
Abstract
Lipid keratopathy is a disease in which fat deposits accumulate in the cornea, leading to opacification and decrease of visual acuity. This condition can be idiopathic without signs of previous corneal disease or secondary to ocular or systemic diseases. Lipid keratopathy is usually associated with abnormal vascularization of the cornea, and the lipid classically deposits adjacent to these vessels. Treatment of this condition usually aims to eliminate or prevent abnormal vessel formation, and several modalities have been described. In this review we summarize the etiology, pathophysiology, and clinical presentation of lipid keratopathy and describe current and emerging treatment regimens.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
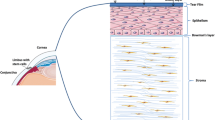
Lipid keratopathy (LK) is a disease characterized by lipid deposition in the cornea, most commonly secondary to corneal neovascularization. |
Corneal neovascularization is the growth of new blood vessels and lymphatic vessels into previously avascular areas of the cornea and can result from ocular trauma, inflammation, or infection. |
Lipid keratopathy can be distinguished from other causes of ocular lipid deposition by the presence of neovascularization and characteristic pathologic findings. |
Secondary LK typically presents unilaterally with cream-colored opacification adjacent to neovascularization, while idiopathic LK is usually bilateral. |
Various pharmacologic and surgical methods have been described in the treatment of LK; however, high costs, risk of recurrence, and treatment complications limit the clinical utility of many of the current regimens. |
Digital Features
This article is published with digital features, including a summary slide, to facilitate understanding of the article. To view digital features for this article go to https://doi.org/10.6084/m9.figshare.12988532.
Introduction
Lipid keratopathy (LK) is characterized by the opacification of the cornea due to lipid deposition. LK may be idiopathic, with no evidence of systemic or local disease, or secondary due to ocular infection, inflammation, or trauma. Most commonly, LK occurs in regions of corneal neovascularization (NV) and scarring [1]. These lesions can be progressive and threaten the visual axis. Treatment modalities for LK vary widely in terms of approach and effectivity. The purpose of this paper is to review the literature and analyze the etiology, pathophysiology, clinical manifestations, differential diagnosis, and treatment of LK.
Methods
A literature search using various databases, including PubMed, Scopus, and ScienceDirect, was conducted for studies published in English. No restrictions were set for the date of publications, which ranged from 1876 to 2019. Key search terms included “lipid keratopathy,” lipid degeneration,” “lipid deposition,” “fat deposition,” “corneal neovascularization,” “angiogenesis,” “lymphangiogenesis,” and a combination of these terms. Inclusion criteria for publications included those that discussed the etiologies, pathogenesis, clinical manifestations, differential diagnosis, and management of corneal NV or LK. References were also acquired from citations in publications found in the original search. Non-English articles were used if abstract information was available in English or a translation was possible.
This article is based on previously conducted studies and does not contain any studies with human participants or animals performed by any of the authors.
Historical Overview
Findings consistent with LK may have been reported as early as 1876 by Baumgarten [2], who described “fatty degeneration” in a patient with sclerosing keratitis. Cogan and Kuwabara [3] were the first to coin the term lipid keratopathy in 1958, which they described as a lipid exudation adjacent to vascularization due to inflammation or trauma. In their review, they note several alternative terms that have been used to describe what is now known as lipid keratopathy, including fatty dystrophy, adiposis of the eye, dystrophia adiposa corneae, lipidosis corneae, xanthomatosis, secondary steatosis, and lipid interstitial keratitis. In the years following the 1958 article, there have been many studies on the biochemical, histopathologic, and pathophysiologic aspects of the disease.
Etiology
Idiopathic LK is characterized by neutral fat, glycoproteins, cholesterol, and lipid deposits in the stromal layer of the cornea and the adjacent limbus [1, 4, 5]. Idiopathic LK may occur without prior vascularization and inflammation, and when serum levels of chylomicrons, very-low-density lipoprotein (VLDL) low-density lipoprotein (LDL), and high-density lipoprotein (HDL) are in the expected ranges. Idiopathic LK usually occurs bilaterally. Lipid deposition in idiopathic LK may be due to excess lipid production or a failure to metabolize fat [6], possibly due to a similar mechanism that causes arcus senilis [7]. Some researchers have hypothesized that these lipids originate from the aqueous humor [8], while others suggest that lipid can be deposited due to incompetent limbal vessels [9]. Another proposed mechanism involves intrinsic metabolic derangements within keratocytes, leading to lipid-rich cells and inflammatory foci with the presence of cholesterol clefts [10]. Undetected low-grade inflammation and subsequent NV may also cause idiopathic LK [1]. The exact mechanism is unclear, however, and understanding of the condition is complicated by the fact that a primary disorder leading to leakage of lipid could then cause secondary inflammation and corneal NV. Table 1 outlines key distinguishing features between idiopathic and secondary LK.
Secondary LK almost always occurs due to the deposition of lipids into a neovascularized area of the cornea; consequently virtually any cause of corneal NV can lead to LK. These etiologies can be inflammatory, traumatic, iatrogenic, or degenerative. Trachoma, an infection caused by the bacteria Chlamydia trachomatis, is the world’s leading cause of blindness, and recurrent episodes can lead to eyelash-induced corneal abrasions, ulcerations, and NV [11]. Onchocerciasis, also called river blindness, is a parasitic infection caused by Onchocerca volvulus and is another common cause of blindness worldwide. Microfilariae produced by adult worms travel to the cornea and cause inflammation and NV [12]. Herpetic corneal infections, which can arise from either herpes simplex virus 1 (HSV-1) or herpes zoster, are also tightly linked to NV and LK [13]. Recurrent infection of HSV-1 causes herpes simplex keratitis, which is characterized by inflammation of the cornea, leading to NV, ulceration, scarring, and lipid deposition [14]. Reactivation of the varicella-zoster virus (shingles) can cause herpes zoster ophthalmicus, which can also cause secondary LK. LK has also been reported as a result of other forms of bacterial keratitis [15] but, in theory, could occur from any infection of the cornea, including fungal or parasitic infections.
Ocular trauma, when severe, can also induce corneal NV. Chemical burns, particularly alkali burns and thermal burns, are known to cause rapid and severe inflammation that promotes NV [16]. Contact lens use has been associated with corneal NV in up to 11–23% of individuals [17], which can also lead to LK [18]. This may be due to hypoxia, mechanical injury, or limbal stem cell deficiency [19, 20]. Corneal transplantation, the most common solid tissue transplantation procedure, can cause corneal NV and subsequent LK via suture-induced inflammation and alloimmune responses [21, 22]. Corneal NV can reduce graft survival after corneal transplant [23]. Intracorneal ring segments were previously used for low to moderate myopia correction and now are used to manage keratoconus, pellucid marginal degeneration, and iatrogenic corneal ectasia [24]. Implantation of these devices may cause deep stromal NV and subsequent lipid deposition into the potential spaces between lamellar channels and the surface of the implant [25, 26]. Indeed, any ocular foreign body could lead to NV and lipid deposition.
