
Abstract
Introduction
Adintrevimab is a fully human immunoglobulin G1 extended half-life monoclonal antibody that was developed to have broad neutralization against SARS-CoV, SARS-CoV-2, and other SARS-like CoVs with pandemic potential. Here we report the safety, pharmacokinetics (PK), serum viral neutralizing antibody (sVNA) titers, and immunogenicity results of the first three cohorts evaluated in the first-in-human study of adintrevimab in healthy adults.
Methods
This is a phase 1, randomized, placebo-controlled, single ascending-dose study of adintrevimab administered intramuscularly (IM) or intravenously (IV) to healthy adults aged ≥ 18–55 years with no current or prior SARS-CoV-2 infection. Participants were randomized 8:2 to adintrevimab or placebo in each of three dose cohorts: adintrevimab 300 mg IM (cohort 1), 500 mg IV (cohort 2), and 600 mg IM (cohort 3). Follow-up was 12 months. Blood samples were taken predose and at multiple time points postdose up to month 12 to assess sVNA, PK, and antidrug antibodies (ADAs).
Results
Thirty participants received a single dose of adintrevimab (n = 24; 8 per cohort) or placebo (n = 6). All except one adintrevimab participant in cohort 1 completed the study. No participants in any treatment arm experienced a study drug-related adverse event. Across adintrevimab-treated participants, 11 (45.8%) experienced at least one TEAE. All but one TEAE were mild in severity, and all were either viral infection or respiratory symptoms. There were no serious adverse events, discontinuations due to adverse events, or deaths. Adintrevimab exhibited a linear and dose-proportional PK profile and extended serum half-life (mean 96, 89, and 100 days in cohorts 1, 2, and 3, respectively). Participants receiving adintrevimab demonstrated dose-dependent increased sVNA titers and breadth across multiple variants.
Conclusion
Adintrevimab at doses of 300 mg IM, 500 mg IV, and 600 mg IM was well tolerated in healthy adults. Adintrevimab demonstrated dose-proportional exposure, rapid development of neutralizing antibody titers, and an extended half-life.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Why carry out this study? |
This Phase 1, randomized, double-blind, single ascending-dose study was carried out to evaluate safety, tolerability, pharmacokinetics (PK), and immunogenicity of a single intramuscular (IM) or intravenous (IV) dose of adintrevimab in healthy participants. |
What was learned from the study? |
Adintrevimab was safe and well tolerated in healthy adults. PK was expected, proportional to dose, and correlated to sVNA titers. Immunogenicity was infrequent and ADA titers low. |
Introduction
Since the end of November 2022, there have been over 632 million confirmed cases of COVID-19 and over 6.5 million COVID-19–related deaths globally [1]. While vaccines are highly effective in preventing severe COVID-19, waning immunity and the emergence of new, highly transmissible variants of concern (VOCs) represent ongoing global public health issues [1,2,3]. There is a continued need for therapies to prevent or treat COVID-19 that are broadly neutralizing and have ease of use in community settings.
Adintrevimab, formerly known as ADG20, is a fully human immunoglobulin (Ig) G1 monoclonal antibody (mAb) derived from a survivor of the SARS 2003 epidemic and engineered to have high potency and broad neutralizing activity against SARS-CoV-2 and other SARS-like CoVs with pandemic potential. Adintrevimab prevents viral entry into host cells by binding to a highly conserved epitope on the receptor-binding domain of the SARS-CoV-2 spike protein [4,5,6]. In addition, the crystallizable fragment region of the heavy chain contains an LA modification (M428L/N434A) to provide an extended half-life [7]. Using a population pharmacokinetic (PK) model, a median half-life of 141–152 days was estimated following 300 mg intramuscular (IM) administration for prevention and treatment, respectively [8]. In vitro, adintrevimab demonstrated potent neutralization activity against clade 1 SARS-like CoVs and most SARS-CoV-2 VOCs tested, including B.1.1.7/Alpha, B.1.351/Beta, P.1/Gamma, B.1.617.1/Delta, and B.1.617.2/Delta [4, 5, 9]. Adintrevimab has shown reduced in vitro neutralization against Omicron BA.1/BA1.1 sublineages and no neutralizing activity against other Omicron sublineages [9]. In both Syrian golden hamster and rhesus macaque in vivo models, prophylactic administration of a single dose of adintrevimab provided protection from SARS-CoV-2 infection in a dose-dependent manner [10].
