
Abstract
Stroke is a leading cause of death worldwide, and about a quarter of stroke patients are dead within 1 month. The prognosis is even worse for those with hemorrhagic stroke because the 1-month mortality approaches 50%. Besides, most patients who survive experience complications such as nausea, vomiting, and chronic pain. These adverse experiences, especially the existence of chronic pain, can lead to a decline in the patient’s quality of life. In order to improve the treatment and prognosis of hemorrhagic stroke, there is an urgent need to understand its pathophysiological mechanism as well as the chronic pain it induces. This paper reviews studies of the molecular mechanisms of hemorrhagic stroke, especially the activation of microglia and the relationship between microglia and pain after stroke, which could shed new light on hemorrhagic stroke treatment.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Chronic pain is one of the most troublesome sequelae of stroke. It is reported that about 11–55% of stroke patients will suffer chronic pain. |
Pain after stroke is related to the activation of microglia; there is an urgent need to understand the associated pathophysiological mechanism. |
This paper reviews studies of the molecular mechanisms of hemorrhagic stroke (HS), especially the activation of microglia and the relationship between microglia and pain after stroke. |
Microglia are a therapeutic target for hemorrhagic stroke and HS-related pain. |
Introduction
Hemorrhagic stroke (HS) has the second highest incidence of any type of acute stroke, accounting for 10–20% of all strokes [1]. Every year, more than a million people around the world are affected by this disease, and this number will increase greatly due to the aging of populations. Hemorrhagic stroke can induce vomiting, dizziness, impaired consciousness, and even hemiplegia. In addition to the abovementioned acute symptoms, most patients will suffer paraesthesia, in which the most unbearable feeling is chronic pain, which affects the prognosis and quality of life of those patients.
Pain is one of the most troublesome sequelae of stroke. It is reported that about 11–55% of stroke patients suffer chronic pain, which is mostly related to anxiety caused by long-term illness [2, 3]. The most common manifestations of chronic pain after stroke are shoulder pain, central post-stroke pain, cramps, and tension-type headaches [4]. The pathogenesis of post-stroke pain (PSP) remains unknown, but it has been suggested that the underlying causes include hyperexcitation in the damaged sensory pathways, central nervous system lesions, or a combination of the two. For central nervous system lesions, the most important factor is microglia dysregulation.
In this review, we provide an overview of microglial function after hemorrhagic stroke and in the process of PSP, especially microglial polarization, modulators, and interactions with other cells. We also address clinical observations and highlight new therapeutic directions relating to microglia for hemorrhagic stroke and PSP.
This article is based on previously conducted studies and does not contain any new studies with human participants or animals performed by any of the authors.
Activation of Microglia Following Hemorrhagic Stroke
When hemorrhage occurs, brain damage, which is mainly caused by edema or hematoma, will develop rapidly within the first few hours. Brain edema and hematoma increase the intracranial pressure sharply, which induces herniation and death [5]. Later on, microglia act as intracranial phagocytes, protecting the brain tissue by eliminating blood cells and other small molecules. In the middle and late stages, microglia transform into inflammatory cells and secrete large amounts of proinflammatory factors to destroy the vascular endothelium. The direct phagocytosis and the indirect effects of proinflammatory factors will aggravate the hemorrhagic injury. Activation and modulation of the microglial phenotype not only help to absorb the hematoma or edema but they also improve white matter integrity, brain repair, and functional recovery [6].
Molecular Mechanisms Underlying Microglial Activation Following Hemorrhagic Stroke
Phenotypic Polarization of Microglia and Markers
In response to CNS inflammation and brain injury, microglia cells can be activated; they then migrate to the lesion and envelop the injury zone. Activated microglia have been found in the perihematomal region as early as 1 h after hemorrhagic stroke [7], where they provide protective effects—clearing the debris and hematoma and promoting neurogenesis. Microglia acutely stimulated by a hemorrhage can be activated to two polarization states: M1, for promoting inflammation, and M2, for suppressing inflammation. A few minutes after the stroke, M1-phenotype microglia cells secrete and recruit inflammation factors such as Toll-like receptor 2 (TLR2) and TLR4 [8,9,10], INF-γ, and complement C3a and C5a. Almost the pro-inflammation factors are induced by the M1 type. The proinflammatory molecules activated by M1 microglia comprise the interleukin (IL) family (IL-1β, IL-6, IL-12, and IL-23), tumor necrosis factor alpha (TNF-α), chemokines (CX3C), redox molecules (NADPH oxidase, phagocyte oxidase, and inducible NO synthase), costimulatory proteins (CD40), and major histocompatibility complex II (MHC-II) [11,12,13,14,15].
On the contrary, M2 microglia play a protective role by promoting anti-inflammatory effects and suppressing oxidative injury [16, 17]. These cells create anti-inflammatory mediators such as IL-10, transforming growth factor beta (TGFβ), glucocorticoids, arginase-1, Ym-1 (chitinase 3-like 3), and CD206 [18,19,20]. M2 microglia are also considered to be nerve-repairing cells, as they secrete anti-inflammatory factors and upregulate neuroprotective factors in CNS disease.
M1 and M2 microglia express different factors, so different molecular markers can be used to distinguish these two phenotypes by double or triple immunofluorescence. Usually, M1 microglia markers can be labeled with MHC-II, CD16, CD32, CD80 (B7-1), CD86 (B7-2), and CD40 (TNFR). M2-cell-specific antigens include Ym-1, CD206 (mannose receptor), CD68, and arginase-1.
Dynamic Changes in Microglial Polarization
It was observed that hemorrhage induced the activation of microglia with high M1 phenotype gene expression at 24 h, while the expression of M2 phenotype genes gradually increased in the first 2 weeks, and the expression time varied slightly in different brain regions [21]. This contrasts with what is seen following ischemic stroke, where the M2 phenotype increases first, followed by the M1 phenotype [22]. These results suggest that the spatiotemporal dynamics of microglia polarization may be related to the degree of damage and the disease model considered.
