
Abstract
Introduction
AOD01 is a novel, fully human immunoglobulin (Ig) G1 neutralizing monoclonal antibody that was developed as a therapeutic against severe acute respiratory syndrome coronavirus type 2 (SARS-CoV-2). This first-in-human study assessed safety, tolerability, pharmacokinetics (PK), and pharmacodynamics of AOD01 in healthy volunteers.
Methods
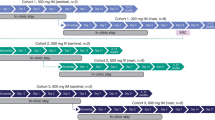
Intravenous doses of AOD01 were evaluated in escalating cohorts [four single-dose cohorts (2, 5, 10, and 20 mg/kg) and one two-dose cohort (two doses of 20 mg/kg, 24 h apart)].
Results
Twenty-three subjects were randomized to receive AOD01 or a placebo in blinded fashion. A total of 34 treatment-emergent adverse events (TEAEs) were reported; all were mild in severity. Related events (headache and diarrhea) were reported in one subject each. No event of infusion reactions, serious adverse event (SAE), or discontinuation due to AE were reported. The changes in laboratory parameters, vital signs, and electrocardiograms were minimal. Dose-related exposure was seen from doses 2 to 20 mg/kg as confirmed by Cmax and AUC0–tlast. The median Tmax was 1.5–3 h. Clearance was dose independent. Study results revealed long half-lives (163–465 h). Antidrug antibodies (ADA) to AOD01 were not detected among subjects, except in one subject of the two-dose cohort on day 92. Sustained ex vivo neutralization of SARS-CoV-2 was recorded until day 29 with single doses from 2 to 20 mg/kg and until day 43 with two doses of 20 mg/kg.
Conclusions
AOD01 was safe and well tolerated, demonstrated dose-related PK, non-immunogenic status, and sustained ex vivo neutralization of SARS-CoV-2 after single intravenous dose ranging from 2 to 20 mg/kg and two doses of 20 mg/kg and show good potential for treatment of SARS-CoV-2 infection.
(Health Sciences Authority identifier number CTA2000119).
Similar content being viewed by others


Avoid common mistakes on your manuscript.
This study was planned to assessed safety, tolerability, pharmacokinetics, and pharmacodynamics of AOD01. |
AOD01 is a novel, fully human immunoglobulin (Ig) G1 neutralizing monoclonal antibody with Fc-mediated antiviral activity, developed as a therapeutic treatment against SARS-CoV-2. |
After single IV dose ranging from 2 to 20 mg/kg and two doses of 20 mg/kg, AOD01 demonstrated dose-related PK, non-immunogenic, and sustained ex vivo neutralization of SARS-CoV-2. |
AOD01 was found to be safe and well tolerated in this study. |
Introduction
The coronavirus disease 2019 (COVID-19) pandemic caused by the novel severe acute respiratory syndrome coronavirus type 2 (SARS-CoV-2) has affected public health for the last 2 years and it continues to spread on a global scale [1]. Its manifestation ranges from respiratory tract infection to severe disease and death. Though the vaccination of the global population is underway, other options to protect the susceptible population and subjects who develop breakthrough infections should be considered. This has underlined the importance of therapeutic strategies which can provide protective immunity and/or treatment against the SARS-CoV virus [2, 3].
Monoclonal antibodies (mAb) are proven effective early treatment options for COVID-19 [4]. Immediate passive humoral immunotherapy with neutralizing monoclonal antibodies plays a very important role in treating infections. Neutralizing monoclonal antibodies bind to the viral spike (S) protein, which mediates virus entry into the susceptible cells, and prevent viral entry into cells by blocking the interaction between S protein and angiotensin-converting enzyme 2 (ACE2) receptor on the host cell surface [5,6,7,8]. Efforts are underway to develop therapeutic antibodies for SARS-CoV-2. To date, a few therapeutic antibodies have been authorized for emergency clinical use, while several antibodies are in clinical development [9,10,11,12,13].
