME properties. Iran J Pharm Res 20(3):254–270. https://doi.org/10.22037/ijpr.2021.114536.14901
Idris MO, Abechi SE, Shallangwa GA (2021) Computational evaluation of some compounds as potential anti-breast cancer agents. Future J Pharm Sci. https://doi.org/10.1186/s43094-021-00315-2
Ikwu FA, Shallangwa GA, Mamza PA, Uzairu A (2020) In silico studies of piperazine derivatives as potent anti-proliferative agents against PC-3 prostate cancer cell lines. Heliyon 6(1):e03273. https://doi.org/10.1016/j.heliyon.2020.e03273
Jafaru SC, Uzairu A, Sallau MS, Ndukwe GI, Ibrahim MT, Danazumi AU (2023) Structure-based design of potential anti-schistosomiasis agent targeting SmHDAC8: an in silico approach utilizing QSAR, MD simulation and ADMET prediction. Chem Afr 7(2):725–745
Khan M, Hashim MJ, King JK, Govender RD, Mustafa H, Al Kaabi J (2020a) Epidemiology of type 2 diabetes - global burden of disease and forecasted trends. J Epidemiol Glob Health 10(1):107–111. https://doi.org/10.2991/jegh.k.191028.001
Khan MAB, Hashim MJ, King JK, Govender RD, Mustafa H, Al Kaabi J (2020b) Epidemiology of type 2 diabetes - global burden of disease and forecasted trends. J Epidemiol Glob Health. 10(1):107–111. https://doi.org/10.2991/jegh.k.191028.001
Kroemer G, López-Otín C, Madeo F, de Cabo R (2018) Carbotoxicity-noxious effects of carbohydrates. Cell 175(3):605–614. https://doi.org/10.1016/j.cell.2018.07.044
Li C, Begum A, Numao S, Kwan HP, Withers SG, Brayer GD (2005) Acarbose rearrangement mechanism implied by the kinetic and structural analysis of human pancreatic α-amylase in complex with analogues and their elongated counterparts. Biochemistry 44(9):3347–3357. https://doi.org/10.1021/bi048334e
Lin AH, Lee BH, Nichols BL, Quezada-Calvillo R, Rose DR, Naim HY, Hamaker BR (2012) Starch source influences dietary glucose generation at the mucosal α-glucosidase level. J Boil Chem 287(44):36917–36921. https://doi.org/10.1074/jbc.M112.378331
Meng XY, Zhang HX, Mezei M, Cui M (2011) Molecular docking: a powerful approach for structure-based drug discovery. Curr Comput Aided Drug Des 7(2):146–157. https://doi.org/10.2174/157340911795677602
Mitra I, Saha A, Roy K (2010) Exploring quantitative structure-activity relationship studies of antioxidant phenolic compounds obtained from traditional Chinese medicinal plants. Mol Simul 36(13):1067–1079. https://doi.org/10.1080/08927022.2010.503326
Nakrani MN, Wineland RH, Anjum F (2021) Physiology, Glucose Metabolism. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2022 https://www.ncbi.nlm.nih.gov/books/NBK560599/
Oboh G, Isaac AT, Akinyemi AJ, Ajani RA (2014) Inhibition of key enzymes linked to type 2 diabetes and sodium nitroprusside induced lipid peroxidation in rats’ pancreas by phenolic extracts of avocado pear leaves and fruit. Int J Biomed Sci 10(3):208–216
Peyrot Des Gachons C, Breslin PA (2016) Salivary amylase: digestion and metabolic syndrome. Curr Diab Rep 16(10):102. https://doi.org/10.1007/s11892-016-0794-7
Ramachandran GN, Sasisekharan V (1968) Conformation of polypeptides and proteins. Adv Protein Chem 23:283–438
Rebollo-Lopez MJ, Lelièvre J, Alvarez-Gomez D, Castro-Pichel J, Martínez-Jiménez F, Papadatos G, Kumar V, Colmenarejo G, Mugumbate G, Hurle M, Barroso V, Young RJ, Martinez-Hoyos M, González del Río R, Bates RH, Lopez-Roman EM, Mendoza-Losana A, Brown JR, Alvarez-Ruiz E, Marti-Renom MA, Overington JP, Cammack N, Ballell L, Barros-Aguire D (2015) Release of 50 new, drug-like compounds and their computational target predictions for open source anti-tubercular drug discovery. PLoS ONE 10(12):e0142293. https://doi.org/10.1371/journal.pone.0142293
Rorsman P, Ashcroft FM (2018) Pancreatic β-cell electrical activity and insulin secretion: of mice and men. Physiol Rev 98(1):117–214. https://doi.org/10.1152/physrev.00008.2017
Saddique FA, Ahmad M, Ashfaq UA, Muddassar M, Sultan S, Zaki MEA (2022) Identification of cyclic sulfonamides with an N-arylacetamide group as α-glucosidase and α-amylase inhibitors: biological evaluation and molecular modeling. Pharmaceuticals 15(1):106. https://doi.org/10.3390/ph15010106
Sivakumar PM, Geetha Babu SK, Mukesh D (2007) QSAR studies on chalcones and flavonoids as anti-tuberculosis agents using genetic function approximation (GFA) method. Chem Pharm Bull 55(1):44–49. https://doi.org/10.1248/cpb.55.44
Thatikayala M, Garige AK, Gadegoni H (2022) Benzimidazole: Pharmacological Profile. In P. Kendrekar, & V. Adimule (Eds.), IntechOpen. https://doi.org/10.5772/intechopen.102091
Umar AB, Uzairu A, Shallangwa GA, Uba S (2021) Ligand-based drug design and molecular
Williams CJ, Headd JJ, Moriarty NW, Prisant MG, Videau LL, Deis LN, Verma V, Keedy DA, Hintze BJ, Chen VB, Jain S, Lewis SM, Arendall WB 3rd, Snoeyink J, Adams PD, Lovell SC, Richardson JS, Richardson DC (2017) MolProbity: more and better reference data for improved all-atom structure validation. Protein Sci 27(1):293–315. https://doi.org/10.1002/pro.3330
Zafar F, Gupta A, Thangavel K, Khatana K, Sani AA, Ghosal A, Tandon P, Nishat N (2020) Physicochemical and pharmacokinetic analysis of anacardic acid derivatives. ACS Omega 5(11):6021–6030. https://doi.org/10.1021/acsomega.9b04398
Acknowledgements
The authors sincerely acknowledge Ahmadu Bello University, Zaria for its support in the course of this research.
Funding
This research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.
Author information
Authors and Affiliations
Contributions
AU and KSA conceptualized the research idea; KSA performed all the computational analysis and wrote the manuscript; AC and NS performed molecular dynamic simulation and analyzed the results; KSA, SEA, GAS, ABU, AC and NS analyses the result to ensure minimal errors, AU supervised and proof read the manuscript. All authors read and approved the manuscript before final submission.
Corresponding author
Ethics declarations
Conflict of interest
The authors have no conflict of interest to declare.
Ethical approval
Not Applicable.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Aminu, K.S., Uzairu, A., Chandra, A. et al. Exploring the potential of 2-arylbenzimidazole scaffolds as novel α-amylase inhibitors: QSAR, molecular docking, simulation and pharmacokinetic studies. In Silico Pharmacol. 12, 29 (2024). https://doi.org/10.1007/s40203-024-00205-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s40203-024-00205-4