Based on existing evidence and studies, once microglia have been activated by acute injury, they can change dynamically; this is especially seen in collagenase-induced [23, 24] and blood-induced [25] models. In mice with collagenase-induced cerebral hemorrhage, the microglia complete the transition from M1 to M2 phenotype within 1–3 days. In mice with blood-induced cerebral hemorrhage, this transition occurs within 1 week. Generally speaking, about 3 days after an intracerebral hemorrhage, microglia polarize into the M1-like phenotype, which is the main reason for the activation of microglia in the acute phase after hemorrhage, whereas M2-like microglia may play a role in the long-term recovery from stroke. Furthermore, studies have shown that in the process of microglial polarization, M2-type cells have multiple subtypes that are involved in damage repair and anti-inflammatory processes, and they play an important role (see Table 1 [26]).
Other Important Factors Related to Microglia Activation
Microglia are activated in the core part of the hemorrhagic stroke lesion and the perihematoma [23, 24]. The activated microglia produce a large number of inflammatory mediators that cause the activation of and changes in various inflammatory signal pathways [27, 28].
NLRP3
The collagenase-induced hemorrhagic stroke model presented an activated NLRP3 (NACHT leucine-rich repeat protein 3) signal pathway. Typically, NLRP3 activation is induced by the inflammatory response caused by bleeding; heme and other blood components in the blood can activate this pathway [29]. In addition, endogenous cytokines and other stimuli (pathogens, etc.) can activate the classical NLRP pathway. Exogenous or endogenous stimuli act through TLR4 or TNFR to activate the NF-κB pathway together with caspase-8 and FADD (FAS-associated death domain protein) [30], and they also initiate the transcription and translation of NLRP3 and pro-IL-1β. After NLRP3 has been activated, lysosomes are destroyed, resulting in the release of cathepsin B. At the same time, active caspase-1 can cleave the precursor IL-1β and the precursor IL-18 to mature [31]. In the nonclassical pathway, LPS directly induces the activation of intracellular caspase-11 (homologous to human caspase-4 and -5) [32], which in turn activates NLRP3 [32].
TREM2
Triggering receptor expressed on myeloid cells 2 (TREM2) is a natural immune receptor that is mainly expressed in myeloid cells. In the nervous system, it is mostly expressed in microglia, with a small amount expressed in astrocytes and neurons [33, 34]. After injury, its protein expression density in microglia is about 300 times that of other glial cells [35, 36]. This type of receptor is related to a variety of cellular processes, such as cell proliferation, regulating the release of proinflammatory factors, and promoting the phagocytosis of microglia [37]. Unlike TREM1, TREM2 is considered to provide negative feedback regulation [38]. This effect has been confirmed in a variety of neurological disease models. For example, in Alzheimer's disease and multiple sclerosis models, it was found that overexpression of TREM2 can inhibit the inflammatory response and play a protective role [39,40,41]. The latest research has shown that the endogenous expression of TREM2 in the ipsilateral/right hemisphere after intracerebral hemorrhage increases in a time-dependent manner, reaching a peak at 24 h after intracerebral hemorrhage [42]. The PI3K/Akt signaling pathway serves as a downstream target of TREM2 [43, 44], which is likely to play a vital role in the process of cerebral hemorrhage. Therefore, the research and development of drugs that can induce high expression of TREM2 have become a broad prospect for clinical therapeutic applications.
VAP1
Vascular adhesion protein 1 (VAP1) is a glycated protein that can regulate inflammation and at the same time provides a molecular basis for the adhesion and migration of white blood cells [45]. A study of subarachnoid hemorrhage in rats found that the use of VAP1 inhibitors can reduce the deterioration of neurological function after injury [46]. Similarly, VAP-1 inhibitors can downregulate the expression of adhesion molecules, thereby reducing the infiltration of neutrophils into the injured site. Subsequently, the proinflammatory cytokines TNF-α and MCP (monocyte chemoattractant protein) were significantly reduced, suppressing the activation of microglia or macrophages and ultimately improving the behavioral neurological function and brain edema of mice [47].
Microglial Crosstalk with Other Cells During Hemorrhagic Stroke
Astrocytes
The main role of astrocytes is to connect neurons to the CNS vasculature and act as a bridge. Anatomically speaking, the unique structure of an astrocyte extends from the cell body of the astrocyte to its end-feet, which attach to the basement membranes of endothelial cells. This structure helps maintain the integrity of the blood–brain barrier and prevents harmful substances in the blood from entering the brain. However, in central nervous system inflammation, microglia receive chemical stimulation and migrate to the blood–brain barrier. In this process, microglia initially play a protective role by secreting endothelial connexin (claudin-5); later, as the inflammation continues, they swallow the ends of astrocytes, breaking the blood–brain barrier and aggravating the inflammation of the nervous system [48,49,50]. More interestingly, it was observed that astrocytes could transform into mature neurons in situ under stroke conditions [51], suggesting that the mature brain is capable of replacing neurons lost after injury [52], even though these newborn neurons from astrocytes have a little function which cortical neurons have.
