
Abstract
Migraine involves brain hypersensitivity with episodic dysfunction triggered by behavioral or physiological stressors. During an acute migraine attack the trigeminal nerve is activated (peripheral sensitization). This leads to central sensitization with activation of the central pathways including the trigeminal nucleus caudalis, the trigemino-thalamic tract, and the thalamus. In episodic migraine the sensitization process ends with the individual act, but with chronic migraine central sensitization may continue interictally. Increased allostatic load, the consequence of chronic, repeated exposure to stressors, leads to central sensitization, lowering the threshold for future neuronal activation (hypervigilance). Ostensibly innocuous stressors are then sufficient to trigger an attack. Medications that reduce sensitization may help patients who are hypervigilant and help to balance allostatic load. Acute treatments and drugs for migraine prevention have traditionally been used to reduce attack duration and frequency. However, since many patients do not fully respond, an unmet treatment need remains. Calcitonin gene-related peptide (CGRP) is a vasoactive neuropeptide involved in nociception and in the sensitization of peripheral and central neurons of the trigeminovascular system, which is implicated in migraine pathophysiology. Elevated CGRP levels are associated with dysregulated signaling in the trigeminovascular system, leading to maladaptive responses to behavioral or physiological stressors. CGRP may, therefore, play a key role in the underlying pathophysiology of migraine. Increased understanding of the role of CGRP in migraine led to the development of small-molecule antagonists (gepants) and monoclonal antibodies (mAbs) that target either CGRP or the receptor (CGRP-R) to restore homeostasis, reducing the frequency, duration, and severity of attacks. In clinical trials, US Food and Drug Administration-approved anti-CGRP-R/CGRP mAbs were well tolerated and effective as preventive migraine treatments. Here, we explore the role of CGRP in migraine pathophysiology and the use of gepants or mAbs to suppress CGRP-R signaling via inhibition of the CGRP ligand or receptor.
Plain Language Summary
Migraine is a neurological disease affecting one in eight people. Symptoms include nausea and/or sensitivity to light and sound, and a throbbing headache. Although certain genes may increase the likelihood of migraine, environmental stimuli and molecules that increase the sensitivity of brain blood vessels and their innervations also play a role. During a migraine attack, nerves in the brain are activated, leading to increased sensitivity to stimuli, lowering the future threshold for activation, and making patients hypervigilant. Chronic, repeated exposure to certain stimuli can also lower this activation threshold, such that relatively innocuous stimuli can trigger an attack. Excessive use of certain migraine treatments can increase headache frequency over time and produce unwanted side effects; thus, selective agents are needed that specifically target the systems and pathways affected in migraine pathophysiology. Calcitonin gene-related peptide (CGRP), produced and released by nerve cells, is important in migraine pathophysiology. CGRP binds smooth muscle cell receptors, dilating blood vessels supplying the brain, and also binds to peripheral nerve cells that transmit pain signals to the spinal cord and brain. CGRP levels are elevated during a migraine attack. Medications targeting the CGRP pathway may decrease sensitivity and potentially normalize responses in hypervigilant patients. Two commercially available oral drugs that block CGRP receptors (‘gepants’) have reduced symptoms during migraine attacks in clinical trials. Four monoclonal antibodies (proteins that bind a specific molecule) have also been developed that target CGRP or the receptor and have been shown to significantly reduce the number of migraine days per month.
Role of CGRP in Migraine
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Migraine is a common neurological disorder affecting approximately 14% of the population. There is an unmet need for new therapeutics because many patients do not respond to treatment. |
Calcitonin gene-related peptide (CGRP) plays a key role in migraine pathogenesis, and recent therapeutic advances are based around targeting CGRP or its canonical receptor (CGRP-R). |
Individuals affected by migraine have a reduced threshold for central sensitization: this is known as hypervigilance. The combination of central sensitization and migraine triggers (allostatic load) results in migraine attacks. Repeated migraine attacks, which involve the release of CGRP to facilitate peripheral and central sensitization, probably contribute to increased allostatic load. |
In this review, the evidence supporting the role of CGRP in migraine is evaluated, including its role in the sensitization of peripheral and central neurons of the trigeminovascular system as well as its implication for hypervigilance and allostatic load. |
By inhibiting the release of pro-inflammatory molecules within the trigeminal ganglion and/or the dura and, therefore, reducing peripheral sensitization, anti-CGRP monoclonal antibodies (mAbs) might indirectly diminish central sensitization, reduce allostatic load, and contribute to migraine regression. |
Use of anti-CGRP and anti-CGRP-R mAbs for the treatment of migraine is promising. A summary of the latest mAb clinical trials and real-world data provides an overview of the therapeutic landscape, including erenumab-aooe, galcanezumab-gnlm, fremanezumab-vfrm, and eptinezumab-jjmr, which are approved by the US Food and Drug Administration (FDA) for the preventive treatment of migraine. |
An update is also provided on clinical trials involving small-molecule CGRP receptor antagonists known as gepants, including ubrogepant and rimegepant, which were recently approved by the FDA for the acute treatment of migraine with or without aura in adults, and two additional therapeutic candidates (atogepant and zavegepant [formerly vazegepant]), which remain in development. |
Interactive Infographic
This article contains an interactive infographic to help to illustrate the role of CGRP in migraine. To view it, click here: https://go.sn.pub/tsAHdE.