Here we report results from the first three dose cohorts of a first-in-human, phase 1 study of adintrevimab in healthy adults. These cohorts evaluated the safety, tolerability, PK, serum-neutralizing titers, and immunogenicity of adintrevimab at doses up to 600 mg IM and 500 mg IV. Higher doses of adintrevimab have been evaluated separately to establish safety margins given that emerging variants may have varying susceptibilities to adintrevimab.
Methods
Study Ethics
The study was performed in accordance with the Declaration of Helsinki with principles consistent with International Council for Harmonisation Good Clinical Practice and applicable regulatory requirements. Institutional Review Board (IRB) approval was obtained for the investigation from Advarra IRB Services. All participants provided IRB-approved written informed consent prior to receiving study drugs.
Study Design and Participants
This was a randomized, double-blind, placebo-controlled, single ascending dose, phase 1 study conducted at a single center in the USA. The doses of adintrevimab selected for this study were based on a quantitative systems pharmacology (QSP) whole-body physiologically based pharmacokinetic (PBPK) model for extended half-life monoclonal antibodies following IV and IM administration [11]. The model was developed based on published literature describing monoclonal antibody distribution kinetics in humans, including binding affinity to Fc receptors and IM bioavailability [12].
Eligible participants were male or female aged ≥ 18 to 55 years with body mass index ≥ 18.5 and < 30.0 kg/m2 and in good health with no clinically significant abnormalities based on medical history, physical examination, vital signs, electrocardiogram, and laboratory values. Participants were required to have a laboratory-confirmed negative SARS-CoV-2 test (as measured by reverse transcription polymerase chain reaction [RT-PCR] and negative SARS-CoV-2 serology [Confirm Biosciences Rapid IgG and IgM Lateral flow immunoassay kit] from a sample taken during the screening period, with an additional negative [repeat] SARS-CoV-2 RT-PCR test on the day prior to randomization [day-1]). Female participants of childbearing potential had to be using an approved form of contraception through 6 months after receipt of study drug and have negative results on a pregnancy test at screening and on day 1, and male participants with partners of child-bearing potential had to use a medically acceptable method of contraception through 6 months after receipt of study drug.
Participants were not eligible if they had a chronic medical condition; known or suspected allergy, intolerance, or hypersensitivity to any component of the study drug; receipt of investigational or licensed vaccine indicated for the prevention of SARS CoV-2 or COVID-19 or expected receipt within 180 days (~ 6 months) of dosing; confirmed or potential SARS-CoV-2 or COVID-19 infection; or symptoms of acute respiratory illness or other febrile illness within 2 weeks before dosing. See supplementary appendix for a full list of exclusion criteria.
Screening assessments were performed up to 28 days before dosing. Study design is shown in Fig. 1. Participants were admitted to the clinical unit on the day before randomization (day-1). The first two participants (sentinel participants) in each dosing cohort were randomized 1:1 to adintrevimab or placebo. The investigator was to review available safety data through 72 h (IM cohorts) or through 24 h (IV cohort) post dose for the sentinel participants. If the investigator had no safety concerns, the remainder of the participants in the cohort were to be dosed. Dose escalation to cohorts 2 and 3 was determined by a safety review committee based on 14-day safety and tolerability data from all participants in cohort 1. Enrollment was planned for 30 participants (10 per cohort). Participants returned to the clinic for assessments on days 7, 14, and 21 and months 3, 6, and 12 (end of study) and were monitored by weekly phone calls from day 21 onward. All participants who completed the study were followed for 12 months. Cohorts 1–3 participated in the study from February 2021 through March 2022.