Systemic dyslipoproteinemias, including familial lecithin-cholesterol acyltransferase (LCAT) deficiency (FLD), Fish-eye disease (FED), and Tangier disease (TD) may also lead to lipid deposition. All three conditions manifest with low plasma HDL levels and corneal clouding. LCAT plays a key role in removing cholesterol from peripheral cells and transporting it to the liver in the form of cholesterol esters on HDL. FLD is a rare disease characterized by complete LCAT deficiency and the accumulation of unesterified cholesterol and lecithin in the plasma. Ocular manifestations include bilateral corneal opacities comprised of granular dots that are limited to the corneal stroma [27]. It is thought that the dots are composed of cholesterol or other lipid materials due to the systemic accumulation of unesterified cholesterol [28]. The corneal opacities classically appear only in homozygotes and present before other systemic manifestations, which include hypertension, hypertriglyceridemia, hemolytic normochromic normocytic anemia, and proteinuria which often progresses to end-stage renal disease [29]. Kidney biopsy will reveal lipoprotein-X deposition in the glomeruli [29]. FED is another rare disease characterized by partial LCAT deficiency. Unlike in true LCAT deficiency, corneal opacities are the only clinical manifestation of FED. They appear as dot-like opacities in all layers of the cornea except the epithelium. Histopathologic studies show vacuoles filling Bowman’s layer and the corneal stroma between collagen fibrils; these vacuoles are believed to contain lipid [27]. Diagnosis of FLD and FED is supported by clinical findings and laboratory tests and confirmed by gene analysis; therefore complete blood count, urinalysis, and lipid panels may be indicated in these patients if these diseases are suspected [29]. Lastly, TD is a disorder characterized by a deficiency of HDL in the plasma leading to cholesterol ester accumulation throughout the body. This condition presents with corneal clouding, neuropathy, hepatosplenomegaly, large yellow-orange tonsils, and cardiovascular disease. Corneal opacities in this condition appear as a dot-like haze on slit-lamp examination and is limited to the stroma on confocal microscopy [30, 31]. Biochemical and histopathologic analyses have proven that the corneal opacities are due to esterified cholesterol and lipid deposition [32]. Diagnosis can be confirmed by molecular genetic testing [33].
Terrien marginal degeneration (TMD) is an uncommon condition that leads to slowly progressive, non-inflammatory thinning of the peripheral cornea. This condition is characterized by furrowing of the cornea that typically begins superiorly, followed by superficial vascularization and lipid deposition at the leading edge of the pannus [34]. TMD will initially appear as white-yellow punctate opacifications that may resemble arcus senilis, but over time will develop into a solid line at the anterior edge. LK has also been described in a patient using nasal continuous positive airway pressure for obstructive sleep apnea where air leakage, lagophthalmos and diminished Bell’s phenomenon led to chronic irritation and lipid deposition [35]. Other etiologies of LK reported in the literature include anterior scleritis [36], corneal hydrops [8], mustard gas exposure [37, 38] and interstitial keratitis [39]. A summary of the proposed etiologies of LK is given in Table 2.
Pathophysiology and Pathology
Angiogenesis is defined as the formation of new vessels from pre-existing ones and plays an important role in most blinding ocular conditions [40]. This process is regulated by growth factors, cytokines, and antiangiogenic molecules. The optical quality of the cornea relies on its transparency and avascularity, two properties that are often tightly associated. As such, the cornea has developed several mechanisms to maintain avascularity by balancing these pro-angiogenic factors with antiangiogenic factors. This dynamic process is termed “angiogenic privilege” [41]. This balance can be disturbed by tissue injury from infection, inflammation, or trauma. When this occurs, angiogenic factors become upregulated, while antiangiogenic factors are downregulated, leading to invasion of new blood vessels (hemangiogenesis) and lymphatic vessels (lymphangiogenesis) into previously avascular areas of the cornea. This is termed corneal NV.
Among the most important factors leading to corneal NV are members of the vascular endothelial growth factor (VEGF) family, including VEGF-A, VEGF-B, VEGF-C, VEGF-D, and placental growth factor. VEGF-A is considered to be the main factor for normal and pathologic blood vessel growth, while VEGF-C and VEGF-D are mostly implicated in lymphangiogenesis [14]. VEGF-A, when bound to the receptor tyrosine kinase VEGF receptor 2 (VEGFR-2), promotes the migration and proliferation of vascular endothelial cells and also increases vascular permeability [14, 42]. Basic fibroblast growth factor (bFGF) includes FGF-1, which is expressed in the normal corneal epithelium, and FGF-2, which is upregulated after injury [43]. bFGF also leads to the migration of endothelial cells as well as matrix remodeling and VEGF overexpression [44]. Together, the bFGF and VEGF signaling pathways may interact to potentiate angiogenesis and recruit monocytes, macrophages, and neutrophils to sites of inflammation [45]. Other important pro-angiogenic factors include platelet-derived growth factor [46], angiopoietins [47], and various inflammatory mediators [14, 48]. The proposed mechanism of LK is illustrated in Fig. 1.

Proposed mechanism of lipid keratopathy. FGF fibroblast growth factor, IFN interferon, IL Interleukin, NK natural killer, PDGF platelet-derived growth factor, TNF tumor necrosis factor, VEGF vascular endothelial growth factor
The cornea, however, possesses multiple mechanisms to maintain avascularity. Some of these antiangiogenic factors come from the corneal epithelium. One such example is the “decoy” VEGF receptors that bind these molecules, a process which serves a critical role in preventing angiogenesis [49, 50]. These receptors include soluble VEGFR-1, which prevents hemangiogenesis by binding VEGF-A [49], and non-vascular VEGFR-3, which prevents lymphangiogenesis by binding VEGF-C and VEGF-D [51]. Membrane-type 1 metalloproteinase (MT1-MMP) appears to be another important antiangiogenic factor expressed in the corneal basal epithelium. MT1-MMP produces other antiangiogenic factors, such as angiostatin and neostatin, via proteolytic activity [43]. The limbal stem cells also play an important role in corneal epithelium renewal, and the limbus itself may prevent corneal NV, as evidenced by the increase in corneal NV with limbal stem cell deficiency and improvement with limbal stem cell transplantation [52]. The mechanism is controversial, but the limbus may act as a physical and physiologic barrier to angiogenesis [53].
Several steps must occur for blood vessels to develop in the cornea. This likely occurs due to a process called sprouting [54]. Upon initial injury due to infection, inflammation, or trauma, damaged corneal epithelial cells release growth factors and interleukin (IL)-α. These molecules recruit innate immune cells, including monocytes, macrophages, neutrophils, and natural killer (NK) cells, which in turn release cytokines such as IL-1, IL-6, and tumor necrosis factor, which activate other immune cells. Macrophages act as another source of pro-angiogenic factors such as VEGF and can be enhanced by NK cells secreting interferon-gamma [45, 55]. When bound to their receptors on nearby vessels, these pro-angiogenic growth factors stimulate vascular endothelial cell proliferation and “activation” [43]. This stimulation leads to the alteration of endothelial cell adhesion molecules and the expression of proteolytic enzymes, allowing the endothelial cells to degrade their basement membrane and migrate from post-capillary venules towards these angiogenic stimuli. Eventually, a lumen will form along with the development of branches, leading to the formation of a loop connecting the tip of one lumen to another [48]. These loops have afferent and efferent limbs and appear to be a basic process in corneal NV [56]. Newly formed endothelial loops are relatively fragile and can lead to small intracorneal hemorrhages. The clearance of blood recruits more macrophages, which secrete VEGF, further supporting the vascularization process [56]. Vascularization most commonly occurs in the superior and middle third of the anterior stroma [57].
Corneal NV can lead to lipid deposition and subsequent LK in several ways. As blood vessels are formed, larger quantities of lipoproteins can reach these areas, especially if accompanied by high levels of serum lipoproteins [58]. Cogan and Kuwabara [3] proposed that most patients with LK have associated elevations in circulating cholesterol levels. These newly formed vascular tissues are unusually permeable due to a lack of pericyte coverage, fewer basement membrane layers, and fewer tight junctions, allowing leakage of lipid and cholesterol [59, 60]. This cholesterol-rich lipid is endocytosed by fibroblasts, transferred to lysosomes, and then hydrolyzed into free cholesterol. Once re-esterified in the Golgi complex, cholesterol is then stored as droplets in the cytoplasm or used in membrane metabolism. Membranous lamellae form, possibly to trap this excess cholesterol in a soluble, non-crystalline form [58]. However, these fibroblasts eventually become overloaded with lipid, leading to necrosis and deposition of these membranous lamellae along with crystalline material in the corneal stroma [58]. Necrosis initiates an inflammatory response, exacerbating the process of NV and furthering lipid deposition.