AOD01 is a novel, fully human immunoglobulin (Ig) G1 SARS-CoV-2 neutralizing monoclonal antibody with Fc-mediated antiviral activity. Its functionality is similar to other therapeutic antibodies [2, 14, 15]. AOD01 was identified in a patient infected with the virus during the initial outbreak in Singapore involving the wild-type virus. AOD01 was identified following isolation and screening of a panel of memory B cells 2 months after the start of the outbreak, with clinical-grade drug product manufactured within 6 months of identification [2]. It has been shown to be efficacious in vitro against both the Delta variant and the wild-type virus. In vitro studies demonstrated that AOD01 inhibits virus interaction with ACE2 receptors. In a direct SARS-CoV-2 neutralization assay, AOD01 had a half-maximal inhibitory concentration of 0.3 μg/mL. The in vivo efficacy of AOD01 was demonstrated in three animal models of COVID-19. In the golden Syrian hamster and Indian rhesus macaque SARS-CoV-2 models, AOD01, at a single dose of 20 mg/kg given 4 to 6 h after the virus challenge, was efficacious in minimizing or preventing disease progression. Reduction in viral load was measured by median tissue culture infectious dose (TCID50) and real-time quantitative polymerase chain reaction (RT-qPCR) analysis in the Indian rhesus macaque SARS-CoV-2 model. Doses of 5 and 10 mg/kg were also found to be equally efficacious based on TCID50 data. Similarly, AOD01 treatment in SARS-CoV-2-challenged k18-human ACE2 (hACE2) transgenic mice was found efficacious at doses ranging from 5 to 20 mg/kg given once, 6 h after the virus challenge. It was seen that the optimal therapeutic efficacy of AOD01 requires Fc-mediated effector functions that promote an IFNγ-driven antiviral immune response along with its neutralization ability. Antibody-dependent enhancement (ADE) was not observed in the mouse model when treated with subtherapeutic doses of AOD01 [2].
On the basis of the favorable data derived from these preclinical experiments, AOD01 has clear potential for treating COVID-19 (AOD01, data on file). Hence, the first-in-human (FIH) study of AOD01 was undertaken in healthy volunteers to study safety, tolerability, pharmacokinetics (PK), and immunogenicity.
Methods
Study Design
This phase I, FIH, ascending dose study was conducted at Tan Tock Seng Hospital, Singapore from October 2020 to April 2021. The primary objective was to evaluate the safety and tolerability of ascending doses of AOD01. Secondary objectives were to characterize the PK profile and to assess the immunogenicity of ascending doses of AOD01. Ex vivo bioassay and immunomonitoring of clinical samples were exploratory objectives of the study.
Ethics
The study was approved by the institutional review board and was conducted in accordance with the Declaration of Helsinki, in compliance with the Council for International Organizations of Medical Sciences International Ethical Guidelines, applicable ICH Good Clinical Practice (GCP) Guidelines, and applicable laws and regulations. Written informed consents were obtained from all participants prior to enrolment in the study.
Study Participants
Healthy male and female subjects aged 21 to 55 years, body weight within 50 to 100 kg, body mass index (BMI) within 18.0 to 35.0 kg/m2, who agreed to use contraceptive methods and women of non-childbearing potential were enrolled in the study.
Subjects with a history of any systemic disorders that may interfere with administration of the drug, laboratory-confirmed SARS-CoV-2, evidence of active or latent tuberculosis (TB), allergies to humanized mAbs, malignancy within the past 5 years, current or chronic history of liver diseases, alanine transaminase (ALT), and bilirubin greater than 1.5 times the upper limit of normal (ULN), corrected QT interval (QTc) greater than 450 ms for men or greater than 470 ms for women, human immunodeficiency virus (HIV), hepatitis B surface antigen (HbsAg), and hepatitis C virus (HCV) infections were excluded from the study. Individuals who had used over the counter or prescription medication within 7 days, live vaccine(s) within 1 month, treatment with biologic agents within 3 months or 5 half-lives prior to dosing, exposure to more than four new chemical entities within 12 months, alcohol consumption, or use of drugs of abuse were also excluded from the study.