Monocyte Macrophages
Similar to microglia, these are innate immune cells and respond earliest to lesions. Interferon γ (IFN-γ) secreted by TH1 cells can convert macrophages into activated histiocytes, which can produce nitrous oxide and other oxygen-derived free radicals to kill pathogens. However, excessive release of inflammatory factors can exacerbate cell damage in hemorrhagic stroke. Macrophages have the same activation pathways and effects as microglia during the hemorrhage process. The markers of these two kinds of cells are almost the same, e.g., IBA1, CD11b, and F4/80 [53]. However, there are at least two differences between microglia and monocyte macrophages. Firstly, some studies have shown that macrophages that are activated by injury and migrate from the blood into the lesion express even more of the M2 phenotype than microglia that proliferate in situ. Macrophages that infiltrate from blood vessels have a important phagocytic function and inflammatory properties, while in situ microglia seem to preferentially remove cell debris [54]. In addition, infiltrating macrophages may be cytotoxic regardless of whether they adopt the M1 or M2 phenotype [55]. These results indicate that although the expression of M2-type macrophages increases, M2 microglia have more advantages than M2-type macrophages in brain protection. Secondly, the marker CD45lowCD11b+ was found to be expressed in microglia but not in macrophages. Similarly, TMEM119 is only expressed in macrophages [56]. Accordingly, the above two markers can be used to distinguish microglia and macrophages.
Different causes and environments of injuries may lead to slight differences in their effects. As we all know, the brain has the ability to self-repair after it has been damaged [57]. In this process, microglia and macrophages work together to surround the injured area and secrete various nutritional factors so that the protruding formation of new granular cells will follow the nutritional gradients toward the injured area, ensuring that the correct shape is formed. Microglia and macrophages participate in the whole process until the synapse is completely formed [56, 58].
Microglial Activation Plays an Important Role in Pain Post Hemorrhagic Stroke
The activation of microglia is closely related to the occurrence of pain after stroke. Microglia are activated in the core part of the hemorrhagic stroke lesion and the perihematoma [23, 24]. The activated microglia produce a large number of inflammatory mediators that activate and change various inflammatory signal pathways [27, 28]. Inflammation is considered the main pathological feature that causes pain. The activation of the P2X4/P2RX7 receptors in microglia [59,60,61] and the activation of NLRP3 in microglia are both related to the occurrence of pain after cerebral hemorrhage and stroke [62, 63]. In the thalamic hemorrhage model, depletion of microglia can effectively prevent the development of analgesia [64].
Nagaka et al., using a macaque model [65], found that microglial activation may play a key role in post-stroke pain, although the associated mechanism remains unclear. At present, it is mainly believed that the increased secretion of chemokines and proinflammatory factors after the nervous system is damaged is the main cause of pain symptoms. Chemokines are small molecular proteins that are often divided into four subtypes: CC, CXC, XC, and CX3C. The CX3C family currently only includes one member: CX3CL1. When damage occurs, the chemokine CX3CL1 is expressed in primary afferent and spinal neurons and induces microglial activation through its microglia receptor, CX3CR1 [66, 67]. CX3CL1 plays an important role in the development of neuropathic pain. In the neuropathic pain model induced by paclitaxel, the expression of the CX3CL1 protein in spinal cord neurons can be upregulated through the NF-κB-dependent H4 acetylation mechanism [68]. When CX3CL1 inhibitor is injected intrathecally, it can reverse the mechanical hyperalgesia and thermal sensitivity induced by cathepsin S [69]. When neuropathic pain occurs, the activation of and functional changes to microglia play an important role [70]. During the transition from immature to mature microglia, the macrophage transcription factor MAFB is greatly upregulated [71]. At the same time, mice that selectively knock out the Mafb gene in microglia do not develop hyperalgesia, which confirms that MafB plays an important role in the process of neural pain [72].
There are many other mechanisms for post-stroke pain, but they are not well understood, and mainly include mitochondrial Ca2+ uptake, NLRP3 activation, and endoplasmic reticulum stress. Most of these are caused by the massive release of proinflammatory factors to the pain-causing effect [62, 73, 74].
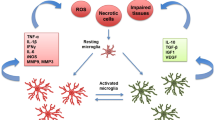
As mentioned above, although the activation of microglia plays an important role in post-stroke pain, no definitive studies have identified the type of microglia that plays a major role in PSP. Moreover, the specific mechanism of M1 and M2 microglia activation in post-stroke pain is not clear. Of course, this also provides possibilities for future research directions (Fig. 1).

Microglial activation plays an important role in pain post hemorrhagic stroke and interactions with other cells. After a stroke, inflammation can cause pain, and pain can also aggravate the inflammatory response. When microglia and macrophages are activated, they can differentiate into two subtypes, M1 (which increase inflammation and pain) and M2 (protective), under the influences of different chemokines. However, the specific functions of the two subtypes of microglia in post-stroke pain have not yet been clarified. At the same time, microglia may have an impact on astrocytes during the ongoing inflammation, thereby changing the functional state of the blood–brain barrier (BBB)
Microglia as a Therapeutic Target for Hemorrhagic Stroke and HS-Related Pain
At present, the clinical treatment of hemorrhagic stroke is mostly based on the cause of treatment. Conventional treatments include hemostasis, lowering the intracranial pressure, preventing complications, and the surgical removal of intracranial hematoma. This shortens the procession of the disease to some extent, and reduces the direct stimulation of physical damage. Thus, when the primary cause is controlled, the continuous damage from inflammation will be greatly reduced.
In recent years, more and more studies have indicated that the interactions of numerous neuroimmune cells in the central nervous system play an essential role in neuropathic pain. Neuropathic pain is not only related to the activation of neurons but also to the response of immune cells [75]. Previous studies have shown that the activation of immune cells, particularly glial cells (including microglia), is involved in neuropathic pain. Glial cells actively communicate with neurons through direct connections, synapses, and the release of neuroregulatory chemicals that affect neuronal firing and signal transmission within the cell [76]. Under neuropathological conditions, microglia express a wide range of neurotransmitter receptors, which leads to intracellular signal transduction (e.g., STATs, NF-κB, and MAPKs) and to a series of cascades that are involved in the development of neuropathic pain. Analgesics targeting the microglia pathway have achieved a certain level of clinical efficacy.