The presence of lipid droplets is characteristic of LK pathology. Cogan and Kuwabara [3] described two types of fatty plaques in the setting of LK: globular and granular lipids. The globular lipids exist intracellularly and appear as bright-red droplets 10–20 μm wide on histochemical staining with oil red-O. The granular lipids are derived from these globular lipids within cells that have necrosed and appear as smaller granules. Jack and Luse [5] observed irregular spaces containing membrane remnants in the corneal stroma on electron microscopy, along with degenerative changes in keratocytes. They also noted an abundance of macrophages near pathologic blood vessels, which may play a role in lipid removal and regression of LK [58]. Silva-Araujo et. al [61] performed biochemical analysis on a patient with bilateral idiopathic LK, which revealed levels of cholesterol and sphingomyelin that were 6- to 11-fold higher in the affected patient’s cornea than in controls.
Interestingly, while NV is almost always a requirement for LK, not all NV leads to lipid deposition. This is particularly apparent in conditions leading to “conjunctivalization” of the cornea, such as in limbal stem cell deficiency (LSCD). LSCD, which is a failure of stem cells to regenerate corneal epithelium, can occur from acquired injuries, such as chemical or thermal ocular burns and Stevens–Johnson syndrome and from inherited conditions such as aniridia and ectodermal dysplasia [62,63,64]. These stem cells are typically located in the basal epithelial layer at the corneal limbus and can be congenitally absent or damaged by ocular injury. When the ability to renew corneal epithelium is lost, the corneal surface will become covered by conjunctival epithelium instead, referred to as “conjunctivalization.” [65] This may lead to persistent inflammation and superficial corneal NV, but it may not lead to lipid deposition [66]. The mechanism of this phenomenon is unknown. We speculate that it could be related to the maturation of newly formed vessels or pericyte recruitment. When vascular tissue is newly formed, high levels of angiogenic factors and decreased numbers of pericytes, tight junctions, and basement membrane layers may lead to increased vessel permeability and lipid deposition. Therefore, the reverse of these circumstances may prevent lipid deposition from occurring. It is thought that corneal pericytes originate from bone marrow-derived cells and pre-existing limbal capillaries [67]. Perhaps conjunctivalization of the cornea leads to faster or greater quantities of limbal pericyte recruitment. It may also be conceivable that VEGF-induced vascular permeability is downregulated sooner in these tissues. Further studies must be pursued to elucidate a mechanism for this phenomenon.
Clinical Manifestations and Differential Diagnosis
Idiopathic LK typically presents bilaterally without previous corneal NV or serum lipid abnormalities. The lens, ocular fundi, and intraocular pressure are normal. Secondary LK will present with findings related to the primary pathology, such as keratitis and dendritic ulcers in herpes simplex infection. The associated NV can appear in three forms, namely, superficial stromal NV, deep stromal vascularization, and vascular pannus, each of which is often associated with a certain etiology. Superficial vascularization occurs when vessels develop from the superficial marginal arcade into the subepithelium as a result of trauma, inflammation, or infection [68]. Deep stromal vascularization occurs anywhere between Bowman’s layer and Descemet’s membrane due to anterior segment injury, scleritis, tuberculosis, and syphilis. Lastly, vascular pannus presents with collagen and vessel growth from the limbus to the peripheral cornea. Pannus usually occurs from ocular surface disease [69]. Common to all forms of NV is a resulting decrease in visual acuity. This can be due to opacity caused by the blood cells themselves, corneal irregularity due to pannus, high-order aberrations from irregular vascular walls, or leakage from vessels [41]. This leakage can be edematous or lipidic, the latter being the case of LK. Lipid deposition near the pupil will cause a marked decrease in visual acuity, but isolated peripheral corneal NV rarely has a substantial effect on visual acuity [41].
On examination, cream-colored opacification or fan-like cholesterol crystals on the corneal stroma may be observed surrounding blood vessels (Figs. 2, 3, 4, 5, 6). A fan-shaped plaque is common in the setting of active keratitis [3]. A disc-shaped distribution may be seen in an otherwise normal eye years after keratitis. In the setting of diffuse corneal NV, corresponding diffuse lipid deposition will be observed instead of a localized plaque. As the disease progresses, lipid deposition can cause NV, which subsequently leads to more lipid deposition, further decreasing visual acuity. Ring-shaped deposits may be present in idiopathic LK [9, 70]. In vivo confocal microscopy (IVCM) can also help in characterizing lipid deposits in LK. IVCM often reveals crystalline structures in the stroma in both idiopathic and secondary LK [9, 13, 18, 26, 31, 38, 71]. Deposits in secondary LK have also been described as amorphous or granular [18, 26, 31]. Animal models have shown several patterns of lipid deposition that can appear in both the peripheral and central cornea either adjacent or non-adjacent to corneal NV and anywhere from the basal epithelial layer to the deep stromal layer [18]. A key differentiating feature is that idiopathic LK will typically show no evidence of inflammation [71].

(Courtesy of Majid Moshirfar)
Yellow-white central corneal opacification representing lipid keratopathy likely due to interstitial keratitis from the herpes virus

(Courtesy of Dean P. Ouano)
Lipid deposition at the ends of neovascular vessels encroaching on the central cornea

(Courtesy of Dean P. Ouano)
Lipid keratopathy in a fan-like pattern surrounding corneal neovascularization

(Courtesy of Dean P. Ouano)
Lipid deposition secondary to neovascularization from intracorneal ring segments that has coalesced into a white-colored central corneal opacification

(Courtesy of Gerald B. Rosen)
Lipid deposition surrounding multiple corneal neovascular vessels
Corneal lipid deposition has been described in other conditions, including Schnyder’s crystalline dystrophy and corneal arcus. Corneal arcus is the most common form of ocular lipid deposition and is characterized by the deposition of cholesterol and phospholipid into the peripheral cornea [58]. Although the mechanism of corneal arcus is unclear, it is thought to be part of the normal aging process and most often appears bilaterally in patients over 50 years of age [27]. Corneal arcus is more common in men than women and can be associated with underlying lipid disorders, such as hypercholesterolemia or familial hyperlipidemia [27]. Corneal arcus first develops at the superior and inferior periphery of the cornea, likely due to the greater perfusion and warmer temperatures in these areas leading to increased capillary permeability and lipoprotein deposition [27]. When the lipid is delivered beyond the reach of the peripheral vascular supply, it cannot be cleared, leading to arcus formation [72]. This deposition begins in the deep layers of the stroma and will eventually involve the entire thickness of the stroma, including Bowman’s layer and Descemet’s membrane [73].
In contrast to LK, lipid deposition in corneal arcus occurs in the absence of inflammation or cellular damage and is primarily extracellular. Corneal arcus will appear as gray peripheral lesions that first develop at the superior and inferior periphery of the cornea and eventually extend around the entire circumference. The arcus is separated from the limbus by an area 0.3–1.0 mm in size (lucid interval of Vogt) [27]. Corneal arcus typically occurs bilaterally, but unilateral arcus may be seen in carotid artery stenosis, possibly due to decreased ocular blood flow to the ipsilateral eye providing protection from arcus formation [74]. Because it is limited to the periphery of the cornea, this lipid deposition does not cause diminished visual acuity [1].
Schnyder corneal dystrophy or crystalline stromal dystrophy is an uncommon condition associated with cholesterol and phospholipid deposition in the cornea and vision loss. This presents typically during the first few decades of life with central corneal haze that progresses peripherally, with the development of corneal arcus at an earlier age than in the general population [75]. These opacities can appear crystalline or diffuse and occur primarily in the anterior stroma into Bowman’s layer [27]. Lipid accumulation within fibroblasts with subsequent necrosis and lipid deposition is found in Schnyder corneal dystrophy, similar to LK [58]. Again, however, there will be no associated inflammation or corneal NV.
Treatment
There are multiple avenues of treatment for LK, most of which are used to eliminate corneal NV. Standard therapy for corneal NV typically begins with corticosteroids due to their ability to suppress the activity of inflammatory cells and mediators that release pro-angiogenic growth factors [76]. Steroids can be useful if the primary etiology of NV is inflammatory, but these agents do not inhibit angiogenesis directly, and NV can form in the absence of inflammation. Prolonged steroid use can also lead to well-known side effects, including cataract and glaucoma. Therefore, multiple steroid-sparing modalities for the treatment of corneal NV and LK have been proposed, including photodynamic therapy (PDT), anti-VEGF antibodies, argon laser treatment, needlepoint cautery, and penetrating keratoplasty.