Study design
Study Endpoints
The primary objective was to evaluate the safety and tolerability of a single dose of adintrevimab, including the incidence of treatment-emergent adverse events (TEAEs) and serious adverse events (SAEs), injection site reactions (ISRs), hypersensitivity reactions, and changes from baseline in clinical laboratory test results and vital signs. Secondary objectives included evaluation of PK (area under the concentration–time curve from time zero extrapolated to infinity [AUC0-inf], maximum concentration [Cmax], time to maximum concentration [Tmax], clearance [CL], half-life [t1/2], apparent volume of distribution during terminal phase [Vz], and volume of distribution at steady state [Vss]) and immunogenicity (development of anti-drug antibodies [ADAs] to adintrevimab) of adintrevimab. Exploratory objectives were to evaluate ex vivo serum-neutralizing activity against SARS-CoV-2 and the potential for antibody-dependent enhancement of COVID-19 infection and/or disease.
Assessments
AEs were reported in accordance with standard medical terminology when possible (verbatim AE terms were coded to standardized system organ class, lower-level terms, and preferred terms using Medical Dictionary for Regulatory Activities). AEs were classified as mild, moderate, or severe and were determined by the investigator as related or unrelated to study drug. ISRs in cohorts 1 and 3 were assessed using US Food and Drug Administration Guidance for Industry: Toxicity Grading Scale for Healthy Adult and Adolescent Volunteers Enrolled in Preventive Vaccine Clinical Trials (2007) [13]. Serious AEs were assessed through 12 months postdosing and solicited AEs through day 4 (defined as ISRs associated with IM administration). Hypersensitivity reactions were recorded as AEs of special interest (AESIs) through 72 h after dosing. Antibody-dependent enhancement was monitored in participants who tested positive for COVID-19 and might have had symptoms of greater severity than what might be expected such as development of complications, need for hospitalization or mechanical ventilation, or death.
Serum adintrevimab concentrations were determined using a validated hybrid ligand-binding liquid chromatography-high resolution mass spectrometry (LC/HRMS/MS) assay. For IM dose groups, blood samples for measuring PK were drawn predose, 8 h post dose on Day 1, 24 h post dose (Day 2), and on days 7, 14, and 21 and months 3, 6, and 12. For IV dose group, blood samples for measuring PK were drawn predose, at end of the infusion, 8 h after the start of infusion on Day 1, 24 h after start of infusion (Day 2), days 7, 14, and 21, and months 3, 6, and 12. Serum samples were evaluated for anti-adintrevimab antibodies predose on day 1 and postdose on days 14 and 21 and months 3, 6, and 12 (PPD BioA,Richmond VA).
Ex vivo neutralizing activity of adintrevimab against SARS-CoV-2 over time was based on plasma samples drawn predose (baseline) on day 1, then postdose on days 1, 2, 7, 14, and 21 and months 3, 6, and 12. Serum virus neutralizing antibody (sVNA) titers were determined using a plaque reduction assay against authentic SARS-CoV-2 D614G (BavPat1/2020; cohorts 1–3) and Beta/B.1.351, Gamma, Delta/B.1.617.2, and Omicron BA.1 variants (cohort 1 only) (Viroclinics, Rotterdam, The Netherlands). Neutralizing antibody titers were reported using a measure of maximum serum dilution that reduced the plaque number by 50% (MN50) and 80% (MN80) compared with virus-only control (i.e., no serum or plasma present). Neutralizing titers were analyzed using methodology detailed by Zielinska et al. [14].
Blood was collected prior to dosing and post-dose at Months 3, 6, and 12 to monitor for asymptomatic SARS-CoV-2 infection based on the detection of antibodies to the SARS-CoV-2 “N” antigen (Eurimmun anti-SARS-CoV-2 NCP ELISA (IgG)). Participants who developed COVID-19-like illness (CLI)-qualifying symptoms with laboratory confirmation of SARS-CoV-2 were monitored daily until resolution of symptoms.