As they are the brain's innate immune cells, microglia can not only differentiate into different subtypes to cope with the development of disease during hemorrhagic stroke, but they can also secrete nutrients to induce neuronal axons to transform damaged regions for regeneration. To a certain extent, the protective effect of M2-type microglia is enhanced while the proinflammatory effect of M1 is weakened. M2 microglia exert their protective effect through the secretion of remodeling factors such as VEGF, BDNF, and matrix metalloproteinase-9 [77, 78]. Therefore, based on the aforementioned effects of microglia, microglia-based therapy and any therapeutic strategy aimed at the regulation of these immune cells may prove to be promising and practical therapeutic methods.
This cell therapy strategy can be divided into two main aspects: the inhibition of M1 proinflammatory type cells and factors or receptors—for example, anti-HMGB1 antibody treatment attenuates the brain injury after stroke [79] and anti-inflammatory cytokines (such as IL-4, IL-10, and TGF-β) can improve the brain recovery process after injury [80]—and the promotion of M2 protective-type cells. Interestingly, activated protective cytokines, such as transforming growth factor-β (TGF-β) and IL-10, may suppress inflammation and promote axonal growth and angiogenesis [7, 81].
Multiple forms of microglial activation and associated inflammatory responses are being considered as new targets for the development of effective treatments for neuropathies. As previously discussed above, the activation pattern of microglia varies greatly during neuropathic pain, with microglial activation peaking in the first few days after injury and declining from 3 days to 2 weeks after injury. Further research is needed on the potential mechanisms through which microglia are involved in neuropathic pain and their role in neuropathic pain during the hemorrhagic stroke recovery phase.
Conclusion and Perspectives
A large number of studies have proved that the activation of microglia plays an important role in the pathophysiological progression of ICH. Different cell phenotypes may contribute differently to neuroinflammation in brain disease models. Regulating the function of microglia can reduce hemorrhage-related brain damage and promote tissue repair and functional recovery. However, given that microglia are widespread in the central nervous system, do microglia in the brain and microglia in the spinal cord play the same role in PSP after intracerebral hemorrhage? And does the regulation of microglia function during certain periods and for certain phenotypes improve pathological pain after stroke? This is not yet clear. Simultaneously, studies have shown that the underlying mechanism may vary with pain phenotype. Therefore, the specific role of microglia in post-stroke pain after intracerebral hemorrhage needs to be further studied.
There are still many limitations of existing animal models of cerebral hemorrhage. Although research into the cellular and molecular mechanism of pathological pain in stroke has become more and more prominent, most studies have failed to make the leap from basic experiment to clinical application. This also highlights the importance of establishing a clinical stroke model. Progress in science and technology is expected to lead to a greater understanding of the role of microglia in pathological neuralgia after intracerebral hemorrhage, and thus to improve our understanding of the mechanism of pathological pain after stroke, allowing the development of an effective treatment for post-stroke pathological neuralgia [82,83,84,85,86,87].
References
Mensah GA, Sacco RL, Vickrey BG, Sampson UKA, Waddy S, Ovbiagele B, Pandian JD, Norrving B, Feigin VL. From data to action: neuroepidemiology informs implementation research for global stroke prevention and treatment. Neuroepidemiology. 2015;45:221–9. https://doi.org/10.1159/000441105.
Bovim MR, Indredavik B, Hokstad A et al. New-onset pain in the early phase and three months following stroke - data from a multicenter study. J Pain Res. 2018;11:1869–76.
Klit H, Finnerup NB, Jensen TS. Central post-stroke pain: clinical characteristics, pathophysiology, and management. Lancet Neurol. 2009;8:857–68. https://doi.org/10.1016/S1474-4422(09)70176-0.
Kong K, Woon V, Yang S. Prevalence of chronic pain and its impact on health-related quality of life in stroke survivors. Arch Phys Med Rehabil. 2004;85:35–40. https://doi.org/10.1016/S0003-9993(03)00369-1.
Xi G, Keep RF, Hoff JT. Mechanisms of brain injury after intracerebral haemorrhage. Lancet Neurol. 2006;5:53–63. https://doi.org/10.1016/S1474-4422(05)70283-0.
Zhang Z, Zhang Z, Lu H, Yang Q, Wu H, Wang J. Microglial polarization and inflammatory mediators after intracerebral hemorrhage. Mol Neurobiol. 2017;54:1874–86. https://doi.org/10.1007/s12035-016-9785-6.
Zhao X, Sun G, Zhang J, Strong R, Song W, Gonzales N, Grotta JC, Aronowski J. Hematoma resolution as a target for intracerebral hemorrhage treatment: role for peroxisome proliferator-activated receptor gamma in microglia/macrophages. Ann Neurol. 2007;61:352–62. https://doi.org/10.1002/ana.21097.
Lively S, Schlichter LC. Age-related comparisons of evolution of the inflammatory response after intracerebral hemorrhage in rats. Transl Stroke Res. 2012;3:132–46. https://doi.org/10.1007/s12975-012-0151-3.
Rodriguez-Yanez M, Brea D, Arias S, Blanco M, Pumar JM, Castillo J, Sobrino T. Increased expression of Toll-like receptors 2 and 4 is associated with poor outcome in intracerebral hemorrhage. J Neuroimmunol. 2012;247:75–80. https://doi.org/10.1016/j.jneuroim.2012.03.019.
Wang YC, Zhou Y, Fang H, Lin S, Wang PF, Xiong RP, Chen J, Xiong XY, Lv FL, Liang QL, Yang QW. Toll-like receptor 2/4 heterodimer mediates inflammatory injury in intracerebral hemorrhage. Ann Neurol. 2014;75:876–89. https://doi.org/10.1002/ana.24159.
Boche D, Perry VH, Nicoll JA. Review: activation patterns of microglia and their identification in the human brain. Neuropathol Appl Neurobiol. 2013;39:3–18. https://doi.org/10.1111/nan.12011.