Study Procedure
The study investigated four single-dose cohorts at dose levels of 2, 5, 10, and 20 mg/kg and one two-dose cohort at the highest safe dose level of 20 mg/kg as determined from the single ascending dose cohorts. A sentinel group of two subjects (one AOD01 and one placebo) was dosed as planned within each of the first, fourth, and fifth cohorts. The remainder of the cohort was dosed 24 h after the dosing of the last subject in the sentinel group. Dosing in the two-dose cohort was done 72 h after the start of dosing of the last subject in the safe single-dose cohort.
The duration of the study was 92 days which includes a drug administration period (1 day for single-dose cohorts and 2 days for the two-dose cohort), an inpatient observation period (5 days, including the day of study drug administrations), and a follow-up period (92 days from day 1).
Treatment
AOD01 was administered as a continuous slow intravenous (IV) infusion over 60 min. A 5-mL vial was used which contained AOD01 at a concentration of 30 mg/mL. AOD01 was diluted with commercially available 500 mL of 0.9% sodium chloride solution to achieve the desired dose level. Commercially available 0.9% sodium chloride was used as the placebo. Treatment duration was 1 day for single-dose cohorts and 2 days for the two-dose cohort. For the two-dose cohort, the second dose was administered 24 h after the first dose.
Randomization and Blinding
The subjects were randomly allocated to receive either AOD01 or a placebo. The randomization list was generated using SAS® Version 9.4. An interactive response technology was used to administer the randomization schedule. This was a blinded study where subjects, investigator staff, persons performing the assessments, and data analysts remained blinded to the identity of study treatments.
Study Endpoints
The primary endpoint was the incidence and severity of treatment-emergent adverse events (TEAEs) in subjects treated with AOD01 or placebo. Secondary endpoints were the assessment of PK parameters (Cmax, AUC0–t, AUC0–inf, Tmax, t1/2) and antidrug antibody (ADA) to AOD01. Ex vivo bioassay for neutralization of SARS-CoV-2 and immunomonitoring of clinical samples for changes in cytokines, immune cells, and RNA sequencing of whole blood were exploratory endpoints.
Safety Assessment
Safety assessments were performed starting from the signing of the informed consent form until the last follow-up visit.
Safety parameters included TEAEs, related TEAEs, TEAEs by severity, TEAEs leading to discontinuation, serious TEAEs, and death. Parameters also included acute or delayed infusion reaction, vital signs, physical examination, electrocardiogram (ECG) changes, and laboratory parameters. Common Terminology Criteria for Adverse Events (CTCAE) Version 5.0 was used for the assessment of severity. Subjects were also evaluated for symptoms of COVID-19 at each follow-up visit.
PK Assessment
Blood collected in BD Vacutainer® serum tubes with silica was used to obtain serum as per the manufacturer’s instruction for PK assessment. For single-dose cohorts, PK sampling was performed on day 1 and through day 5 discharge at the following time points: pre-infusion, at 0.5, 1, 2, 4, 6, 8, 12, 24, 48, and 72 h after the start of the infusion, at the follow-up visits on days 6, 8, 15, 22, 29, 43, 57, 71, and 92.
For the two-dose cohort, PK sampling was performed on day 1 at the following time points: pre-infusion, at 0.5, 1, 2, 4, 6, 8, 10, and 12 h after the start of infusion. On day 2, sampling was performed pre-infusion, at 0.5, 1, 2, 4, 6, 8, 10, 12, 24, 48, 60, 72, 96, and 120 h after the start of infusion. Sampling was done on follow-up visits on days 8, 9, 15, 22, 29, 43, 57, 71, and 92. PK parameters (AUC0–tlast, AUC0–inf, AUC0–tau, t1/2, CL/F, V/F, Cmax, Ctrough, and Tmax) were estimated.
A precise, sensitive, and reproducible method was validated for the quantification of AOD01 in human serum used in an enzyme-linked immunosorbent assay (ELISA). In brief, isolated spike protein from COVID-19 virus was used as the coating protein; 1 μg/mL spike protein was made up in 1× phosphate buffered saline (PBS) and 30 μL per well was left overnight at 2–8 °C. The plates were then blocked with 1% BSA in PBS for 1.5 h. Serum samples were then diluted to minimum required dilution (MRD) (1:100 and subsequently 1:50 to get a total dilution of 1:5000) and added to the wells. The wells were then incubated at room temperature for 1 h and anti-horse reddish peroxidase (HRP) was used as a secondary antibody. 3,3′,5,5′-Tetramethylbenzidine (TMB) is then added 1 h later for 10 min and quenched with 1 M HCl solution. The plate is read with the HIDEX Sense reader for OD (450 nm) values.