Statistical Analysis
The sample size was determined based on having a reasonable number of participants to provide meaningful statistical estimates (no formal power calculation for hypothesis testing). The safety population comprised all participants who received any amount of study drug; the PK population included all participants in the safety population who had adintrevimab concentration data for at least two postdose time points. The immunogenicity population included all participants in the safety population who had a valid immunogenicity test result predose and at least one postdose time point.
Participant baseline characteristics, safety, and immunogenicity data were summarized using descriptive statistics. Placebo participants were pooled across dose groups for safety analyses.
Serum PK parameters were estimated for adintrevimab using non-compartmental analysis methods (WinNonLin software, Phoenix 8.2). Descriptive statistics of AUC0-inf, Cmax, Tmax, CL(/F), t1/2, Vz(/F), and Vss (/F) were provided for each dose level.
Results
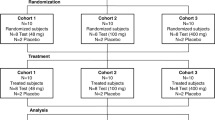
Thirty participants (10 in each of 3 cohorts) were randomized to adintrevimab (n = 24) or placebo (n = 6; Fig. 2). All participants received the full dose of study drug (or placebo) and completed the study with 12 months of follow-up, except one participant in the adintrevimab arm of cohort 1 who withdrew from the study 190 days after dosing.

Participant disposition. IM intramuscular, IV intravenous, PK pharmacokinetic
Baseline characteristics were well balanced among cohorts and participants who received adintrevimab or placebo in each cohort (Table 1). All participants were asymptomatic and had negative COVID-19 IgG and IgM serology tests and RT-PCR tests at time of screening.
Safety and Tolerability
The incidence of TEAEs among participants who received adintrevimab was 25.0% (2/8), 75.0% (6/8), and 37.5% (3/8) in cohorts 1, 2, and 3, respectively, and 16.7% (1/6) in the pooled placebo group (Table 2). No deaths, SAEs, hypersensitivity or infusion reaction, or solicited ISRs were reported. All TEAEs were mild or moderate in severity and considered unrelated to study drug. The most frequently reported TEAS among the adintrevimab-treated participants were influenza-like illness (n = 4), COVID-19 (n = 3), and nasopharyngitis (n = 2). All remaining TEAEs in the adintrevimab group were mild respiratory symptoms or viral rash reported in one participant each. Placebo groups reported one TEAE, which was chest pain.
Participants testing positive for COVID-19 had mild or moderate disease. As the cases were primarily mild in nature and did not worsen, no cases were considered attributable to antibody-dependent enhancement. All cases of COVID-19 occurred approximately 9 months post dose and after Omicron became the dominant variant, to which adintrevimab had reduced susceptibility.
Anti-Drug Antibodies
All participants were negative at baseline, and all participants in the placebo arm remained ADA-negative through Month 12. Treatment-emergent ADAs were relatively infrequent, occurring in four (16.7%) participants treated with adintrevimab, including three in the 300 mg IM group and one in the 600 mg IM group, and were observed at month 3 (n = 1 participant), month 6 (n = 1), and month 12 (n = 3). Overall detected ADA titers were low. Based on a minimum required dilution of 90, the lowest reportable titer was 90. Three participants with treatment-emergent ADA at Month 12 visit had a median titer of 90 (range 90–180). One participant had a titer of 720 at Month 3 but was ADA negative at later time points.
Pharmacokinetic Profile
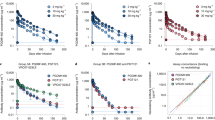
The observed PK profile for a single dose of adintrevimab 300 mg IM, 500 mg IV, or 600 mg IM was linear and dose proportional (Fig. 3). The mean AUC0-inf (SD) was 166,941.88 (38,392.98) hr*μg/ml, 343,843.48 (62,370.89) hr*μg/ml, and 366,186.19 (117,163.75) hr*μg/ml for the 300 mg IM, 500 mg IV, and 600 mg IM dose, respectively (Table 3). Adintrevimab demonstrated an extended half-life with both IM and IV modes of administration, with a mean (SD) t1/2 of 96 (14), 89 (9), and 100 (25) days in cohorts 1, 2, and 3, respectively. The median Tmax was 311.65 h (143.6–483.1) [13 days (range: 6–20)] after a single 300 mg IM dose.