PDT is used to eliminate cancer cells and has also proven useful in the field of ophthalmology. A vascular-selective light-sensitive substance, such as verteporfin or dihematoporhyrin, is injected into the cornea, followed by irradiation of the area with a low-power laser directed towards the area of interest. The interaction of light, oxygen, and photosensitizers creates reactive oxygen species, leading to endothelial damage, microvascular thrombosis, and the occlusion of blood vessels in the desired area with little effect on surrounding tissues. In the setting of LK, this method can counteract NV and the deposition of cholesterol and glycoproteins in the cornea, restoring visual acuity [77].
Multiple studies have reported improved visual acuity after treatment with PDT for LK due to herpes simplex keratitis [78], contact lens keratitis [79], and corneal transplantation [80]. In one prospective study, Yoon et al. [78] describe an approach using PDT to treat 18 eyes with corneal vascularization, of which eight eyes were noted to have LK due to herpetic keratitis or trauma. Lipid-formulated verteporfin prepared at a dose of 6 mg/m2 based on body surface area was diluted with 5% dextrose in water to a volume of 30 mL and then administered intravenously over 10 min. A 689-nm non-thermal laser light was delivered at a power intensity of 600 mW/cm2 over several spots ranging in size from 3 to 5 mm, delivering a total light dose of 150 J/cm2. After 1 year, corneal NV was reduced in 77.8% of eyes, with complete vascular occlusion achieved in 50% of eyes. Visual acuity improved in 44.4% of eyes [78]. Goh et al. [13] reported a similar approach in a patient with a history of herpes zoster ophthalmicus. IVCM after treatment demonstrated a marked reduction in the density of LK. Advantages of PDT are that it is minimally invasive and can be repeated if lesions are recurrent. A major limitation to the use of verteporfin is its high cost. A less expensive option is dihematoporphyrin; however, it is associated with significant side effects, such as iritis, transient angioedema requiring treatment, delayed phototoxic reaction, and delayed cutaneous reaction [13].
Other treatment methods involve topical, subconjunctival, and intracorneal administration of anti-VEGF monoclonal antibodies, such as bevacizumab and ranibizumab. Bevacizumab is currently the only medication approved by the US Food and Drug Administration for intravenous administration for the treatment of several neoplastic conditions, but it is often used off-label to treat age-related macular degeneration associated with choroidal NV [81], diabetic retinopathy [82], neovascular glaucoma [83], and central retinal vein occlusion [84]. VEGF plays a crucial role in angiogenesis; thus, the ability of these antibodies to block VEGF-mediated vessel formation underlies their clinical utility in treating neovascular conditions of the eye. VEGF also increases vessel permeability, possibly contributing to lipid leakage from NV and the development of LK. Therefore, anti-VEGF therapy is a potential therapeutic strategy for LK. Indeed, several studies have shown beneficial results from treating corneal NV with lipid deposition, which are outlined in Table 3 [85,86,87,88,89,90]. Bevacizumab has been shown to decrease the neovascularized corneal area and improve visual acuity [91]. Subconjunctival injection can induce involution of new vessels and restore corneal function [92]. Because bevacizumab is a full-length immunoglobulin with a molecular weight of 149 kDa, it is thought that its size could prevent topical formulations from penetrating the corneal epithelium, limiting efficacy. However, NV can lead to a damaged epithelial barrier, allowing entry of topical bevacizumab [93]. Indeed, topical bevacizumab appears to have comparable efficacy with subconjunctival injection [91].
Combining subconjunctival injection of bevacizumab with intrastromal injection may allow for increased drug concentrations in deeper corneal layers. Oh et al. [85] describe an approach to treating three eyes of three patients with extensive superficial and deep corneal NV and consequent LK. A single subconjunctival injection of bevacizumab at a concentration of 1.25 mg/0.05 mL was performed at the limbus adjacent to the pathologic vessels. Another injection was performed in the stroma at the site of NV. These injections were repeated at 1-month intervals until their condition stabilized or vessels became occluded. Each patient had a reduction in NV of the cornea, and one had a reduction in lipid deposition. There were no adverse effects, except one patient had a minor intracorneal hemorrhage, which rapidly resolved and cleared.
Ranibizumab is another anti-VEGF antibody that has been proposed as a treatment for corneal NV. This medication offers several potential advantages over bevacizumab: it is one-third of the size of bevacizumab, which could allow better corneal penetration, and it has a higher affinity for VEGF-A [14]. Some studies show no difference between the two when administered as a subconjunctival injection, while others suggest that bevacizumab may lead to less inflammation and NV as well as decreased length of blood vessels [69]. Administration of 1.0% topical ranibizumab 4 times daily for 3 weeks was shown to be superior to 1.0% topical bevacizumab administered with the same treatment regimen in reducing neovascular area and vessel caliber, especially if administered earlier in the treatment course [14]. However, the differences between the groups were not statistically significant, and more head-to-head trials are needed.
If bevacizumab passes into the systemic circulation, it may remain there longer than ranibizumab due to its larger size, leading to local and systemic overdosage [91]. Systemic side effects may be related to inhibition of VEGF-mediated physiologic wound healing and angiogenesis and can lead to hypertension, proteinuria, and cardiovascular events [94, 95]. Subconjunctival bevacizumab has rarely been associated with corneal epitheliopathy [96]. Topical treatment is generally safe and well-tolerated but it can inhibit corneal wound healing and nerve regeneration [91]. Care must be taken in patients with neurotrophic keratopathy or epithelial defects.
Although studies have shown beneficial effects of anti-VEGF treatment, it appears that the efficacy of bevacizumab is not always promising. This lack of efficacy could be related to the maturity of corneal NV; it is thought that bevacizumab may play a more important role in acute NV than in chronic NV [97, 98], possibly due to the downregulation of angiogenic factors as newly formed vessels mature; maintenance of chronic NV appears to be less dependent on VEGF. The subsequent recruitment of pericytes may act as a physical barrier, which typically occurs within 2 weeks [60, 86]. Both of these mechanisms can potentially decrease the efficacy of anti-VEGF antibodies in treating chronic NV [99]. While there appears to be no strict definition of chronic corneal NV, a time frame of 3–6 months has been used in the literature [60, 86, 87, 100]. Another major disadvantage of bevacizumab is the risk of recurrence and the need for repeated treatments to maintain effects. Proposed mechanisms include residual subclinical inflammation and rebound release of VEGF after cessation of treatment [87]. Additionally, while VEGF plays a major role in angiogenesis, other factors are involved. Therefore, inhibiting VEGF alone may be insufficient for permanently reducing corneal NV and LK [87, 101]. Combining anti-VEGF antibodies with other procedures may be superior in efficacy compared to using either alone [102,103,104]. Newer anti-VEGF therapeutics include VEGF traps (aflibercept), VEGFR tyrosine kinase inhibitors, and RNA aptamers (pegaptanib) [69, 105, 106]. However, more long-term studies must be performed to evaluate the effectiveness and safety of anti-VEGF therapy for the treatment of LK before providers can fully integrate this strategy into their clinical practice.
Another potential pharmacologic target is substance P (SP), which is a neuropeptide secreted from nerve endings and immune cells during inflammation [107]. When bound to the neurokinin-1 receptor (NK-1R), SP promotes wound healing by stimulating epithelial cells to proliferate and migrate, but also leads to the release of inflammatory mediators that can initiate and maintain corneal NV [108]. Higher levels of SP in tears of patients affected by corneal NV have been associated with more severe vascularization [108]. NK-1R antagonists such as fosaprepitant have been shown to reduce corneal NV, corneal opacity, and inflammation in animal models [109].
Argon laser treatment causes heat-induced vessel occlusion by directing a beam of light onto the pathologic vessels, which can be assisted by digital fluorescein angiography (FA) [14, 110, 111]. Marsh [110, 112] has authored several reports of argon laser treatment for LK using an aperture of 50 μm with 0.1-s exposure and 0.2- to 0.8-W power. This treatment led to a reduction in the extent and density of LK in 62 and 49% of eyes, respectively. Laser-induced tissue destruction, however, can worsen NV by causing inflammation and the release of angiogenic factors and can cause corneal hemorrhage, corneal thinning, and iris atrophy [14].