Digital Features
This article is published with digital features, including a plain language summary, key summary points, interactive infographic, and video to facilitate understanding of the article. To view digital features for this article go to https://doi.org/10.6084/m9.figshare.14061551.
Introduction
Migraine is among the most disabling disorders globally; in 2016, an estimated 1 billion people were affected [1]. The global age-standardized prevalence was approximately 14%, with approximately twice as many women than men being affected [1]. Estimates in the USA indicate an annual cost of approximately $36 billion [2,3,4]. Prevalence increases between the ages of 12 and 40 years and accounts for at least 1 day of school or work lost every 3 months among up to one-third of individuals [2, 4]. Social perceptions of migraine mean that patients may be dismissed as not having a serious disease. This perception is shared by some physicians [5], which exacerbates the significant negative impact migraine has on patients’ quality of life and ability to work [6]. Migraine features include moderate-to-severe throbbing headache, nausea and/or vomiting, and hypersensitivity to light and noise [7]. There is an important and complex genetic component to the disorder, but it remains unclear which genes and loci are implicated in its pathogenesis [8]. Migraine may be triggered by stress [9], exposure to bright light [9], pungent or strong odors [10], and barometric pressure changes [11]. Prodromal symptoms (e.g., fatigue, irritability, difficulty concentrating, mood change, yawning, stiff neck, phonophobia, and nausea) and postdromal symptoms (e.g., tiredness, weakness, cognitive difficulties, and mood change) are characteristic [12]. Migraine is classified as either episodic migraine (EM; < 15 days of headache per month) or chronic migraine (CM; for > 3 months, headache for ≥ 15 days per month with features of a migraine headache on ≥ 8 days per month) [13, 14].
Acute migraine management aims to reduce pain and symptoms and to restore physical and psychosocial function [6]. Possibly owing to a lack of adequate preventive treatments, acute therapies are frequently overused, which can lead to medication-overuse headache. Therefore, attempts are made to limit use of these treatments to 2–3 days per week [15]. Preventive migraine therapies are used for patients who have frequent and/or debilitating attacks, in whom acute medication fails to provide relief, or in whom there is a risk of overuse of acute medication [15]. The goals of preventive therapies are to reduce migraine frequency, duration, and/or severity of attacks, to improve responsiveness to acute therapy, to decrease use of acute therapy, and to increase the patient’s functional ability [16].
Traditional preventive therapies for migraine are inadequate, with up to 86% of patients discontinuing treatment early [17,18,19]. A retrospective US claims analysis shows that persistence with oral migraine preventive medications decreases over time, with only 14% of patients remaining on initial preventive treatment at 12 months [18]. Additionally, in a US-based survey, 39% of patients with migraine reported that they had used preventive medication at some time, with only 12% indicating that they were current users [20]. Thus, there remains a considerable unmet need for efficacious, well-tolerated preventive therapies with low potential for side effects.
The neuropeptide calcitonin gene-related peptide (CGRP) is a vasodilator with multiple sites of action that is involved in nociception and sensitization of peripheral and central neurons in the trigeminovascular system, which is implicated in migraine pathophysiology [7, 21,22,23,24]. Levels of CGRP in jugular venous blood and cerebrospinal fluid are elevated in patients with CM or EM [25,26,27], and these levels fall as migraine pain subsides upon triptan treatment, supporting a relationship between CGRP and migraine [28]. In rodents, application of CGRP to the cranial meninges triggered migraine-like signs of allodynia and pain in females but not in males: this hypersensitivity may contribute to the higher prevalence of migraine in females [29]. The involvement of CGRP in migraine has been substantiated by preclinical and clinical trials of drugs that block the activity of CGRP [30,31,32,33,34]. Additionally, CGRP infusion can trigger an attack in susceptible patients who experience migraine [35]. This review explores the role of CGRP in migraine, including its involvement in allostatic overload (i.e., when the beneficial acute stress response to external stimuli becomes maladaptive owing to chronic exposure to stressors) [36] and the use of monoclonal antibodies (mAbs) that suppress CGRP receptor (CGRP-R) signaling, potentially rebalancing allostatic load (Fig. 1).