Mean adintrevimab concentration over time by dose. Data are reported as mean ± standard error of the mean. SEM standard error of the mean
Serum Viral Neutralizing Antibody Titers
Adintrevimab serum concentration strongly correlated with sVNA titers (Fig. 4). In the adintrevimab IM treatment groups, MN80 titers against SARS-CoV-2 (BavPat1/2020/D614G variant) peaked at 7 to 14 days post dose, whereas titers peaked immediately post infusion in the IV treatment group. The MN80 titers against BavPat1/2020/D614G were sustained through 6 months and remained detectable across all adintrevimab treatment groups through Month 12 (Fig. 5A).

Adintrevimab serum concentration/sVNA relationship. Blue line = mean linear regression; blue shading = 95% CI; blue circles = individual time matched ADG20 serum concentration and MN80 sVNA titer measurements. Linear regression excluded samples that were taken following SARS-CoV-2 vaccination from participants who received vaccination during the trial

sVNA titers for adintrevimaba A MN80 for all cohorts, B MN80 for cohort 1 against Beta, Delta, Gamma, and Omicron, C MN50 Omicron BA.1. asVNA data from participants that were vaccinated or confirmed to be infected with SARS-CoV-2 (based on positive RT-PCR or “N” seroconversion) post-dosing were excluded from the geometric mean summary analyses at time points following the exposure (vaccination or infection)
MN80 and MN50 titers against other variants were analyzed using an exploratory research assay for samples from participants in cohort 1. Following 300 mg adintrevimab IM, MN80 titers were detected through 6 months post dose against multiple variants, including BavPat1/2020/D614G, Beta, Gamma, and Delta variants, with the highest titers against Delta. MN80 titers against Omicron were below the limit of the assay at most time points, as anticipated based on the reduced activity of adintrevimab against the Omicron variant. All participants treated with adintrevimab demonstrated detectable MN50 titers against Omicron at two or more early time points (between Days 1 and 21), ranging from 25 to 189, but only three had detectable titers at Month 3 (Fig. 5B, C).
Discussion
This study reports first-in-human data of the fully human IgG1 monoclonal antibody adintrevimab, studied for the treatment and prevention of COVID-19. In this dose-escalation study, a single dose of adintrevimab administered at 300 mg IM, 600 IM, or 500 mg IV was well tolerated by healthy adults with no serious AEs, discontinuations due to study treatment, or deaths. AEs that did occur were mild, except one case of moderate COVID-19. There were no ISRs or hypersensitivity reactions. Adintrevimab’s safety profile was consistent with other SARS-CoV-2 neutralizing mAbs authorized for use or in clinical development [4, 15,16,17,18].
Development of ADA was infrequent among participants, and time-dependent nonlinear elimination, which typically indicates ADA-mediated elimination for therapeutic monoclonal antibodies, was not observed in adintrevimab-treated participants in any cohort through Day 360. Generally, sVNA titers were not impacted by the low titer ADA.
The PK profile for adintrevimab was linear and dose proportional in this study. The PK profile of adintrevimab was linear and dose proportional, similar to other SARS-CoV-2 neutralizing mAbs in healthy and hospitalized adults [4, 16, 18, 19]. The PK profile was also consistent with an extended half-life monoclonal antibody. The typical range of serum half-life for non-half life extended COVID-19 IgG1 mAbs is approximately 16–30 days [4, 16, 18, 19]. The serum half-life of 89–100 days we observed in this study was comparable to the median 118 days estimated for healthy volunteers given 300 mg IM adintrevimab using a population PK model and was also similar to the 90 days observed with the extended–half-life mAb combination tixagevimab-cilgavimab following either IM or IV administration in healthy adults [8, 20]. Durability of sVNA titers was noted across susceptible variants; however, against Omicron variants, noticeably lower titers were observed.