Hanisch UK, Kettenmann H. Microglia: active sensor and versatile effector cells in the normal and pathologic brain. Nat Neurosci. 2007;10:1387–94. https://doi.org/10.1038/nn1997.
Henkel JS, Beers DR, Zhao W, Appel SH. Microglia in ALS: the good, the bad, and the resting. J Neuroimmune Pharmacol. 2009;4:389–98. https://doi.org/10.1007/s11481-009-9171-5.
Ponomarev ED, Veremeyko T, Weiner HL. MicroRNAs are universal regulators of differentiation, activation, and polarization of microglia and macrophages in normal and diseased CNS. Glia. 2013;61:91–103. https://doi.org/10.1002/glia.22363.
Varnum MM, Ikezu T. The classification of microglial activation phenotypes on neurodegeneration and regeneration in Alzheimer’s disease brain. Arch Immunol Ther Exp (Warsz). 2012;60:251–66. https://doi.org/10.1007/s00005-012-0181-2.
Donnelly RP, Dickensheets H, Finbloom DS. The interleukin-10 signal transduction pathway and regulation of gene expression in mononuclear phagocytes. J Interferon Cytokine Res. 1999;19:563–73. https://doi.org/10.1089/107999099313695.
Yoshimura A, Naka T, Kubo M. SOCS proteins, cytokine signalling and immune regulation. Nat Rev Immunol. 2007;7:454–65. https://doi.org/10.1038/nri2093.
Butovsky O, Ziv Y, Schwartz A, Landa G, Talpalar AE, Pluchino S, Martino G, Schwartz M. Microglia activated by IL-4 or IFN-gamma differentially induce neurogenesis and oligodendrogenesis from adult stem/progenitor cells. Mol Cell Neurosci. 2006;31:149–60. https://doi.org/10.1016/j.mcn.2005.10.006.
Hu X, Leak RK, Shi Y, Suenaga J, Gao Y, Zheng P, Chen J. Microglial and macrophage polarization-new prospects for brain repair. Nat Rev Neurol. 2015;11:56–64. https://doi.org/10.1038/nrneurol.2014.207.
Orihuela R, McPherson CA, Harry GJ. Microglial M1/M2 polarization and metabolic states. Br J Pharmacol. 2016;173:649–65. https://doi.org/10.1111/bph.13139.
Zheng ZV, Lyu H, Lam S, Lam PK, Poon WS, Wong G. The dynamics of microglial polarization reveal the resident neuroinflammatory responses after subarachnoid hemorrhage. Transl Stroke Res. 2020;11:433–49. https://doi.org/10.1007/s12975-019-00728-5.
Hu X, Li P, Guo Y, Wang H, Leak RK, Chen S, Gao Y, Chen J. Microglia/macrophage polarization dynamics reveal novel mechanism of injury expansion after focal cerebral ischemia. Stroke. 2012;43:3063–70. https://doi.org/10.1161/STROKEAHA.112.659656.
Lan X, Han X, Li Q, Li Q, Gao Y, Cheng T, Wan J, Zhu W, Wang J. Pinocembrin protects hemorrhagic brain primarily by inhibiting Toll-like receptor 4 and reducing M1 phenotype microglia. Brain Behav Immun. 2017;61:326–39. https://doi.org/10.1016/j.bbi.2016.12.012.
Lan X, Han X, Li Q, Yang QW, Wang J. Modulators of microglial activation and polarization after intracerebral haemorrhage. Nat Rev Neurol. 2017;13:420–33. https://doi.org/10.1038/nrneurol.2017.69.
Taylor RA, Chang CF, Goods BA, Hammond MD, Mac GB, Ai Y, Steinschneider AF, Renfroe SC, Askenase MH, McCullough LD, Kasner SE, Mullen MT, Hafler DA, Love JC, Sansing LH. TGF-beta1 modulates microglial phenotype and promotes recovery after intracerebral hemorrhage. J Clin Invest. 2017;127:280–92. https://doi.org/10.1172/JCI88647.
Xue Y, et al. Microglial polarization: novel therapeutic strategy against ischemic stroke. Aging Dis. 2021;12(2):466–79.
Popiolek-Barczyk K, Mika J. Targeting the microglial signaling pathways: new insights in the modulation of neuropathic pain. Curr Med Chem. 2016;23:2908–28. https://doi.org/10.2174/0929867323666160607120124.
Trang T, Beggs S, Salter MW. Brain-derived neurotrophic factor from microglia: a molecular substrate for neuropathic pain. Neuron Glia Biol. 2011;7:99–108. https://doi.org/10.1017/S1740925X12000087.
Dutra FF, Alves LS, Rodrigues D, Fernandez PL, de Oliveira RB, Golenbock DT, Zamboni DS, Bozza MT. Hemolysis-induced lethality involves inflammasome activation by heme. Proc Natl Acad Sci USA. 2014;111:E4110–8. https://doi.org/10.1073/pnas.1405023111.
Gurung P, Anand PK, Malireddi RK, Vande WL, Van Opdenbosch N, Dillon CP, Weinlich R, Green DR, Lamkanfi M, Kanneganti TD. FADD and caspase-8 mediate priming and activation of the canonical and noncanonical Nlrp3 inflammasomes. J Immunol. 2014;192:1835–46. https://doi.org/10.4049/jimmunol.1302839.
Lamkanfi M, Dixit VM. Mechanisms and functions of inflammasomes. Cell. 2014;157:1013–22. https://doi.org/10.1016/j.cell.2014.04.007.
Casson CN, Copenhaver AM, Zwack EE, Nguyen HT, Strowig T, Javdan B, Bradley WP, Fung TC, Flavell RA, Brodsky IE, Shin S. Caspase-11 activation in response to bacterial secretion systems that access the host cytosol. PLoS Pathog. 2013;9: e1003400. https://doi.org/10.1371/journal.ppat.1003400.