Role of CGRP in migraine. Some individuals have a genetic predisposition to hypervigilance, thus having a heightened awareness of sensory stimuli, which may have provided an evolutionary advantage [151]. Association with too many risk factors leads to increased CGRP levels, which results in allostatic overload, making individuals more sensitive to sensory triggers that initiate a migraine attack [69]. mAbs targeting CGRP or the CGRP-R inhibit CGRP signaling, potentially rebalancing allostatic load and reducing the likelihood of a migraine attack. It remains to be seen if there is a direct effect of anti-CGRP therapeutics in helping to balance allostatic load. CGRP calcitonin gene-related peptide, CGRP-R CGRP receptor, mAb monoclonal antibody
This article is based on previously conducted studies and does not contain any studies with human participants or animals performed by any of the authors.
Migraine Pathophysiology
Migraine consists of distinct phases; in ~ 30% of patients it may be associated with aura (visual, sensory, motor, and language or brain stem disturbances) [37, 38]. Phases include the prodromal phase, probably mediated by the hypothalamus with multiple neurotransmitters believed to be involved (e.g., serotonin, orexin) and a potential aura phase, associated with a slow wave of neural depolarization called cortical spreading depression (CSD) that may activate trigeminal afferents (although the precise mechanism is unknown) [9, 12]. These are followed by the headache phase, which is mediated by activation of the trigeminovascular system, and the postdromal phase, which is associated with hyperperfusion in patients with aura and with bilateral posterior cortical hypoperfusion that persists after treatment in patients without aura [12]. The final (interictal) phase is characterized by reduced activation of the spinal trigeminal nuclei (Fig. 2) [9].

Migraine phases. Migraine consists of different phases characterized by physiological changes in the brain and trigeminovascular system. The headache phase is associated with activation of the trigeminovascular system and may last for ~ 4 to 72 h
CSD is associated with an increase in proinflammatory molecules and neuropeptides, including CGRP, and can activate the trigeminovascular system [39, 40]. The relationship between CSD and CGRP may be a contributory factor in migraine [39]. Drugs targeting CGRP-R signaling, including mAbs, reduce the activation of trigeminovascular neurons by CSD [41, 42].
Migraine is regarded as a neurovascular disease, driven by both neurovascular changes and modulation of nociceptive input [2]. Activation of the trigeminovascular system leads to an inflammatory cascade involving the meninges, meningeal blood vessels, and subsequent neural dysfunction (Fig. 3) [37]. The headache phase of migraine is thought to stem from activation of nociceptors that innervate the intracranial vasculature [21]. Nociceptive innervation of this vasculature consists of axons that originate in the trigeminal ganglion and release vasoactive neuropeptides such as CGRP and substance P [21]. Axonal projections of neurons in the trigeminovascular system transmit nociceptive signals via second-order neurons in the trigeminal nucleus caudalis and ascending fiber tracts to nuclei of the brain stem, hypothalamus, and basal ganglia and are thus potentially relevant to many symptoms of migraine (e.g., nausea, vomiting, fatigue, anxiety, and depression). Furthermore, relay trigeminovascular thalamic neurons projecting into various regions of the cortex could account for the location and intensity of migraine pain as well as transient symptoms such as clumsiness, loss of concentration and memory, photophobia, and phonophobia [40]. Perivascular CGRP in the periphery and central nervous system (CNS) may therefore act via the vessels innervated by the trigeminovascular system, the thalamus, and eventually the cortex to cause migraine symptoms [2].