Immunogenicity Assessment
Blood collected in BD Vacutainer® serum tubes with silica was used to obtain serum as per the manufacturer’s instruction. For immunogenicity analysis, blood samples were collected pre-infusion (day 1) and on days 15, 29, 57, and 92 from subjects in single-dose cohorts. For subjects in a two-dose cohort, samples were collected pre-infusion (day 1) and on days 9, 15, 29, 57, and 92. Antidrug antibodies were measured with an assay developed and validated in-house utilizing the receptor-binding domain of the SARS-CoV-2 virus’s spike protein as a surrogate antigen.
Biomarker Assessment
Immunomonitoring Assessment
Blood collected in BD Vacutainer® CPT™ with sodium citrate was used to obtain plasma as per the manufacturer’s instruction for cytokine analysis. Blood samples for measurement of plasma cytokines, chemokines, acute phase reactants, and RNA sequencing were collected pre-infusion, and 1 and 4 h after the start of infusion on day 1, and on days 2, 3, and 8 for subjects in single-dose cohorts. For the two-dose cohort, samples were collected pre-infusion (day 1), at 1 and 4 h after the start of infusion on days 1 and 2 and on days 3, 4, and 9.
Ex Vivo Bioassay Assessment
Blood samples to evaluate ex vivo neutralization of SARS-CoV-2 in single-dose cohorts were collected pre-infusion (day 1), and on day 2, 5, 8, 15, 29, 43, and 92. Samples were collected pre-infusion (day 1), and on day 3, 5, 9, 15, 29, 43, and 92 in subjects who received two doses of AOD01.
Statistical Analysis
All statistical analyses were conducted using SAS version 9.4. Descriptive statistics were provided in the summary tables as per the dose cohorts. Continuous variables were summarized using N, mean, standard deviation (SD), median, and range. Categorical variables were summarized using frequency counts and percentages. Dose proportionality for AOD01 in PK analysis was evaluated from single-dose cohorts for Cmax, AUC0–tlast, and AUC0–inf. PK calculations were performed using Phoenix® WinNonlin® (Certara L.P., Princeton, NJ) Version 8.0 or SAS Version 9.4.
No formal sample size calculation was performed for this study. The sample size was chosen on the basis of experience from previous studies of a similar nature. All subjects who received at least one dose of the study drug were included in the safety analysis. Subjects in whom serum concentration data facilitate the derivation of at least one PK parameter and in whom the time of drug administration on the day of sampling was known were included in PK analysis.
Results
Subjects Disposition and Baseline Characteristics
A total of 24 subjects were randomly assigned to five study cohorts; of these, 23 subjects (95.8%) received the study drug (AOD01 or placebo). One subject randomized to a 5 mg/kg single-dose cohort withdrew his consent and was replaced by another subject. All 23 subjects completed the study (see Table S1 in the electronic supplementary material for details). The demographic characteristics were similar across the study cohorts. Nineteen subjects (82.6%) in the study were male. Twenty-two subjects (95.7%) were not Hispanic or Latino by ethnicity. All subjects were Asian, mostly Chinese (16 subjects [69.6%]) or Malay (5 subjects [21.7%]). The mean age was 32.9 years, the mean weight was 69.7 kg, and the mean BMI was 24.0 kg/m2. All subjects had negative viral serology, no findings on chest X-ray, and negative RT-PCR results on screening. For all study cohorts, the median infusion time was 60 min (range 60–67 min). The infusion was not interrupted in any subject (Table 1).
Safety
A total of 34 TEAEs were reported in 17 subjects (73.9%) in the study. The most common TEAE was catheter site rash which was reported in four subjects (17.4%). Other commonly reported TEAEs included diarrhea, rhinorrhea, and headache reported in three subjects (13.0%) each. Vessel puncture site bruises, musculoskeletal chest pain, and rash were reported in two subjects (8.7%) each.