Kober DL, Brett TJ. TREM2-ligand interactions in health and disease. J Mol Biol. 2017;429:1607–29. https://doi.org/10.1016/j.jmb.2017.04.004.
Ulrich JD, Holtzman DM. TREM2 function in Alzheimer’s disease and neurodegeneration. ACS Chem Neurosci. 2016;7:420–7. https://doi.org/10.1021/acschemneuro.5b00313.
Ford JW, McVicar DW. TREM and TREM-like receptors in inflammation and disease. Curr Opin Immunol. 2009;21:38–46. https://doi.org/10.1016/j.coi.2009.01.009.
Mracsko E, Veltkamp R. Neuroinflammation after intracerebral hemorrhage. Front Cell Neurosci. 2014;8:388. https://doi.org/10.3389/fncel.2014.00388.
Takahashi K, Rochford CD, Neumann H. Clearance of apoptotic neurons without inflammation by microglial triggering receptor expressed on myeloid cells-2. J Exp Med. 2005;201:647–57. https://doi.org/10.1084/jem.20041611.
Sharif O, Knapp S. From expression to signaling: roles of TREM-1 and TREM-2 in innate immunity and bacterial infection. Immunobiology. 2008;213:701–13. https://doi.org/10.1016/j.imbio.2008.07.008.
Jay TR, von Saucken VE, Landreth GE. TREM2 in neurodegenerative diseases. Mol Neurodegener. 2017;12:56. https://doi.org/10.1186/s13024-017-0197-5.
Raha AA, Henderson JW, Stott SR, Vuono R, Foscarin S, Friedland RP, Zaman SH, Raha-Chowdhury R. Neuroprotective effect of TREM-2 in aging and Alzheimer’s disease model. J Alzheimers Dis. 2017;55:199–217. https://doi.org/10.3233/JAD-160663.
Takahashi K, Prinz M, Stagi M, Chechneva O, Neumann H. TREM2-transduced myeloid precursors mediate nervous tissue debris clearance and facilitate recovery in an animal model of multiple sclerosis. PLoS Med. 2007;4: e124. https://doi.org/10.1371/journal.pmed.0040124.
Chen S, Peng J, Sherchan P, Ma Y, Xiang S, Yan F, Zhao H, Jiang Y, Wang N, Zhang JH, Zhang H. TREM2 activation attenuates neuroinflammation and neuronal apoptosis via PI3K/Akt pathway after intracerebral hemorrhage in mice. J Neuroinflamm. 2020;17:168. https://doi.org/10.1186/s12974-020-01853-x.
Zhang H, Sheng L, Tao J, Chen R, Li Y, Sun Z, Qian W. Depletion of the triggering receptor expressed on myeloid cells 2 inhibits progression of renal cell carcinoma via regulating related protein expression and PTEN-PI3K/Akt pathway. Int J Oncol. 2016;49:2498–506. https://doi.org/10.3892/ijo.2016.3740.
Zhu M, Li D, Wu Y, Huang X, Wu M. TREM-2 promotes macrophage-mediated eradication of Pseudomonas aeruginosa via a PI3K/Akt pathway. Scand J Immunol. 2014;79:187–96. https://doi.org/10.1111/sji.12148.
Watcharotayangul J, Mao L, Xu H, Vetri F, Baughman VL, Paisansathan C, Pelligrino DA. Post-ischemic vascular adhesion protein-1 inhibition provides neuroprotection in a rat temporary middle cerebral artery occlusion model. J Neurochem. 2012. https://doi.org/10.1111/j.1471-4159.2012.07950.x.
Xu HL, Garcia M, Testai F, Vetri F, Barabanova A, Pelligrino DA, Paisansathan C. Pharmacologic blockade of vascular adhesion protein-1 lessens neurologic dysfunction in rats subjected to subarachnoid hemorrhage. Brain Res. 2014;1586:83–9. https://doi.org/10.1016/j.brainres.2014.08.036.
Ma Q, Manaenko A, Khatibi NH, Chen W, Zhang JH, Tang J. Vascular adhesion protein-1 inhibition provides antiinflammatory protection after an intracerebral hemorrhagic stroke in mice. J Cereb Blood Flow Metab. 2011;31:881–93. https://doi.org/10.1038/jcbfm.2010.167.
Haruwaka K, Ikegami A, Tachibana Y, Ohno N, Konishi H, Hashimoto A, Matsumoto M, Kato D, Ono R, Kiyama H, Moorhouse AJ, Nabekura J, Wake H. Dual microglia effects on blood brain barrier permeability induced by systemic inflammation. Nat Commun. 2019;10:5816. https://doi.org/10.1038/s41467-019-13812-z.
Haruwaka K, Ikegami A, Tachibana Y, Ohno N, Konishi H, Hashimoto A, Matsumoto M, Kato D, Ono R, Kiyama H, Moorhouse AJ, Nabekura J, Wake H. Dual microglia effects on blood brain barrier permeability induced by systemic inflammation. Nat Commun. 2019. https://doi.org/10.1038/s41467-019-13812-z.
Zhu X, Hill RA, Dietrich D, Komitova M, Suzuki R, Nishiyama A. Age-dependent fate and lineage restriction of single NG2 cells. Development. 2011;138:745–53. https://doi.org/10.1242/dev.047951.
Magnusson JP, Goritz C, Tatarishvili J, Dias DO, Smith EM, Lindvall O, Kokaia Z, Frisen J. A latent neurogenic program in astrocytes regulated by Notch signaling in the mouse. Science. 2014;346:237–41. https://doi.org/10.1126/science.346.6206.237.