Components of the trigeminovascular system. The trigeminovascular system consists of sensory nerves with their cell bodies in the trigeminal ganglion that innervate the meningeal blood vessels. Activation of the trigeminovascular system leads to increased release of CGRP and vasodilation of the meningeal vessels. Pain in the face and head is primarily mediated by nociceptive afferents of the ophthalmic division of the trigeminal nerve. Activation of the meningeal afferents leads to activation of the trigeminal nucleus caudalis and subsequently rostral brain structures involved in pain perception. CGRP calcitonin gene-related peptide
CGRP
CGRP is a member of the calcitonin peptide family, which includes calcitonin, amylin, adrenomedullin, and adrenomedullin 2/intermedin [43, 44]. There are two human isoforms of CGRP, α and β, which differ by three amino acids and share similar biological activities, but are synthesized from two different genes on chromosome 11 [45]. CGRP is widely expressed [22, 33] and is best known for its vasodilatory effects. Most relevant to its role in the pathophysiology of migraine is its expression in peripheral components (sensory neurons of trigeminal and cervical dorsal root ganglia) and in central components (e.g., parabrachial nucleus, amygdala, and hypothalamus) [3, 45]. CGRP is primarily expressed in small, unmyelinated sensory C fibers and myelinated Aδ fibers in the peripheral nervous system [22]. In contrast to amino acid neurotransmitters, which act on ionotropic receptors to alter postsynaptic membrane potentials quickly, neuropeptides are slower-acting neuromodulators that act through G protein-coupled receptors (GPCRs) [46]. CGRP acts as a modulator of postsynaptic gain when released from trigeminal nociceptors [46] and indirectly increases vascular permeability by promoting the release of substance P [47]. CGRP is stored in axonal dense core vesicles and can be released from the axon terminal or along the length of the axon, unlike neurotransmitters, which are stored in clear vesicles and released only at the axon terminals. Neuropeptides such as CGRP also differ from neurotransmitters in that they can function locally or more distally via diffusion in the vasculature [48].
CGRP Receptors
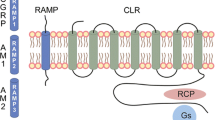
The calcitonin receptor family comprises two GPCRs: the calcitonin receptor and the calcitonin-like receptor (CLR). Additional receptor diversity is provided by heterodimerization of each of these GPCRs with one of three receptor activity-modifying proteins (RAMPs), which modify receptor binding characteristics. The calcitonin receptor preferentially responds to calcitonin but also associates with one of the three RAMPs to form amylin receptors (AMY1–3) [43]. CLR binds to its ligands (CGRP, adrenomedullin, or adrenomedullin 2) only when associated with RAMPs [43]. Thus, the functional CGRP-R complex consists of two subunits: CLR and RAMP1. An additional protein, known as receptor component protein, can also enhance signaling by this receptor complex [49]. Although the RAMP1-CLR complex is the CGRP-R, recent studies have established that AMY1 acts as a second CGRP-R and is present in the trigeminovascular system (Fig. 4) [43, 50, 51].

The calcitonin receptor family subunit composition. The histograms are indicative of relative ligand activity upon binding for human receptors. AM adrenomedullin, AM2 adrenomedullin 2, AMY amylin receptor, CGRP calcitonin gene-related peptide, CLR calcitonin-like receptor, CT calcitonin, CTR calcitonin receptor, RAMP receptor activity-modifying protein
CGRP-R is expressed on the trigeminal afferents that project into the meninges; cell bodies of trigeminal neurons, and satellite glial cells located in the ganglion; astrocytes and microglia within the CNS; and mast cells in the meninges [3, 24]. In cell-based assays, CGRP-R has high affinity for CGRP and lower affinity for other calcitonin family ligands [43]. This promiscuity in binding within the receptor family may have important consequences for therapies targeting CGRP-R signaling [43]. For example, AMY1 is equally well activated by both CGRP and amylin and may or may not be involved in migraine [26, 43]. Furthermore, in rat trigeminal neurons, CGRP has been shown to act at AMY1 in addition to CGRP-R, potentially implicating both receptors in CGRP ligand signal processing [50]. However, to date, a role for AMY1 in migraine has not been confirmed. The role of amylin in migraine is unclear, and an amylin and CGRP head-to-head provocation study is ongoing (NCT03598075) [52].
CGRP Receptor Signaling Pathway
GPCR signaling in the CGRP-R-mediated pathway is transduced via adenylate cyclase either to modulate vasodilation in cerebral and meningeal arteries or to transmit nociceptive signals centrally via the brain stem and midbrain to the thalamus and higher cortical regions [23]. Activation of CGRP-R may also trigger alternative signaling pathways dependent on the G proteins and other signaling proteins expressed [26, 53, 54]. Upon ligand binding to CGRP-R, the ligand-receptor complex is internalized into the cell [43, 55, 56]; however, the cell-type specificity and significance of CGRP-R internalization are not fully understood [43], and cell signaling may continue once the receptor is internalized [43, 57]. AMY1 signaling in response to CGRP and amylin is similar to that of the CGRP-R, but a recent study suggests that AMY1 regulation is distinct, because the receptor does not internalize in response to CGRP or amylin agonists [53, 55, 58, 59].