Fine needle diathermy (FND) was first developed in 2000 by Pillai et al. [113]. This treatment involves attaching a stainless steel 3/8 single-armed needle to a 10-0 monofilament black nylon suture held by a microsurgical needle holder. After the needle is inserted near the limbus adjacent to a pathologic vessel, a grounding electrode is strapped to the foot of the patient. A diathermy probe in coagulating mode at its lowest setting (0.5–1.0 mA) is then brought into contact with the needle. This leads to cauterization and elimination of the abnormal vessel, typically in under 1 s. In the original study, 100% occlusion of vessels was achieved in two of two eyes with LK. In a more recent case series, 82.3% of eyes with LK treated with FND showed reduced corneal lipid deposition [114]. Complications include transient whitening of the cornea and intrastromal hemorrhage. The procedure may also need to be repeated due to recanalization or collateral vessel development [113].
A more recent adaptation to the FND technique uses an electrolysis needle (electrolysis-needle cauterization [ENC]). ENC involves direct thermal cautery instead of electrical currents as in FND [115]. The electrolysis needle is also finer and more flexible than the diathermy needle (0.125 vs. 0.15 mm), possibly posing an advantage for treating fine vessels. Vessel occlusion was accomplished in all three eyes treated for LK with a subtle decrease in lipid [115]. Two eyes required repeat ENC, with one requiring a penetrating keratoplasty.
Lastly, a penetrating keratoplasty, commonly referred to as a corneal transplant, can be effective in severe cases of LK. Complications with the surgery include corneal thinning, hypoesthesia, sustained vascularization, and graft rejection [116]. In idiopathic LK, the treatments mentioned earlier in this article should be implemented before considering surgery. If there is no resolution, then surgery is a good option. For secondary LK, the underlying condition should be initially treated. If there is no resolution or the effects of the LK cannot be reversed with treatment of the primary disease, then the treatments, as mentioned earlier, including surgery, should be explored to provide relief for the patient. However, outcomes of keratoplasty may be worse when used to treat secondary LK compared to the idiopathic form, as corneal NV has been shown to lead to a worse prognosis after keratoplasty [23, 76].
The future for the management of LK is of great interest, as recurrence, cost, and complications limit the clinical utility of many current treatment regimens. The ideal treatment should destroy existing abnormal corneal vessels and prevent further vascularization. Newer treatment modalities may include mitomycin intravascular chemoembolization, or “MICE,” as pioneered by Dean Ouano of Coastal Eye Clinic in New Bern, North Carolina (USA) (in preliminary stages of research). Transcatheter arterial chemoembolization (TACE) has been used for years in hepatocellular carcinoma as a method to reduce tumor size by preventing its blood supply [117]. This procedure combines an intra-arterial infusion of a chemotherapeutic agent with an artificial embolus that leads to vessel occlusion. A common antineoplastic agent used is mitomycin-C, a DNA cross-linking agent that also inhibits protein and RNA synthesis. In theory, this method could be used to occlude pathologic vessels in corneal NV, preventing or halting the development of LK. Challenges include accurately identifying afferent and efferent blood vessels for intravascular cannulation. Afferent vessels should be targeted if possible, as chemoablation of efferent vessels alone may lead to worsened lipid exudation. Previous studies have shown that the combination of FA and indocyanine green angiography (ICGA) can better delineate vessels compared with biomicroscopy [118]. However, this process is invasive, time-consuming, and carries a small risk of severe anaphylaxis [119]. Recently, anterior segment optical coherence tomography angiography (OCTA) has demonstrated the potential for identifying small vessels, especially in the setting of corneal opacities that limit visualization by slit-lamp [120]. Another challenge is employing a needle that is thin enough to cannulate these newly formed vessels. Presently, 33-gauge needles are readily available; however, newer options include 36-gauge and even 41-gauge subretinal injection needles. One may even consider micro-pipettes for the delivery of intravascular chemoembolization. Further studies should be pursued to identify the safety, efficacy, and proper technique of corneal intravascular chemoembolization as a treatment for LK secondary to corneal NV.
Conclusion
Lipid keratopathy is a disease of the eye that is classically characterized by NV of the cornea, cholesterol deposits, opacification, and decrease of visual acuity. The disease progresses slowly and causes a steady decline in vision. The cause of idiopathic LK is unkown, and this form occurs bilaterally. Secondary LK occurs secondary to a disease or trauma to the eye and is typically unilateral. Although many treatment options are available, further studies must be performed to identify more robust, reliable, and precise treatment methods that are accessible and affordable to patients.
References
Alfonso E, Arrellanes L, Boruchoff SA, Ormerod LD, Albert DM. Idiopathic bilateral lipid keratopathy. Br J Ophthalmol. 1988. 72(5):338–43.
Baumgarten P. Ophthalmologisch-histologische Mittheilungen. Graefe’s Arch für Ophthalmol. 1876;22(2):185–203. https://doi.org/10.1007/BF01705018.
Cogan DG, Kuwabara T. Lipid keratopathy and atheroma. Circulation. 1958;18(4):519–25.
Croxatto JO, Dodds CM, Dodds R. Bilateral and massive lipoidal infiltration of the cornea (secondary lipoidal degeneration). Ophthalmology. 1985;92(12):1686–90.
Jack RL. Lipid keratopathy. Arch Ophthalmol. 1970;83(6):678. https://doi.org/10.1001/archopht.1970.00990030678002.
Silva-Araútjo A, Tavares MA, Lemos MM, Soares MI, Castro-Correia J, Salgado-Borges J. Primary lipid keratopathy: a morphological and biochemical assessment. Br J Ophthalmol. 1993;77:248–50.
Fine BS, Townsend WM, Zimmerman LE, Lashkari MH. Primary lipoidal degeneration of the cornea. Am J Ophthalmol. 1974;78(1):12–23.
Shapiro LA, Farkas TG. Lipid keratopathy following corneal hydrops. Arch Ophthalmol. 1977;95(3):456–8.
Ghanem RC, Ghanem VC, Victor G, Alves MR. Bilateral progressive idiopathic annular lipid keratopathy. Case Rep Ophthalmol Med. 2012;2012:1–4.
Loeffler KU. Unusual idiopathic lipid keratopathy: a newly recognized entity? Arch Ophthalmol. 2005;123(10):1435.
World Health Organization (WHO). Future approaches to trachoma control: report from a global scientific meeting, Geneva, 17–20 June 1996. Geneva: WHO; 1996. https://apps.who.int/iris/handle/10665/63413. Accessed July 9 2020.
Allen JE, Adjei O, Bain O, et al. Of mice, cattle, and humans: the immunology and treatment of river blindness. LoS Negl Trop Dis. 2008;2(4):e217.
Goh YW, Mcghee CN, Patel DV, Barnes R, Misra S. Treatment of herpes zoster related corneal neovascularisation and lipid keratopathy by photodynamic therapy. Clin Exp Optom. 2014;97(3):274–7.
Stevenson W, Cheng SF, Dastjerdi MH, Ferrari G, Dana R. Corneal neovascularization and the utility of topical VEGF inhibition: ranibizumab (Lucentis) vs bevacizumab (Avastin). Ocul Surf. 2012;10(2):67–83.
Li EY, Jhanji V. Massive lipid keratopathy after Elizabethkingia meningosepticum keratitis. Contact Lens Anterior Eye. 2014;37(1):55–6. https://doi.org/10.1016/j.clae.2013.07.011.
Hamill CE, Bozorg S, Peggy Chang H-Y, Lee H, Sayegh RR, Shukla AN, et al. Corneal alkali burns. Int Ophthalmol Clin. 2013;53(4):185–94. https://doi.org/10.1097/IIO.0b013e31829ceefa.
Cursiefen C, Kruse FE. New aspects of angiogenesis in the cornea. In: Reinhard T, Larkin D, editors. Cornea and external eye disease. Essentials in ophthalmology. Berlin Heidelberg: Springer; 2006. p. 83–99.