Preliminary data from this phase 1 study were used to optimize the QSP whole-body PBPK model for dose selection in a phase 2/3 clinical trial of adintrevimab for both the treatment and prevention of COVID-19 in both the pre- and post-exposure settings [21], 22. Modeling data suggested that the single 300 mg IM dose of adintrevimab had a projected ability to rapidly exceed the IC90 target for susceptible variants in the majority of simulated participants to maintain effective concentrations for up to 12 months and to provide efficacy margins for coverage against non-Omicron SARS-CoV-2 variants. This innovative modeling and simulation approach was a key element in the rapid advancement of the adintrevimab clinical development program during the COVID-19 pandemic.
This trial was conducted in a small number of participants and had several limitations. The participants were healthy adults aged between 18 to 55 years; therefore, data presented may not reflect the safety, PK, serum viral neutralizing, or immunogenicity profiles of older populations or other individuals with increased risk of severe COVID-19 such as those with comorbidities or who are immunocompromised. Furthermore, the completion of cohorts 1, 2 and 3 overlapped with the emergence of the Omicron BA.1 and BA.1.1 variant in which adintrevimab had reduced susceptibility.
Conclusion
These results demonstrated a favorable safety and tolerability profile for adintrevimab and provided the initial PK, sVNA titer, and immunogenicity data that supported the continued development of a single 300 mg IM dose of adintrevimab in phase 2/3 trials: EVADE (ClinicalTrials.gov identifier: NCT04859517) for pre- and post-exposure prophylaxis and STAMP (ClinicalTrials.gov identifier: NCT04805671) for the treatment of ambulatory participants with mild to moderate COVID-19. Given that adintrevimab has reduced in vitro neutralizing activity against Omicron BA.1 and BA.1.1 and lacks activity against more recent Omicron sublineages, both STAMP and EVADE have been terminated.
Development of additional SARS-CoV-2 monoclonal antibodies can be supported by this work. Monoclonal antibodies targeting the receptor binding domain (RBD) of the spike protein have been shown to be safe and effective in volunteers and in participants diagnosed with or at high risk of COVID-19. There is also evidence that sVNA titers can be used as a correlate of protection against COVID-19 disease [7]. Adintrevimab demonstrated a strong correlation between pharmacokinetics and sVNA titers. Using this information coupled with IC50 data from newly arising variants of concern, dose selection and modification may be possible for RBD targeted monoclonal antibody programs. Therefore, having safety data at a range of doses in similar phase 1 studies will be key to rapidly advancing therapies to address the ever-evolving COVID-19 landscape.
References
World Health Organization. Coronavirus disease (COVID-19) Weekly Epidemiological Update and Weekly Operational Update 2022 [Available from: https://www.who.int/emergencies/diseases/novel-coronavirus-2019/situation-reports.
Andrews N, Stowe J, Kirsebom F, Toffa S, Rickeard T, Gallagher E, et al. Covid-19 vaccine effectiveness against the omicron (B.1.1.529) variant. N Engl J Med. 2022;386(16):1532–46.
Liu L, Iketani S, Guo Y, Chan JF, Wang M, Liu L, et al. Striking antibody evasion manifested by the Omicron variant of SARS-CoV-2. Nature. 2022;602(7898):676–81.
Dejnirattisai W, Zhou D, Supasa P, Liu C, Mentzer AJ, Ginn HM, et al. Antibody evasion by the P.1 strain of SARS-CoV-2. Cell. 2021;184(11):2939–54.
Liu C, Ginn HM, Dejnirattisai W, Supasa P, Wang B, Tuekprakhon A, et al. Reduced neutralization of SARS-CoV-2 B.1.617 by vaccine and convalescent serum. Cell. 2021;184(16):4220–36.