The action of a ligand can be subject to functional selectivity at a given receptor towards a specific signaling pathway (biased agonism) [60]. Receptor conformations are not binary; multiple subunit conformations can exist, and different ligands may stabilize different receptor conformations that interact with various cellular signaling pathways. Thus, different ligands acting at the same receptor (e.g., CGRP or adrenomedullin at the CGRP-R) could activate distinct downstream cellular responses, a phenomenon not related to their binding affinity or efficacy [43]. If other ligands prove to be involved in migraine pathophysiology, this may have implications for the importance of targeting the CGRP-R rather than the CGRP ligand.
CGRP in Migraine
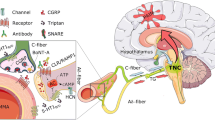
CGRP-Rs are ubiquitous in the smooth muscle of the vasculature (human middle meningeal, middle cerebral, pial, and superficial temporal arteries) and in the trigeminal afferent terminals (Fig. 5a). CGRP initiates and maintains peripheral and central sensitization by modulating neurons, glia, and immune cells in the trigeminal pain signaling pathway (Fig. 5b) [3]. During migraine, CGRP is released at various sites in the trigeminovascular system, including trigeminal afferent terminals that innervate the meninges, trigeminal cell bodies in the ganglion, and trigeminal efferent fibers that terminate in the medulla and cervical spinal cord (Fig. 5c) [3, 24]. At synapses outside the CNS, CGRP causes vasodilation via receptors on smooth muscle cells, whereas, at synapses inside the CNS, CGRP acts on second-order neurons to transmit pain signals to higher cortical pain regions [7, 61].

CGRP receptor and ligand expression sites in the trigeminovascular system. (a) Localization of the CGRP-R has been confirmed in the smooth muscle of the vasculature and in the trigeminal afferents. Some studies have suggested the CGRP-R may also be present in macrophages and fibroblasts. (b) CGRP released in the trigeminal ganglion acts on satellite glia triggering neurogenic inflammation. This initiates peripheral sensitization and, as a result, downstream central sensitization. (c) CGRP is released at various sites in the trigeminovascular system where it is involved in nociception and vasodilation. CGRP calcitonin gene-related peptide, CGRP-R CGRP receptor
In the dura, CGRP mediates neurogenic inflammation, immune cell recruitment (such as mast cells and macrophages), and activation of sensory neurons; it also activates neurons in the trigeminal ganglion and signals in a retrograde manner from the spinal cord to neurons and glia in the trigeminal ganglion, sensitizing peripheral nociceptors [24, 62,63,64]. Neuron activation activates second-order neurons and glia, which promote central sensitization in the dorsal horn and spinal trigeminal nucleus caudalis [2, 62].
In sensory neurons within the trigeminal ganglion, CGRP may increase its expression by a paracrine mechanism involving release of proinflammatory mediators from satellite cells [3, 22]. Although CGRP released in the trigeminal ganglion is unlikely to cross the blood-brain barrier (BBB), it may influence central expression of pro-nociceptive compounds and receptors [24]. Neurotrophic factors such as brain-derived neurotrophic factor are stimulated by CGRP, which may act to potentiate central nociceptive neurotransmission [24]. Neurons expressing CGRP project from the trigeminal ganglion to the trigeminal nucleus caudalis, where CGRP induces release of glutamate from trigeminal afferents, further facilitating nociception [24]. Together, these effects provide a mechanism by which drugs acting on CGRP-R signaling in the peripheral trigeminal ganglion may modulate both peripheral and central pain signaling through a peripheral site of action, highlighting the potential for the use of these drugs for migraine therapy. It is well established that when peripheral activation is switched off, central activation also decreases [65].
CGRP mediates neuron-glial signaling via gap junctions within the trigeminal ganglion [24, 61]. Regional nonsynaptic cross-excitation occurs within the trigeminal ganglion following neuronal and glial activation. This may allow for coordinated responses to noxious stimuli that may also, however, sensitize neighboring neurons [61]. Neuron-glial signaling probably contributes to peripheral sensitization, which drives central sensitization [40], facilitating a lower threshold for neuronal activation in patients with migraine. Peripheral and central sensitization in patients may therefore lead to hypersensitivity to sensory stimuli.
Hypervigilance, Anxiety-Like State, and Allostatic Overload
Anxiety-like states and a persistent state of hypervigilance are contributory risk factors for migraine [66]. Individuals affected by migraine have a hypervigilant or hyperexcitable nervous system that is characterized by a lower sensory activation threshold and hypersensitivity towards sensory stimuli [67]. The resulting sensitization and activation of trigeminal nerves and the nociceptive pathway are thought to play an important role in the pathology of migraine [67]. The increased sensitivity to sensory stimuli is caused by a state of sensory neuron hyperresponsiveness or hyperexcitability, which lowers pain threshold and tolerance levels [38, 67]. In essence, a state of hypervigilance is the same as a state of central sensitization, in which the sensory activation threshold is reduced.