All TEAEs were mild in severity. Two subjects (8.7%) reported two TEAEs that were related to study drugs: headache in one subject (33.3%) in AOD01 20 mg/kg (single-dose cohort) and diarrhea in one subject (33.3%) in AOD01 20 mg/kg (two-dose cohort). Both TEAEs were mild in severity and resolved in a day. Headache was treated with paracetamol and no treatment was given for diarrhea. None of the subjects reported TEAEs leading to study treatment discontinuation, acute or delayed infusion reactions, serious adverse events (SAEs), or AEs leading to death (Table 2).
Eight subjects reported COVID-19 signs and symptoms during the study. Of these eight subjects, the coronavirus PCR test was performed in only one subject in the placebo group with a headache on day 57 and the test result was negative. All other subjects were judged as not requiring COVID-19 testing by the investigator (see Table S2 in the electronic supplementary material for details). For the laboratory parameters, vital signs, and ECG parameters, the mean changes from baseline were minimal and not clinically significant. Physical examination findings were also normal in most of the subjects.
Pharmacokinetics
The mean serum concentration–time profiles for all single-dose cohorts followed a similar trend. Serum concentrations peaked rapidly with median Tmax values observed at 1.9, 1.5, 1.5, and 2.9 h relative to the start of the infusion for 2, 5, 10, and 20 mg/kg dose levels, respectively. Thereafter, concentrations declined gradually reaching below the limit of quantification (BLQ) by 43 days for the 2 and 5 mg/kg dose levels and to concentrations of about 2% (and below) of the respective peak concentrations observed for the 10 and 20 mg/kg dose levels by day 92 (end of study, EOS) (Fig. 1a).

Mean serum AOD01 concentration versus time profiles by dose. a Single-dose cohort. b Two-dose cohort
The mean serum concentration–time profiles for the two-dose cohort were similar on both day 1 and day 2 of dosing. Serum concentrations peaked at a median Tmax value of about 2 h on both days. After the median Tmax value, the concentration dropped gradually across the sampling period and finally reached a concentration that was about 0.5% of the peak by day 92 (EOS) (Fig. 1b).
The geometric mean peak (Cmax) and total (AUC0–tlast) exposures increased with increasing the dose from 2 to 20 mg/kg. The mean t1/2 values for the 2, 5, 10, and 20 mg/kg dose levels were 163, 126, 199, and 465 h, respectively. The mean clearance was dose independent (approximately the same and between 0.035 and 0.04 mL/h/kg across all dose levels tested). The volume of distribution at 20 mg/kg was higher (23 mL/kg) when compared to lower dose levels that were between 7 and 11 mL/kg. Over the dose range of 2–10 mg/kg, AUC0–inf was contained within the criteria for dose proportionality, but the other parameters, Cmax and AUC0–tlast were not (Fig. 2; Table 3 and see Table S3 in the electronic supplementary material for details).

Individual and mean AOD01 pharmacokinetic parameters versus dose
Immunogenicity
Antidrug antibodies to AOD01 were not detected among subjects, except in one subject (33.3%) in a two-dose cohort on day 92. There was no impact of the ADAs on AOD01 PK or safety. No AEs that could be attributed to ADAs were reported in this subject.
Biomarker Analysis
The flow cytometry and RNA sequencing results showed that single-dose or two-dose infusion of AOD01 did not lead to immune activation or cytokine release. The derived immunoscores, from the mean of healthy donor reference cytokine concentration, were zero at most time points. A transient rise was seen in a few cytokines, in the placebo group and AOD01 20 mg/kg (single-dose and two-dose) cohorts, which recovered within a day. No AEs were reported that could be attributed to the minimal rise in cytokine levels.
The scatter plots of normalized RNA Tempus count by time point showed that in both single-dose and two-dose AOD01 groups and the placebo group, the log2-CPM of normalized RNA Tempus counts at all post-infusion time points clustered around the diagonal line. This indicates that there was minimal difference in the gene expressions at pre-infusion and post-infusion time points (Fig. 3).