Rappazzo CG, Tse LV, Kaku CI, Wrapp D, Sakharkar M, Huang D, et al. Broad and potent activity against SARS-like viruses by an engineered human monoclonal antibody. Science. 2021;371(6531):823–9.
Schmidt P, Narayan K, Li Y, Kaku CI, Brown ME, Champney E, et al. Antibody-mediated protection against symptomatic COVID-19 can be achieved at low serum neutralizing titers. Sci Transl Med. 2023;15(688):eadg2783. https://doi.org/10.1126/scitranslmed.adg2783
Rubino C, Cammarata A, Ambrose P, Schmidt P, Popejoy M, Gong J, et al. Poster 591: Adintrevimab population pharmacokinetics in phase 1 and phase 2/3 COVID-19 prevention and treatment study participants. IDWeek 2022, October 19–23, 2022; October 19–23, 2022; Washington, DC2022.
Kaku CIN, Schmidt P, Engler F, Li Y, Walker LM. ADG20, a half-life–extended monoclonal antibody in development for the prevention and treatment of COVID-19, demonstrated broad in vitro neutralisation against SARS-CoV-2 variants. 32nd European Congress of Clinical Microbiology & Infectious Diseases (ECCMID); April 23–26, 2022; Lisbon, Portugal.
Narayan KK, Walker LM, Connolly L, Hershberger E, Yalcin I, Magyarics Z, Herbert AS, Zumbrun EE, O'Brien CM, Bakken RR, Zak SE, Dye JM. Prophylactic administration of the monoclonal antibody ADG20 provides potent protection against SARS-CoV-2 in rodent and non-human primate models of COVID-19. 31st European Congress of Clinical Microbiology & Infectious Diseases; July 9–12, 2021.
Paguntalan H, Magyarics Z, Connolly LE, Hershberger E, Narayan K, Gupta D, et al. 633. Preliminary results from a phase 1 single ascending-dose study assessing safety, serum viral neutralizing antibody titers (sVNA), and pharmacokinetic (PK) profile of ADG20: an extended half-life monoclonal antibody being developed for the treatment and prevention of coronavirus disease (COVID-19). Open Forum Infect Dis. 2021;8(Suppl 1):420.
Shah DK, Betts AM. Towards a platform PBPK model to characterize the plasma and tissue disposition of monoclonal antibodies in preclinical species and human. J Pharmacokinet Pharmacodyn. 2012;39(1):67–86.
Administration FaD. Toxicity Grading Scale for Healthy Adult and Adolescent Volunteers Enrolled in Preventive Vaccine Clinical Trials 2007 [Available from: https://www.fda.gov/media/73679/download.
Zielinska E, Liu D, Wu HY, Quiroz J, Rappaport R, Yang DP. Development of an improved microneutralization assay for respiratory syncytial virus by automated plaque counting using imaging analysis. Virol J. 2005;2:84.
Bian H, Zheng ZH, Wei D, Wen A, Zhang Z, Lian JQ, et al. Safety and efficacy of meplazumab in healthy volunteers and COVID-19 patients: a randomized phase 1 and an exploratory phase 2 trial. Signal Transduct Target Ther. 2021;6(1):194.
Chen P, Datta G, Grace Li Y, Chien J, Price K, Chigutsa E, et al. First-in-human study of Bamlanivimab in a randomized trial of hospitalized patients with COVID-19. Clin Pharmacol Ther. 2021;110(6):1467–77.
Lanini S, Milleri S, Andreano E, Nosari S, Paciello I, Piccini G, et al. Safety and serum distribution of anti-SARS-CoV-2 monoclonal antibody MAD0004J08 after intramuscular injection. Nat Commun. 2022;13(1):2263.
Wu X, Li N, Wang G, Liu W, Yu J, Cao G, et al. Tolerability, safety, pharmacokinetics, and immunogenicity of a novel SARS-CoV-2 neutralizing antibody, etesevimab, in Chinese healthy adults: a randomized, double-blind, placebo-controlled, first-in-human phase 1 study. Antimicrob Agents Chemother. 2021;65(8): e0035021.