Parent JM, Vexler ZS, Gong C, Derugin N, Ferriero DM. Rat forebrain neurogenesis and striatal neuron replacement after focal stroke. Ann Neurol. 2002;52:802–13. https://doi.org/10.1002/ana.10393.
Ginhoux F, Greter M, Leboeuf M, Nandi S, See P, Gokhan S, Mehler MF, Conway SJ, Ng LG, Stanley ER, Samokhvalov IM, Merad M. Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science. 2010;330:841–5. https://doi.org/10.1126/science.1194637.
Yamasaki R, Lu H, Butovsky O, Ohno N, Rietsch AM, Cialic R, Wu PM, Doykan CE, Lin J, Cotleur AC, Kidd G, Zorlu MM, Sun N, Hu W, Liu L, Lee JC, Taylor SE, Uehlein L, Dixon D, Gu J, Floruta CM, Zhu M, Charo IF, Weiner HL, Ransohoff RM. Differential roles of microglia and monocytes in the inflamed central nervous system. J Exp Med. 2014;211:1533–49. https://doi.org/10.1084/jem.20132477.
Girard S, Brough D, Lopez-Castejon G, Giles J, Rothwell NJ, Allan SM. Microglia and macrophages differentially modulate cell death after brain injury caused by oxygen-glucose deprivation in organotypic brain slices. Glia. 2013;61:813–24. https://doi.org/10.1002/glia.22478.
Paolicelli RC, Bolasco G, Pagani F, Maggi L, Scianni M, Panzanelli P, Giustetto M, Ferreira TA, Guiducci E, Dumas L, Ragozzino D, Gross CT. Synaptic pruning by microglia is necessary for normal brain development. Science. 2011;333:1456–8. https://doi.org/10.1126/science.1202529.
Arvidsson A, Collin T, Kirik D, Kokaia Z, Lindvall O. Neuronal replacement from endogenous precursors in the adult brain after stroke. Nat Med. 2002;8:963–70. https://doi.org/10.1038/nm747.
Batchelor PE, Porritt MJ, Martinello P, Parish CL, Liberatore GT, Donnan GA, Howells DW. Macrophages and microglia produce local trophic gradients that stimulate axonal sprouting toward but not beyond the wound edge. Mol Cell Neurosci. 2002;21:436–53. https://doi.org/10.1006/mcne.2002.1185.
Lu HF, Xu CY, Zhang L, Gan L, Chen C, Yan MY, Guo XN, Fang Q, Xu GY, Zhang YB, Ni JQ, Zhao HR. A new central post-stroke pain rat model: autologous blood injected thalamic hemorrhage involved increased expression of P2X4 receptor. Neurosci Lett. 2018;687:124–30. https://doi.org/10.1016/j.neulet.2018.09.023.
Saab CY, Hains BC. Remote neuroimmune signaling: a long-range mechanism of nociceptive network plasticity. Trends Neurosci. 2009;32:110–7. https://doi.org/10.1016/j.tins.2008.11.004.
Volonte C, Apolloni S, Skaper SD, Burnstock G. P2X7 receptors: channels, pores and more. CNS Neurol Disord Drug Targets. 2012;11:705–21. https://doi.org/10.2174/187152712803581137.
Li SJ, Zhang YF, Ma SH, Yi Y, Yu HY, Pei L, Feng D. The role of NLRP3 inflammasome in stroke and central poststroke pain. Medicine (Baltimore). 2018;97: e11861. https://doi.org/10.1097/MD.0000000000011861.
Luo Y, Reis C, Chen S. NLRP3 Inflammasome in the pathophysiology of hemorrhagic stroke: a review. Curr Neuropharmacol. 2019;17:582–9. https://doi.org/10.2174/1570159X17666181227170053.
Hiraga SI, Itokazu T, Hoshiko M, Takaya H, Nishibe M, Yamashita T. Microglial depletion under thalamic hemorrhage ameliorates mechanical allodynia and suppresses aberrant axonal sprouting. JCI Insight. 2020. https://doi.org/10.1172/jci.insight.131801.
Nagasaka K, Takashima I, Matsuda K, Higo N. Late-onset hypersensitivity after a lesion in the ventral posterolateral nucleus of the thalamus: a macaque model of central post-stroke pain. Sci Rep. 2017;7:10316. https://doi.org/10.1038/s41598-017-10679-2.
Hesselgesser J, Horuk R. Chemokine and chemokine receptor expression in the central nervous system. J Neurovirol. 1999;5:13–26. https://doi.org/10.3109/13550289909029741.
Schall T. Fractalkine—a strange attractor in the chemokine landscape. Immunol Today. 1997;18:147. https://doi.org/10.1016/s0167-5699(97)84655-5.
Li D, Huang ZZ, Ling YZ, Wei JY, Cui Y, Zhang XZ, Zhu HQ, Xin WJ. Up-regulation of CX3CL1 via nuclear factor-kappaB-dependent histone acetylation is involved in paclitaxel-induced peripheral neuropathy. Anesthesiology. 2015;122:1142–51. https://doi.org/10.1097/ALN.0000000000000560.
Clark AK, Yip PK, Grist J, Gentry C, Staniland AA, Marchand F, Dehvari M, Wotherspoon G, Winter J, Ullah J, Bevan S, Malcangio M. Inhibition of spinal microglial cathepsin S for the reversal of neuropathic pain. Proc Natl Acad Sci USA. 2007;104:10655–60. https://doi.org/10.1073/pnas.0610811104.
Inoue K, Tsuda M. Microglia and neuropathic pain. Glia. 2009;57:1469–79. https://doi.org/10.1002/glia.20871.