Sun YC, Yang LC, Hu FR, Lin CT, Chen YM, Chen WL. In vivo confocal microscopic study of hard contact lens-induced lipid keratopathy secondary to corneal neovascularization in a rabbit hypercholesterolemic model. Eye Contact Lens. 2018;44:S325–32.
Afsharkhamseh N, Movahedan A, Gidfar S, et al. Stability of limbal stem cell deficiency after mechanical and thermal injuries in mice. Exp Eye Res. 2016;145:88–92.
Abdelfattah NS, Amgad M, Zayed AA, et al. Clinical correlates of common corneal neovascular diseases: a literature review. Int J Ophthalmol. 2015;8(1):182–93.
Dana MR, Schaumberg DA, Kowal VO, et al. Corneal neovascularization after penetrating keratoplasty. Cornea. 1995;14(6):604–9.
Shah R, Puranik C, Mohamed A, Sangwan VS. Cultivated limbal epithelial transplantation and penetrating keratoplasty postchemical injury: a 14-year follow-up. BMJ Case Rep. 2017;2017:bcr2016217372.
Bachmann B, Taylor RS, Cursiefen C. Corneal neovascularization as a risk factor for graft failure and rejection after keratoplasty. Ophthalmology. 2010;117(7):1300–1305.e7.
Piñero DP, Alio JL. Intracorneal ring segments in ectatic corneal disease—a review. Clin Exp Ophthalmol. 2010;38(2):154–67. https://doi.org/10.1111/j.1442-9071.2010.02197.x.
Bautista-Llamas M-J, Sánchez-González MC, López-Izquierdo I, et al. Complications and explantation reasons in intracorneal ring segments (ICRS) implantation: a systematic review. J Refract Surg. 2019;35(11):740–7. https://doi.org/10.3928/1081597X-20191010-02.
Ly LT, McCulley JP, Verity SM, Cavanagh HD, Bowman RW, Petroll WM. Evaluation of intrastromal lipid deposits after intacs implantation using in vivo confocal microscopy. Eye Contact Lens. 2006;32(4):211–5.
Barchiesi BJ, Eckel RH, Ellis PP. The cornea and disorders of lipid metabolism. Surv Ophthalmol. 1991;36(1):1–22.
Gjone E. Familial lecithin: cholesterol acyltransferase (LCAT) deficiency: an updated review spring 1988. Ophthalm Genet. 1988;9(3):167–9.
Saeedi R, Li M, Frohlich J. A review on lecithin: cholesterol acyltransferase deficiency. Clin Biochem. 2015;48(7–8):472–5.
Chu FC, Kuwabara T, Cogan DG, Schaefer EJ, Brewer HB. Ocular manifestations of familial high-density lipoprotein deficiency (Tangier disease). Arch Ophthalmol. 1979;97(10):1926–8. http://archopht.jamanetwork.com/article.aspx?articleid=633168.
Herrmann WA, von Mohrenfels CW, Lohmann CP. Confocal microscopy and corneal sensitivity in a patient with corneal manifestations of tangier disease. Cornea. 2004;23(8):825–7. https://doi.org/10.1097/01.ico.0000126331.84852.f6.
Winder AF, Alexander R, Garner A, et al. The pathology of cornea in Tangier disease (familial high density lipoprotein deficiency). J Clin Pathol. 1996;49(5):407–10. https://doi.org/10.1136/jcp.49.5.407.
Burnett JR, Hooper AJ, McCormick SP, Hegele RA. Tangier disease. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993–2020.
Chan AT, Ulate R, Goldich Y, Rootman DS, Chan CC. Terrien marginal degeneration: Clinical characteristics and outcomes. Am J Ophthalmol. 2015;160(5):867–872.e1. https://doi.org/10.1016/j.ajo.2015.07.031.
Oikonomakis K, Petrelli M, Andreanos K, et al. Corneal neovascularization with associated lipid keratopathy in a patient with obstructive sleep apnea-hypopnea syndrome using a continuous positive airway pressure machine. Case Rep Ophthalmol. 2017;8(2):416–20.
Morisawa M, Yamagami S, Inoki T, Ochiai M, Tsuru T. Bilateral centripetal lipid keratopathy with diffuse anterior scleritis. Acta Ophthalmol Scand. 2003;81(2):202–3. https://doi.org/10.1034/j.1600-0420.2003.00025.x.
Mann I, Pullinger BD. The pathology of cholesterin and fat deposition in mustard gas injuries of the cornea. Br J Ophthalmol. 1942;26(11):503–7. https://doi.org/10.1136/bjo.26.11.503.
Jafarinasab M-R, Zarei-Ghanavati S, Kanavi MR, Karimian F, Soroush M-R, Javadi M-A. Confocal microscopy in chronic and delayed mustard gas keratopathy. Cornea. 2010;29(8):889–94. https://doi.org/10.1097/ICO.0b013e3181ca324c.
Miserocchi E, Baltatzis S, Foster CS. A case of atypical Cogan’s syndrome with uncommon corneal findings. Cornea. 2001;20(5):540–2. https://doi.org/10.1097/00003226-200107000-00021.
Folkman J. Angiogenesis in cancer, vascular, rheumatoid and other disease. Nat Med. 1995;1(1):27–30. http://www.nature.com/articles/nm0195-27.
Azar D. Corneal angiogenic privilege: angiogenic and antiangiogenic factors in corneal avascularity, vasculogenesis, and wound healing. Trans Am Ophthalmol Soc. 2006;104(2):264–302.
Notara M, Lentzsch A, Coroneo M, Cursiefen C. The role of limbal epithelial stem cells in regulating corneal (lymph)angiogenic privilege and the micromilieu of the limbal niche following UV exposure. Stem Cells Int. 2018;2018:1–15. https://doi.org/10.1155/2018/8620172.
Ellenberg D, Azar DT, Hallak JA, et al. Novel aspects of corneal angiogenic and lymphangiogenic privilege. Prog Retin Eye Res. 2010;29(3):208–48. https://doi.org/10.1016/j.preteyeres.2010.01.002.
Pintucci G, Moscatelli D, Saponara F, et al. Lack of ERK activation and cell migration in FGF-2-deficient endothelial cells. FASEB J. 2002;16(6):598–600. https://doi.org/10.1096/fj.01-0815fje.
Lee HS, Schlereth SL, Park EY, Emami-Naeini P, Chauhan SK, Dana R. A novel pro-angiogenic function for interferon-γ-secreting natural killer cells. Investig Ophthalmol Vis Sci. 2014;55(5):2885–92.
Hoch RV. Roles of PDGF in animal development. Development. 2003;130(20):4769–84. https://doi.org/10.1242/dev.00721.
Augustin HG, Young Koh G, Thurston G, Alitalo K. Control of vascular morphogenesis and homeostasis through the angiopoietin–Tie system. Nat Rev Mol Cell Biol. 2009;10(3):165–77. http://www.nature.com/articles/nrm2639.
Abdelfattah NS, Amgad M, Zayed AA. Host immune cellular reactions in corneal neovascularization. Int J Ophthalmol. 2016;9(4):625–33.
Ambati BK, Nozaki M, Singh N, et al. Corneal avascularity is due to soluble VEGF receptor-1. Nature. 2006;443(7114):993–7. http://www.nature.com/articles/nature05249.
Ambati BK, Patterson E, Jani P, et al. Soluble vascular endothelial growth factor receptor-1 contributes to the corneal antiangiogenic barrier. Br J Ophthalmol. 2007;91(4):505–8. https://doi.org/10.1136/bjo.2006.107417.
Cursiefen C, Chen L, Saint-Geniez M, et al. Nonvascular VEGF receptor 3 expression by corneal epithelium maintains avascularity and vision. Proc Natl Acad Sci USA. 2006;103(30):11405–10. https://doi.org/10.1073/pnas.0506112103.
Tsai RJ-F, Tseng SCG. Human allograft limbal transplantation for corneal surface reconstruction. Cornea. 1994;13(5):389–400. https://doi.org/10.1097/00003226-199409000-00003.