The role of anxiety and hypervigilance in migraine may be potentiated by CGRP-R signaling. CGRP promotes sensitization of primary and secondary trigeminal nociceptive neurons, leading to an enhanced state of neuronal excitability [67]. CGRP has also been implicated in migraine-associated allodynia (a nociceptive response to normally non-painful stimuli, such as hair brushing) [67, 68]. Thus, it is possible that CGRP plays a role in hypervigilance. Cutaneous allodynia is considered to be a marker of central sensitization and can manifest both during and between attacks in CM. Brains of patients with CM remain hyperexcitable in the interictal phase [69].
Over 50% of people with migraine experience an anxiety disorder over their lifetime [66]. Infusion with CGRP has been demonstrated to cause anxiety-like behavior and to induce neuronal activation in the bed nucleus of the stria terminalis (BNST) [70]. Furthermore, it has been hypothesized that reciprocal connections between the BNST and CGRP-containing cells in the parabrachial nucleus may induce a persistent state of anxiety via positive feedback [71]. The BNST is continuous with the amygdala and integrates physiological and behavioral responses to stress and anxiety by acting as a relay between limbic structures and the hypothalamus [3, 72]. Neurons expressing CGRP in the parabrachial nucleus relay pain signals from the periphery to the amygdala, thus encoding the affective component of pain [73]. In rats, CGRP antagonism has been shown to suppress grooming and shaking behaviors induced by CSD, which could indicate that CGRP is implicated in anxiety and/or pain perception [74].
Migraine is a disorder of allostatic load [75], triggered by risk factors including stress, sleep deprivation, light, odors, and noise (Fig. 1). Allostasis is the body’s ability to respond to stressful events and it involves hormonal and neural mediator activation and behavior modification. When behavioral or physiological stressors become more severe or frequent, the prolonged exposure dysregulates allostasis, leading to allostatic overload [76]. Structural and functional brain networks are altered, and the responses to stressors become abnormal [69].
People with migraine have a high allostatic load, which is more prevalent in EM (50%) than CM (33%) [75]. Allostatic load is moderate or high in most patients, and higher than in people without migraine [75]. In migraine, four main processes are involved in allostatic overload: (1) repeated stress; (2) failure to habituate to repeated stressors; (3) inability to stop the stress response normally; and (4) altered stress response that ultimately leads to compensatory responses, such as photophobia [69]. Central sensitization and factors that trigger a migraine attack both contribute to an individual’s allostatic load and, as sensitization increases, the need for an attack trigger decreases. Over time, sensitization levels and exposure to triggers will vary within an individual and between individuals [40]. For migraine treatments to be effective, they need to inhibit or stop persistent central sensitization.
Migraine may provide an evolutionary mechanism that enforces a rebalancing of allostatic load in response to a hypersensitive/hypervigilant state, thus relieving hypothalamic and cortical overload [69]. According to this model, it may be that CNS-driven changes in descending inhibitory pain modulation pathways enable more peripheral sensory information to reach the brain via the trigeminal-thalamus ascending pain pathway, potentially improving survival prospects by increasing vigilance, but also increasing allostatic load. CGRP may play a key role in migraine by increasing allostatic load mediated by risk factors; therefore, rebalancing CGRP-mediated signaling may treat or prevent attacks (Fig. 1) [40, 77]. As a key player in migraine pathophysiology, CGRP-R signaling was recognized as a potential therapeutic target, leading to the development of small-molecule antagonists and mAbs that target either CGRP or CGRP-R [2, 23, 26]. By inhibiting the release of pro-inflammatory molecules within the trigeminal ganglion and/or the dura and, therefore, reducing peripheral sensitization, CGRP mAbs might indirectly diminish central sensitization, reducing allostatic load and contributing to migraine regression.
Development of CGRP Antagonists
Several therapeutics are used to treat migraine: acute treatment includes nonsteroidal anti-inflammatory drugs and oral serotonin (5-hydroxytryptamine receptor 1B/1D) agonists known as triptans [15], whereas drugs such as anticonvulsants, antidepressants, antihypertensives, and botulinum toxins are used for migraine prevention [78]. Many of these drugs, which were not designed specifically for migraine, can have multiple side effects [15] and have poorly understood mechanisms of action [79]. Furthermore, poor adherence and frequent treatment-switching limit the effective use of these medications [17,18,19,20].