Matcovitch-Natan O, Winter DR, Giladi A, Vargas AS, Spinrad A, Sarrazin S, Ben-Yehuda H, David E, Zelada GF, Perrin P, Keren-Shaul H, Gury M, Lara-Astaiso D, Thaiss CA, Cohen M, Bahar HK, Baruch K, Deczkowska A, Lorenzo-Vivas E, Itzkovitz S, Elinav E, Sieweke MH, Schwartz M, Amit I. Microglia development follows a stepwise program to regulate brain homeostasis. Science. 2016;353:8670. https://doi.org/10.1126/science.aad8670.
Tozaki-Saitoh H, Masuda J, Kawada R, Kojima C, Yoneda S, Masuda T, Inoue K, Tsuda M. Transcription factor MafB contributes to the activation of spinal microglia underlying neuropathic pain development. Glia. 2019;67:729–40. https://doi.org/10.1002/glia.23570.
Rizzuto R, De Stefani D, Raffaello A, Mammucari C. Mitochondria as sensors and regulators of calcium signalling. Nat Rev Mol Cell Biol. 2012;13:566–78. https://doi.org/10.1038/nrm3412.
Rossol M, Pierer M, Raulien N, Quandt D, Meusch U, Rothe K, Schubert K, Schoneberg T, Schaefer M, Krugel U, Smajilovic S, Brauner-Osborne H, Baerwald C, Wagner U. Extracellular Ca2+ is a danger signal activating the NLRP3 inflammasome through G protein-coupled calcium sensing receptors. Nat Commun. 2012;3:1329. https://doi.org/10.1038/ncomms2339.
Rojewska E, Korostynski M, Przewlocki R, Przewlocka B, Mika J. Expression profiling of genes modulated by minocycline in a rat model of neuropathic pain. Mol Pain. 2014;10:1744–8069.
Beitz AJ, Lee JH. Intrathecal injection of carbenoxolone, a gap junction decoupler, attenuates the induction of below level neuropathic pain after spinal cord injury in rats. Exp Neurol. 2010;224:123–32.
Crain JM, Nikodemova M, Watters JJ. Microglia express distinct M1 and M2 phenotypic markers in the postnatal and adult central nervous system in male and female mice. J Neurosci Res. 2013;91:1143–51. https://doi.org/10.1002/jnr.23242.
Rosenberg GA. Matrix metalloproteinases and their multiple roles in neurodegenerative diseases. Lancet Neurol. 2009;8:205–16. https://doi.org/10.1016/S1474-4422(09)70016-X.
Shichita T, Hasegawa E, Kimura A, Morita R, Sakaguchi R, Takada I, Sekiya T, Ooboshi H, Kitazono T, Yanagawa T, Ishii T, Takahashi H, Mori S, Nishibori M, Kuroda K, Akira S, Miyake K, Yoshimura A. Peroxiredoxin family proteins are key initiators of post-ischemic inflammation in the brain. Nat Med. 2012;18:911–7. https://doi.org/10.1038/nm.2749.
Iadecola C, Anrather J. The immunology of stroke: from mechanisms to translation. Nat Med. 2011;17:796–808. https://doi.org/10.1038/nm.2399.
Zhao H, Garton T, Keep RF, Hua Y, Xi G. Microglia/macrophage polarization after experimental intracerebral hemorrhage. Transl Stroke Res. 2015;6:407–9. https://doi.org/10.1007/s12975-015-0428-4.
Bowsher D. Stroke and central poststroke pain in an elderly population. J Pain. 2001;2:258–61. https://doi.org/10.1054/jpai.2001.24549.
Bowsher D. Allodynia in relation to lesion site in central post-stroke pain. J Pain. 2005;6:736–40. https://doi.org/10.1016/j.jpain.2005.06.009.
Hansen AP, Marcussen NS, Klit H, Andersen G, Finnerup NB, Jensen TS. Pain following stroke: a prospective study. Eur J Pain. 2012;16:1128–36. https://doi.org/10.1002/j.1532-2149.2012.00123.x.
Wang J. Preclinical and clinical research on inflammation after intracerebral hemorrhage. Prog Neurobiol. 2010;92:463–77. https://doi.org/10.1016/j.pneurobio.2010.08.001.
Yabluchanskiy A, Sawle P, Homer-Vanniasinkam S, Green CJ, Foresti R, Motterlini R. CORM-3, a carbon monoxide-releasing molecule, alters the inflammatory response and reduces brain damage in a rat model of hemorrhagic stroke. Crit Care Med. 2012;40:544–52. https://doi.org/10.1097/CCM.0b013e31822f0d64.
Zhou Y, Wang Y, Wang J, Anne SR, Yang QW. Inflammation in intracerebral hemorrhage: from mechanisms to clinical translation. Prog Neurobiol. 2014;115:25–44. https://doi.org/10.1016/j.pneurobio.2013.11.003.
Acknowledgements
Authorship
All named authors meet the International Committee of Medical Journal Editors (ICMJE) criteria for authorship for this article, take responsibility for the integrity of the work as a whole, and have given their approval for this version to be published.
Author Contributions
All authors have contributed to the preparation of this manuscript and revised and approved the submitted version.
Disclosures
Chen He, Renhuai Liu, Zhongming Fan, Yi Li, Manping Yang, Hou Wugang, Zhihong Lu; Zongping Fang, Binxiao Su have nothing to disclose.
Compliance with Ethics Guidelines
This article is based on previously conducted studies and does not contain any new studies with human participants or animals performed by any of the authors.
Data Availability
Data sharing is not applicable to this article as no datasets were generated or analyzed during the current study.
Funding
Project 81870961 supported by National Natural Science Foundation of China provided funding for both the study and the journal's Rapid Service Fee.
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
He, C., Liu, R., Fan, Z. et al. Microglia in the Pathophysiology of Hemorrhagic Stroke and the Relationship Between Microglia and Pain After Stroke: A Narrative Review. Pain Ther 10, 927–939 (2021). https://doi.org/10.1007/s40122-021-00288-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40122-021-00288-3