Scatter plot of normalized RNA Tempus counts by time point
Ex Vivo Bioassay
In single-dose cohorts, ex vivo bioassay for neutralization of SARS-CoV-2 was recorded as positive from day 2 until day 8 in all 12 subjects who received AOD01. The neutralization capacity was sustained until day 15 in all three subjects (100.0%) of the AOD01 20 mg/kg cohort and one subject (33.3%) of the AOD01 2 mg/kg cohort and until day 29 in two subjects (66.7%) of the AOD01 10 mg/kg cohort. By day 43, neutralization was negative for all subjects receiving the single dose (Table 4).
In subjects who received two doses of AOD01 20 mg/kg, neutralization was recorded as positive from day 3 until day 15 in all three subjects (100.0%). The neutralization capacity was sustained until day 43 in one subject (33.3%), and by day 92 neutralization was negative for all subjects (Table 5).
Discussion
In this randomized, placebo-controlled, FIH study, doses of AOD01 2, 5, 10, and 20 mg/kg and two doses of AOD01 20 mg/kg were found to be safe and well tolerated. The second dose was administered to subjects on day 2 to demonstrate the preliminary safety data necessary to justify the administration of a second dose to subjects with COVID-19 in the event where treating physicians needed an option to administer a second dose. Inclusion of the healthy subjects without any comorbid disease allowed for an unbiased assessment of the safety and tolerability of AOD01.
The safety profile in subjects who received AOD01 was similar to that in placebo-treated subjects. All the reported AEs were mild in severity and resolved with or without medication. There were no AEs leading to treatment discontinuation or treatment interruption, SAEs, and deaths, thereby assuring the safety of AOD01.
Though eight subjects reported COVID-19 signs and symptoms during the study, the PCR test was performed in only one subject. The other subjects had individual symptoms which were actively elicited from a COVID-19 questionnaire, but had no overall flu-like presentation, which the investigator evaluated as very unlikely to be related to a viral infection and hence the COVID-19 testing was not performed in these subjects.
The study showed that AOD01 was non-immunogenic as no antidrug antibodies were detected except in one subject which did not result in any adverse event and change in PK profile. This safety profile of AOD01 was in contrast to other monoclonal antibody products which have reported immune-related AEs like pruritis and rash. Immediate hypersensitivity reactions were reported in nearly 2% of subjects in a study of neutralizing antibodies LY-CoV555 and LY-CoV016 [10, 16]. Allergies, pruritus, and rash were also reported in the MW33 neutralizing antibody trial [17].
AOD01 demonstrated sustained ex vivo neutralization of SARS-CoV-2 until at least day 29 with single doses from 2 to 20 mg/kg and until day 43 with two doses of 20 mg/kg suggesting its potential in COVID-19 treatment. Preliminary evidence suggests that AOD01 retains activity against the Delta variant which was the predominant strain during the second wave of the pandemic (internal data).
The study also demonstrated favorable PK characteristics. Pharmacokinetic analysis showed that AOD01 geometric mean Cmax and AUC0–tlast values increased with increasing doses from 2 to 20 mg/kg. The mean and individual concentration–time profiles for AOD01 increased with increasing doses and were consistent with linear PK for single IV doses. Over the dose range of 2–20 mg/kg, dose proportionality could not be concluded; however, this may be due to the small sample size (n = 3 per dose level) in this study. In the 2 mg/kg and 10 mg/kg cohorts prolonged Tmax was observed. This was due to the long half-life of the drug and the lack of target-mediated drug clearance in healthy volunteers, the minimal differences that were observed in Tmax in some subjects seem to be less clinically significant.
The mean t1/2 values of 2, 5, 10, and 20 mg/kg doses of AOD01 were 163 h (6.7 days), 126 h (5.2 days), 199 h (8.2 days), and 465 h (19.3 days), respectively. The mean t1/2 of bamlanivimab after a single IV dose of 700 mg in patients with COVID-19 was 17.6 days [18]. Similarly, the half-lives of REGN10933 (casirivimab) were 24 and 25 days and 21 and 18 days for REGN10987 (imdevimab) after a single IV dose of 1.2 and 4 g of the study drug [19]. Thus, the half-life of AOD01 is similar to other monoclonal antibodies.