Until recently, first-line treatment for migraine focused on the relief of acute pain with triptans [80]. Triptans may provide effective pain relief during acute migraine; however, a substantial proportion of patients remain unresponsive [26]. Triptans act to constrict cerebral blood vessels and to inhibit activation of peripheral nociceptors on trigeminal afferents to reduce CGRP release [80,81,82]. In clinical trials, it was shown that plasma CGRP levels were reduced in patients whose migraine pain subsided following treatment with sumatriptan, but not in those who did not experience pain relief [31]. CGRP levels also correlated positively with pain intensity [31].
Most migraine preventives inhibit fast synaptic transmission, whereas drugs targeting neuropeptide pathways such as CGRP modulate neuronal activity [46]. CGRP release may be initiated by a Ca2+-dependent process involving soluble N-ethylamide-sensitive factor attachment protein receptor (SNARE), which promotes vesicle docking and membrane fusion or, alternatively, by a Ca2+- and SNARE-independent process [83]. Although triptans may inhibit Ca2+ signaling and although botulinum toxin type A prevents vesicular fusion through cleavage of the SNARE protein SNAP-25, CGRP release at the proximal axon of sensory neurons independent of these mechanisms remains unaffected [83]. This may explain why some patients with migraine do not respond to these therapies and supports the use of agents that block CGRP-R signaling rather than CGRP release.
Consequently, the ‘gepants’ were developed; these are mostly oral, small-molecule CGRP-R antagonists [84], six of which have been studied clinically for the treatment of acute migraine: olcegepant, telcagepant, MK-3207, BI 44370 TA, rimegepant, and ubrogepant [85]. A seventh gepant, atogepant, is in development for EM prevention [85]. Gepants do not directly cause vasoconstriction [86, 87], lending further weight to the argument that migraine is not purely a vascular disorder [26]. In clinical trials, the first-generation gepant, telcagepant, demonstrated efficacy for the treatment of acute migraine compared with placebo and triptans [88, 89]. However, liver toxicity limited its use as a preventive treatment. Other first-generation gepants (olcegepant, MK-3207, and BI 44370 TA) also showed promising results in clinical trials [90,91,92], but were discontinued owing to liver toxicity and to difficulties in developing oral formulations [85]. Elevated aminotransferase levels were attributable to off-target effects by a toxic metabolite not produced by second-generation gepants [84, 93], unleashing the potential of these agents as preventive treatments for migraine. Ubrogepant has recently been approved in the USA for the acute treatment of migraine with or without aura in adults [94], based on positive results from Phase 3 studies, including a recent trial in which significantly more patients taking ubrogepant were symptom-free 2 h after taking the drug compared with patients taking placebo (21.2% vs. 11.8%, respectively, p < 0.001) [95]. The efficacy of rimegepant has been demonstrated in Phase 3 clinical trials in acute migraine, with patients experiencing freedom from pain at 2 h after dosing, sustained pain relief for up to 48 h, and a reduction in associated symptoms such as photophobia and nausea [84, 96]. A further Phase 3 trial reported results with a new formulation of rimegepant as an orally disintegrating tablet (ODT), which was superior to placebo when comparing proportions of patients free from pain and bothersome symptoms 2 h post dose (21% vs. 11%, respectively, p < 0.0001) [97]. Rimegepant ODT was recently approved by the US Food and Drug Administration (FDA) for the acute treatment of migraine with or without aura in adults [98].
Two gepants remain in development: atogepant and zavegepant (formerly known as vazegepant). Atogepant is the only gepant with positive Phase 3 trial data for migraine prevention, with statistically significant reductions in monthly headaches from baseline versus placebo (least squares mean differences: − 0.7 to − 1.4 days; p < 0.05) and no reported signal for hepatotoxicity [99, 100].
Positron emission tomography has shown central CGRP-R occupancy by gepants (telcagepant) is not required for their therapeutic effect in migraine, suggesting that large molecules such as mAbs, which cross the BBB very inefficiently, are a viable option [101]. Areas of the hypothalamus, pituitary, and circumventricular organs, such as the area postrema, are outside of the BBB, and so are accessible to mAbs. Despite their efficacy, early issues with tolerability of gepants drove the search for alternative agents with improved safety profiles, with a focus on mAbs [26].