Lim P, Fuchsluger TA, Jurkunas UV. Limbal stem cell deficiency and corneal neovascularization. Semin Ophthalmol. 2009;24(3):139–48. https://doi.org/10.1080/08820530902801478.
Carmeliet P. Mechanisms of angiogenesis and arteriogenesis. Nat Med. 2000;6(4):389–95.
Chung E-S, Chauhan SK, Jin Y, et al. Contribution of macrophages to angiogenesis induced by vascular endothelial growth factor receptor-3-specific ligands. Am J Pathol. 2009;175(5):1984–92.
Faraj LA, Said DG, Al-Aqaba M, Otri AM, Dua HS. Clinical evaluation and characterisation of corneal vascularisation. Br J Ophthalmol. 2016;100(3):315–22.
Cursiefen C, Maruyama K, Jackson DG, Streilein JW, Kruse FE. Time course of angiogenesis and lymphangiogenesis after brief corneal inflammation. Cornea. 2006;25(4):443–7. https://doi.org/10.1097/01.ico.0000183485.85636.ff.
Crispin S. Ocular lipid deposition and hyperlipoproteinaemia. Prog Retin Eye Res. 2002;21(2):169–224.
Friedman M, Byers SO. Excess lipid leakage: a property of very young vascular endothelium. Br J Exp Pathol. 1962;43:363–72.
Cursiefen C, Hofmann-Rummelt C, Küchle M, Schlötzer-Schrehardt U. Pericyte recruitment in human corneal angiogenesis: an ultrastructural study with clinicopathological correlation. Br J Ophthalmol. 2003;87(1):101–6.
Silva-Araútjo A, Tavares MA, Lemos MM, Soares MI, Castro-Correia J, Salgado-Borges J. Primary lipid keratopathy: a morphological and biochemical assessment. Br J Ophthalmol. 1993;77(4):248–50.
Mayer KL, Nordlund ML, Schwartz GS, Holland EJ. Keratopathy in congenital aniridia. Ocul Surf. 2003;1(2):74–9.
Di Iorio E, Kaye SB, Ponzin D, et al. Limbal stem cell deficiency and ocular phenotype in ectrodactyly-ectodermal dysplasia-clefting syndrome caused by p63 mutations. Ophthalmology. 2012;119(1):74–83.
Koizumi N, Inatomi T, Suzuki T, Sotozono C, Kinoshita S. Cultivated corneal epithelial transplantation for ocular surface reconstruction in acute phase of Stevens-Johnson syndrome. Arch Ophthalmol. 2001;119(2):298–300.
Dua HS, Joseph A, Shanmuganathan VA, Jones RE. Stem cell differentiation and the effects of deficiency. Eye. 2003;17(8):877–85. http://www.nature.com/articles/6700573.
Hatch KM, Dana R. The structure and function of the limbal stem cell and the disease states associated with limbal stem cell deficiency. Int Ophthalmol Clin. 2009;49(1):43–52. https://doi.org/10.1097/IIO.0b013e3181924e54.
Ozerdem U, Alitalo K, Salven P, Li A. Contribution of bone marrow-derived pericyte precursor cells to corneal vasculogenesis. Investig Ophthalmol Vis Sci. 2005;46(10):3502–6. https://doi.org/10.1167/iovs.05-0309.
Lee P, Wang CC, Adamis AP. Ocular neovascularization: an epidemiologic review. Surv Ophthalmol. 1998;43(3):245–69.
Hsu CC, Chang HM, Lin TC, et al. Corneal neovascularization and contemporary antiangiogenic therapeutics. J Chin Med Assoc. 2015;78(6):323–30. https://doi.org/10.1016/j.jcma.2014.10.002.
Durán JA, Rodriguez-Ares MT. Idiopathic lipid corneal degeneration. Cornea. 1991;10(2):166–9.
Pang Y, Zhang Q, Bao Y. A CARE-compliant article: ipsilateral progressive idiopathic lipid keratopathy: a case report. Medicine (Baltimore). 2019;98(35):e16589.
Fielder AR, Winder AF, Sheraidah GA, Cooke ED. Problems with corneal arcus. Trans Ophthalmol Soc UK. 1981;101(1):22–6.
Cogan DG. Arcus senilis. AMA Arch Ophthalmol. 1959;61(4):553–60. https://doi.org/10.1001/archopht.1959.00940090555009.
Smith JL. Unilateral arcus senilis: sign of occlusive disease of the carotid artery. JAMA. 1973;226(6):676. https://doi.org/10.1001/jama.1973.03230060052030.
Weiss JS, Khemichian AJ. Differential diagnosis of schnyder corneal dystrophy. In: corneal dystrophies. Basel: Karger; 2011:67–96. https://doi.org/10.1159/000324078.
Chang J-H, Gabison EE, Kato T, Azar DT. Corneal neovascularization. Curr Opin Ophthalmol. 2001;12(4):242–9. https://doi.org/10.1097/00055735-200108000-00002.
Igarashi T, Takahashi H. Photodynamic therapy for neovascularization in lipid keratopathy. J Nippon Med Sch. 2010;77(2):66. https://doi.org/10.1272/jnms.77.66.
Yoon KC, You IC, Kang IS, Im SK, Ahn JK, Park YG, et al. Photodynamic therapy with verteporfin for corneal neovascularization. Am J Ophthalmol. 2007;144(3):390–5.e1. https://doi.org/10.1016/j.ajo.2007.05.028.
Al-Abdullah AA, Al-Assiri A. Resolution of bilateral corneal neovascularization and lipid keratopathy after photodynamic therapy with verteporfin. Optom–J Am Optom Assoc. 2011;82(4):212–4.
Brooks BJ. Photodynamic therapy for corneal neovascularisation and lipid degeneration. Br J Ophthalmol. 2004;88(6):840. https://doi.org/10.1136/bjo.2003.035071.
Spaide RF, Laud K, Fine HF, et al. Intravitreal bevacizumab treatment of choroidal neovascularization secondary to age-related macular degeneration. Retina. 2006;26(4):383–90. https://doi.org/10.1097/01.iae.0000238561.99283.0e.
Avery RL, Pearlman J, Pieramici DJ, et al. Intravitreal bevacizumab (avastin) in the treatment of proliferative diabetic retinopathy. Ophthalmology. 2006;113(10):1695–1705.e6.
Olmos LC, Sayed MS, Moraczewski AL, et al. Long-term outcomes of neovascular glaucoma treated with and without intravitreal bevacizumab. Eye. 2016;30(3):463–72.
Costa RA, Jorge R, Calucci D, Melo LA, Cardillo JA, Scott IU. Intravitreal bevacizumab (avastin) for central and hemicentral retinal vein occlusions. Retina. 2007;27(2):141–9. https://doi.org/10.1097/IAE.0b013e31802eff83.
Oh JY, Kim MK, Wee WR. Subconjunctival and intracorneal bevacizumab injection for corneal neovascularization in lipid keratopathy. Cornea. 2009;28(9):1070–3.
Chu HS, Hu FR, Yang CM, et al. Subconjunctival injection of bevacizumab in the treatment of corneal neovascularization associated with lipid deposition. Cornea. 2011;30(1):60–6.
Chu HS, Chen TC, Hu FR, Chen WL. Recurrence of corneal neovascularization associated with lipid deposition after subconjunctival injection of bevacizumab. Cornea. 2013;32(11):1446–53.
Castro-Rebollo M, Montes-Mollón MA, Pérez-Rico C, Teus MA. Effectiveness of topical bevacizumab in bilateral primary lipid keratopathy [Eficacia de bevacizumab tópico en queratopatía lipoidea bilateral primaria]. Arch Soc Esp Oftalmol. 2011;86(11):374–6. https://doi.org/10.1016/j.oftale.2011.05.006.
Elbaz U, Mireskandari K, Shen C, Ali A. Corneal fine needle diathermy with adjuvant bevacizumab to treat corneal neovascularization in children. Cornea. 2015;34(7):773–7.
Hussain R, Savant V. Fine-needle diathermy with simultaneous subconjunctival bevacizumab. Semin Ophthalmol. 2017;32(5):550–2. https://doi.org/10.3109/08820538.2015.1123738.