Though AOD01 has been identified from a patient infected with the wild-type virus and has been found to be efficacious in vitro against the Delta variant, its efficacy against Omicron and other newer variants is yet to be studied.
Conclusion
In this FIH study, single doses of AOD01 2, 5, 10, and 20 mg/kg and two doses of AOD01 20 mg/kg were safe and well tolerated and demonstrated dose-related PK, non-immunogenic status, and sustained ex vivo neutralization of SARS-CoV-2. Concentration–time profiles were consistent with linear PK for single IV doses. The data showed that AOD01 has good potential as a treatment for SARS-CoV-2 infection and has paved a path to elucidate the efficacy and safety of AOD01 in patients with COVID-19.
References
WHO. Weekly epidemiological update on COVID-19. 2022. https://www.who.int/publications/m/item/weekly-epidemiological-update-on-covid-19---25-january-2022. Accessed 25 Jan 2022.
Chan CEZ, Seah SGK, Chye DH, et al. The Fcmediated effector functions of a potent SARS-CoV-2 neutralizing antibody, SC31, isolated from an early convalescent COVID-19 patient, are essential for the optimal therapeutic efficacy of the antibody. PLoS ONE. 2021;16:e0253487.
Benani A, Ben MS. Mechanisms underlying potential therapeutic approaches for COVID-19. Front Immunol. 2020;11:1841.
Gaudinski MR, Coates EE, Novik L, et al. Safety, tolerability, pharmacokinetics, and immunogenicity of the therapeutic monoclonal antibody mAb114 targeting Ebola virus glycoprotein (VRC 608): an open-label phase 1 study. Lancet. 2019;393:889–98.
Lu R, Zhao X, Li J, et al. Genomic characterisation and epidemiology of 2019 novel coronavirus: implications for virus origins and receptor binding. Lancet. 2020;395:565–74.
Shi R, Shan C, Duan X, et al. A human neutralizing antibody targets the receptor-binding site of SARS-CoV-2. Nature. 2020;584:120–4.
Wang S, Peng Y, Wang R, et al. Characterization of neutralizing antibody with prophylactic and therapeutic efficacy against SARS-CoV-2 in rhesus monkeys. Nat Commun. 2020;11:5752.
Pinto D, Park YJ, Beltramello M, et al. Cross-neutralization of SARS-CoV-2 by a human monoclonal SARS-CoV antibody. Nature. 2020;583:290–5.
NIH. Anti-SARS-CoV-2 monoclonal antibodies. 2022. https://www.covid19treatmentguidelines.nih.gov/therapies/anti-sars-cov-2-antibody-products/anti-sars-cov-2-monoclonal-antibodies. Accessed 1 Feb 2022.
Chen P, Nirula A, Heller B, et al. SARS-CoV-2 neutralizing antibody LY-CoV555 in outpatients with Covid-19. N Engl J Med. 2021;384:229–37.
Lundgren JD, Grund B, Barkauskas CE, et al. A neutralizing monoclonal antibody for hospitalized patients with covid-19. N Eng J Med. 2021;384:905–14.
Weinreich DM, Sivapalasingam S, Norton T, et al. REGN-COV2, a neutralizing antibody cocktail, in outpatients with Covid-19. N Eng J Med. 2021;384:238–51.
Tuccori M, Ferraro S, Convertino I, et al. Anti-SARS-CoV-2 neutralizing monoclonal antibodies: clinical pipeline. MAbs. 2020;12:1854149.
Jones BE, Brown-Augsburger PL, Corbett KS, et al. The neutralizing antibody, LY-CoV555, protects against SARS-CoV-2 infection in nonhuman primates. Sci Transl Med. 2021;13(593):eabf1906.
Hansen J, Baum A, Pascal KE, et al. Studies in humanized mice and convalescent humans yield a SARS-CoV-2 antibody cocktail. Science. 2020;369(6506):1010–4.
Gottlieb RL, Nirula A, Chen P, et al. Effect of bamlanivimab as monotherapy or in combination with etesevimab on viral load in patients with mild to moderate COVID-19. JAMA. 2021;325(7):632–44.