Meng X, Wang P, Xiong Y, Wu Y, Lin X, Lu S, et al. Safety, tolerability, pharmacokinetic characteristics, and immunogenicity of MW33: a Phase 1 clinical study of the SARS-CoV-2 RBD-targeting monoclonal antibody. Emerg Microbes Infect. 2021;10(1):1638–48.
Loo YM, McTamney PM, Arends RH, Abram ME, Aksyuk AA, Diallo S, et al. The SARS-CoV-2 monoclonal antibody combination, AZD7442, is protective in nonhuman primates and has an extended half-life in humans. Sci Transl Med. 2022;14(635):8124.
Van Wart SA, Tarbell ED, Narayan K, Walker LM, Connolly LE, Ambrose PG. 1089. Use of a whole-body quantitative system pharmacology physiologically-based pharmacokinetic (QSP/PBPK) model to support dose selection of ADG20: an extended half-life monoclonal antibody being developed for the prevention of coronavirus disease (COVID-19). Open Forum Infect Dis. 2021;8(1):635–6.
Tarbell ED, Van Wart SA, Shah DK, Walker LM, Santulli A, Connolly LE, et al. 1088. A whole-body quantitative system pharmacology physiologically-based pharmacokinetic (QSP/PBPK) model to support dose selection of ADG20: an extended half-life monoclonal antibody being developed for the treatment of coronavirus disease (COVID-19). Open Forum Infect Dis. 2021;8(1):635.
Acknowledgements
The authors thank the study participants and their families, the clinical site investigators and personnel, and Debbie Caldwell, who provided consultancy work as the clinical operations study lead for Invivyd, Inc. (Waltham, MA, US). Altascience (formerly WCCT LLC), Cypress, CA, USA, conducted the study, and Veranex Solutions (formerly Quartesian LLC), Princeton, NJ, USA, was responsible for programming and data management.
Funding
This study was funded by Invivyd, Inc., Waltham, MA, USA. Invivyd, Inc., also funded the rapid service fee for publication.
Medical Writing Assistance
Writing assistance was provided by Russell Craddock, PhD, and Jean Turner of Parexel and was funded by Invivyd, Inc.
Author Contributions:
Pete Schmidt, Jean Gong, Kristin Narayan, and Ed Campanero made substantial contributions to concept and design, analysis of data, and writing, review and editing of manuscript. Deepali Gupta and Yong Li made substantial contributions to statistical analysis, analysis of data and writing, review and editing of manuscript. Amanda Copans made significant contributions to the analysis and interpretation of data and writing, editing and review of manuscript.
Disclosures
Pete Schmidt, Jean Gong, Kristin Narayan, Deepali Gupta, Yong Li, Amanda Copans, and Ed Campanaro are employees of and may own stock or stock options in Invivyd. Frank Engler has received consulting fees from Invivyd.
Compliance with Ethics Guidelines
The study was performed in accordance with the Declaration of Helsinki (1964) with principles consistent with International Council for Harmonisation Good Clinical Practice and applicable regulatory requirements. IRB approval was obtained for the investigation [Advarra IRB Services, number MOD00844324]. All participants provided IRB-approved written informed consent which included granting use of de-identified data for publication.
Data availability
The datasets generated during and/or analyzed during the current study are not publicly available due to resource limitations.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Schmidt, P., Gong, J., Narayan, K. et al. Safety, Pharmacokinetics, Serum Neutralizing Titers, and Immunogenicity of Adintrevimab, a Monoclonal Antibody Targeting SARS-CoV-2: A Randomized, Double-Blind, Placebo-Controlled, Phase 1 Dose-escalation Study in Healthy Adults. Infect Dis Ther 12, 1365–1377 (2023). https://doi.org/10.1007/s40121-023-00794-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40121-023-00794-1