Papathanassiou M, Theodoropoulou S, Analitis A, Tzonou A, Theodossiadis PG. Vascular endothelial growth factor inhibitors for treatment of corneal neovascularization: a meta-analysis. Cornea. 2013;32(4):435–44.
Saravia M, Zapata G, Ferraiolo P, Racca L, Berra A. Anti-VEGF monoclonal antibody-induced regression of corneal neovascularization and inflammation in a rabbit model of herpetic stromal keratitis. Graefe’s Arch Clin Exp Ophthalmol. 2009;247(10):1409–16.
Dastjerdi MH, Sadrai Z, Saban DR, Zhang Q, Dana R. Corneal penetration of topical and subconjunctival bevacizumab. Investig Opthalmol Vis Sci. 2011;52(12):8718.
Dastjerdi MH, Al-Arfaj KM, Nallasamy N, Hamrah P, Jurkunas UV, Pineda R, et al. Topical bevacizumab in the treatment of corneal neovascularization; results of a prospective, open-label, noncomparative study. Arch Ophthalmol. 2009;127(4):381–9.
Gillies MC. What we don’t know about avastin might hurt us. Arch Ophthalmol. 2006;124(10):1478.
Kim SW, Ha BJ, Kim EK, Tchah H, Kim T. The effect of topical bevacizumab on corneal neovascularization. Ophthalmology. 2008;115(6):e33–8.
Chen W-L, Chen Y-M, Chu H-S, et al. Mechanisms Controlling the effects of bevacizumab (avastin) on the inhibition of early but not late formed corneal neovascularization. PLoS ONE. 2014;9(4):e94205. https://doi.org/10.1371/journal.pone.0094205.
Chen W-L, Lin C-T, Lin N-T, et al. Subconjunctival injection of bevacizumab (avastin) on corneal neovascularization in different rabbit models of corneal angiogenesis. Investig Opthalmol Vis Sci. 2009;50(4):1659. https://doi.org/10.1167/iovs.08-1997.
Lin CT, Hu FR, Kuo KT, et al. The different effects of early and late bevacizumab (Avastin) injection on inhibiting corneal neovascularization and conjunctivalization in rabbit limbal insufficiency. Investig Ophthalmol Vis Sci. 2010;51(12):6277–85.
Koenig Y, Bock F, Kruse FE, Stock K, Cursiefen C. Angioregressive pretreatment of mature corneal blood vessels before keratoplasty: fine-needle vessel coagulation combined with Anti-VEGFs. Cornea. 2012;31(8):887–92.
Krizova D, Vokrojova M, Liehneova K, Studeny P. Treatment of corneal neovascularization using anti-VEGF bevacizumab. J Ophthalmol. 2014;2014:1–8.
Yoon HJ, Kim MK, Seo KY, Ueta M, Yoon KC. Effectiveness of photodynamic therapy with verteporfin combined with intrastromal bevacizumab for corneal neovascularization in Stevens-Johnson syndrome. Int Ophthalmol. 2019;39(1):55–62.
Kim RY, Chung SK, Kim MS, Ra H. Effects of combined photodynamic therapy and topical bevacizumab treatment on corneal neovascularization in rabbits. Cornea. 2016;35(12):1615–20. https://doi.org/10.1097/ICO.0000000000001016.
Gerten G. Bevacizumab (avastin) and argon laser to treat neovascularization in corneal transplant surgery. Cornea. 2008;27(10):1195–9.
Al-Debasi T, Al-Bekairy A, Al-Katheri A, Al Harbi S, Mansour M. Topical versus subconjunctival anti-vascular endothelial growth factor therapy (Bevacizumab, Ranibizumab and Aflibercept) for treatment of corneal neovascularization. Saudi J Ophthalmol. 2017;31(2):99–105. https://doi.org/10.1016/j.sjopt.2017.02.008.
Voiculescu OB, Voinea LM, Alexandrescu C. Corneal neovascularization and biological therapy. J Med Life. 2015;8(4):444–8.
O’Connor TM, O’Connell J, O’Brien DI, Goode T, Bredin CP, Shanahan F. The role of substance P in inflammatory disease. J Cell Physiol. 2004;201(2):167–80. https://doi.org/10.1002/jcp.20061.
Barbariga M, Fonteyne P, Ostadreza M, Bignami F, Rama P, Ferrari G. Substance P modulation of human and murine corneal neovascularization. Investig Ophthalmol Vis Sci. 2018;59(3):1305–12.
Bignami F, Lorusso A, Rama P, Ferrari G. Growth inhibition of formed corneal neovascularization following fosaprepitant treatment. Acta Ophthalmol. 2017;95(7):e641–8.
Marsh RJ. Argon laser treatment of lipid keratopathy. Br J Ophthalmol. 1988;72(12):900–4.
Peter J, Fraenkel G, Goggin M, Drew A. Fluorescein angiographic monitoring of corneal vascularization in lipid keratopathy. Clin Exp Ophthalmol. 2004;32(1):78–80.
Marsh RJ, Marshall J. Treatment of lipid keratopathy with the argon laser. Br J Ophthalmol. 1982;66(2):127–35.
Pillai CT, Dua HS, Hossain P. Fine needle diathermy occlusion of corneal vessels. Invest Ophthalmol Vis Sci. 2000;41(8):2148–53.
Faraj LA, Elalfy MS, Said DG, Dua HS. Fine needle diathermy occlusion of corneal vessels. Br J Ophthalmol. 2014;98(9):1287–90.
Wertheim MS, Cook SD, Knox-Cartwright NE, Van D Le, Tole DM. Electrolysis-needle cauterization of corneal vessels in patients with lipid keratopathy. Cornea. 2007;26(2):230–1. https://doi.org/10.1097/01.ico.0000248383.09272.ee.
Vanathi M, Panda A, Vengayil S, Chaudhuri Z, Dada T. Pediatric keratoplasty. Surv Ophthalmol. 2009;54(2):245–71. https://doi.org/10.1016/j.survophthal.2008.12.011.
Chuang VP. Chemoembolization: transcatheter management of neoplasms. JAMA 1981;245(11):1151–2. https://doi.org/10.1001/jama.1981.03310360043023.
Spiteri N, Romano V, Zheng Y, et al. Corneal angiography for guiding and evaluating fine-needle diathermy treatment of corneal neovascularization. Ophthalmology. 2015;122(6):1079–84.
Bearelly S, Rao S, Fekrat S. Anaphylaxis following intravenous fluorescein angiography in a vitreoretinal clinic: report of 4 cases. Can J Ophthalmol. 2009;44(4):444–5.
Chan SY, Pan CT, Feng Y. Localization of corneal neovascularization using optical coherence tomography angiography. Cornea. 2019;38(7):888–95.
Acknowledgements
Funding
This review was funded by an unrestricted grant from Research to Prevent Blindness (RPB), 360 Lexington Avenue, 22nd Floor New York, NY 10017 (USA). No support was received for the publication of this article.
Editorial Assistance
Assistance in the preparation of this article was provided by Gerald B. Rosen of Horizon Eye Care, Charlotte, NC (USA).
Authorship
All named authors meet the International Committee of Medical Journal Editors (ICMJE) criteria for authorship for this article, take responsibility for the integrity of the work as a whole, and have given their approval for this version to be published.
Disclosures
MacGregor N Hall, Majid Moshirfar, Armaan Amin-Javaheri, Dean P Ouano, Gerald B Rosen, Yasmyne Ronquillo, and Phillip C Hoopes declare that they have no conflict of interest.
Compliance with Ethics Guidelines
This article is based on previously conducted studies and does not contain any studies with human participants or animals performed by any of the authors.
Data Availability
Data sharing is not applicable to this article as no datasets were generated or analyzed during the current study.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Hall, M.N., Moshirfar, M., Amin-Javaheri, A. et al. Lipid Keratopathy: A Review of Pathophysiology, Differential Diagnosis, and Management. Ophthalmol Ther 9, 833–852 (2020). https://doi.org/10.1007/s40123-020-00309-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40123-020-00309-y