Meng X, Wang P, Xiong Y, et al. Safety, tolerability, pharmacokinetic characteristics, and immunogenicity of MW33: a phase 1 clinical study of the SARS-CoV-2 RBD-targeting monoclonal antibody. Emerg Microbes Infect. 2021;10:1638–48.
Wang C, Horby PW, Hayden FG, et al. A novel coronavirus outbreak of global health concern. Lancet. 2020;395:470–3.
Regeneron COVID-19 program update. Regeneron Pharmaceuticals Inc. 2020. https://investor.regeneron.com/events/event-details/regeneron-covid-19-program-update. Accessed 29 Sept 2020.
Acknowledgements
Funding
This work was funded by the Ministry of Defence, Singapore. Role of funding source: The funder of the study had no role in study design, study execution, data collection, data analysis, or data interpretation. National Centre for Infectious Diseases will fund the journal’s Rapid Service Fees.
Medical Writing and Other Assistance
We thank PPD Singapore for their assistance in conducting this study. We thank Marius Jones and Xavier Camous for their assistance in data analysis. We thank all from the Immuno-oncology and Cell Therapy lab in IMCB for their assistance in sample processing. We thank Med Indite Communications Pvt. Ltd. Pune for their support in drafting the manuscript.
Authorship
All named authors meet the International Committee of Medical Journal Editors (ICMJE) criteria for authorship for this article, take responsibility for the integrity of the work as a whole, and have given their approval for this version to be published.
Author Contributions
Venkateshan S. Prativadibhayankaram, David Lye, Lawrence Soon-U Lee, Najwa S. N. Talib, Nivashini Kaliaperumal, Kantharaj Ethirajulu, Brendon J. Hanson, John E. Connolly, Vishal Pendharkar, Damian O'Connell, and Conrad E. Z. Chan contributed to the study design. Wei Yee Ong and Zi Xin Wong contributed to the pharmacokinetics assay. Najwa S. N. Talib contributed to the immunogenicity assay. Veonice B. Au and Anshula Alok contributed to the biomarker assay. Venkateshan S. Prativadibhayankaram, David Lye, Lawrence Soon-U Lee, Kantharaj Ethirajulu, Vishal Pendharkar, Damian O'Connell, Conrad E. Z. Chan, and Hannes Hentze contributed to data analysis and interpretation. Lawrence Soon-U Lee, David Lye, and Xu Xiaoying led the clinical conduct as the investigators of the clinical site and contributed to clinical assessment and data collection. Shirley G. K. Seah and De Hoe Chye contributed to ex vivo neutralization assays. Jerome D. Boyd-Kirkup, Siyu Guan, Benjamin J. Ayers, and Piers J. Ingram contributed clinical trial material. Venkateshan S. Prativadibhayankaram, Ranjani Nellore, Brendon J. Hanson, Kantharaj Ethirajulu, Damian O'Connell, and Conrad E. Z. Chan contributed to study management and execution. Venkateshan S. Prativadibhayankaram and Conrad E. Z. Chan were involved in primary manuscript writing and all authors commented on previous versions of the manuscript. All authors read and approved the final manuscript.
Disclosures
All named authors confirm that they have no competing interests to declare.
Compliance with Ethics Guidelines
The study was approved by the institutional review board of Tan Tock Seng Hospital, Singapore. The study was conducted in accordance with the Declaration of Helsinki, in compliance with the Council for International Organizations of Medical Sciences International Ethical Guidelines, applicable ICH Good Clinical Practice (GCP) Guidelines, and applicable laws and regulations. Written informed consents were obtained from all participants prior to enrolment in the study.
Data Availability
The datasets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Prativadibhayankaram, V.S., Lee, L.SU., Lye, D. et al. First-in-Human Study to Evaluate Safety, Tolerability, Pharmacokinetics, and Pharmacodynamics of a Rapidly Developed SARS-CoV-2 Therapeutic Antibody, AOD01, in Healthy Adults. Infect Dis Ther 11, 1999–2015 (2022). https://doi.org/10.1007/s40121-022-00681-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40121-022-00681-1