Anti-CGRP and Anti-CGRP-R Monoclonal Antibodies
Therapeutic mAbs are highly selective for their target molecule, binding a single epitope with high affinity [26, 102] and not binding to other antibodies or targets. This largely overcomes the problem of off-target effects and toxicities associated with small-molecule drugs and their metabolites [26]. Therapeutic mAbs are used to treat several diseases, including cancer and autoimmune disorders [103]. They can trigger target cell destruction, as in some types of cancer treatment where the objective is to stimulate the patient’s immune system to attack the cancer cells [104]. Other types of mAb do not cause cell destruction, but instead bind to either a ligand or a receptor, modifying cell signaling pathways [104]; mAbs targeting CGRP-R signaling belong to this group. To reduce the likelihood of an immune reaction to therapeutic antibodies, which is characteristic of antibodies generated in mice, antibody engineering techniques have allowed the development of chimeric, humanized, or fully human mAbs [104].
mAbs targeting CGRP-R signaling are well suited for migraine prevention, not only because they target a pathway relevant to the disease, but also because they require infrequent administration relative to small-molecule drugs [26]. As only a small proportion of any circulating mAb will typically reach the CNS [105], targets within the CGRP-R signaling pathway are likely to be peripheral (vasculature, dura, or trigeminal ganglion) [106]. For CGRP-R signaling, one antibody reversibly targeting the CGRP-R, where it occupies the CGRP ligand-binding site (erenumab-aooe [26, 107]), and three targeting the CGRP ligand (fremanezumab-vfrm, galcanezumab-gnlm, and eptinezumab-jjmr) have been approved by the FDA for migraine prevention (Table 1, Fig. 6) [108]. Although many trials are ongoing, thus far the evidence for the clinical efficacy and tolerability profile of erenumab-aooe has been most comprehensively reported. Anti-CGRP-R signaling mAbs are efficacious in EM and CM in clinical trials of up to 6 months’ duration (Table 2) and are generally well tolerated. Erenumab-aooe has also been shown to be effective and well tolerated longer term in both EM and CM. In a 52-week open-label extension study in patients with CM who continued treatment after a 12-week double-blind period, treatment with erenumab-aooe (70 mg or 140 mg once monthly) was associated with changes in monthly migraine days (MMD; mean [95% confidence interval, CI]) from parent study baseline of − 9.3 [− 10.0 to − 8.6] at week 52; MMD were reduced to 8.8 from 18.1 at parent study baseline. At week 52, respective reductions in MMD of ≥ 50%, ≥ 75%, or 100% from parent-study baseline were observed in 59.0%, 33.2%, and 8.9% of patients still on study [109]. In an analysis of 5-year data from patients with EM who continued open-label treatment following a 12-week double-blind period, treatment with erenumab-aooe (70 mg or 140 mg once monthly) was associated, on average, with 5.3 fewer MMD at year 5, an average reduction of 62.3% from baseline. Moreover, reductions in MMD of ≥ 50%, ≥ 75%, or 100% at 5 years, based on the last 4-week period of assessment, were observed in 71.0%, 47.1%, and 35.5% of patients, respectively [110]. Notably, erenumab-aooe is efficacious in patients with EM in whom multiple preventive treatments have been unsuccessful (LIBERTY trial) [17], a patient population under investigation by others (FOCUS and CONQUER trials; patients with CM or EM). In the LIBERTY trial, which included patients with EM in whom two to four previous preventive treatments had been unsuccessful, 30% of patients treated with erenumab-aooe 140 mg had ≥ 50% reduction from baseline in mean number of MMD at week 12 compared with 14% of patients in the placebo group (odds ratio [OR] [95% CI]: 2.7 [1.4–5.2]; p = 0.002) [17]. Post hoc analyses of pivotal, double-blind, placebo-controlled trials of erenumab-aooe in patients with CM or EM are consistent with these findings. Thus, 41% of patients with CM in whom two or more prior preventive medications failed had ≥ 50% reduction from baseline in mean number of MMD at 12 weeks when treated with erenumab-aooe 140 mg compared with 14% of patients receiving placebo (OR [95% CI]: 4.2 [2.2–7.9]) [111]. Similarly, in patients with EM in whom two or more prior preventive medications failed, 36% of patients treated with erenumab-aooe 140 mg had ≥ 50% reduction from baseline in mean number of MMD (averaged over months 4 to 6) compared with 11% of patients in the placebo group (OR [95% CI]: 4.5 [1.7–12.4]) [112].

mAbs targeting CGRP or CGRP-R. CGRP-R signaling may be reduced by mAbs that target either CGRP or the CGRP-R. Although the CGRP-R is depicted as a CLR-RAMP1 complex, it is not known if all CGRP binding sites are formed by this complex. The CGRP-responsive receptor AMY1 has been found in the trigeminal ganglion and brainstem, although a role of AMY1 in migraine has not been confirmed [50]. AMY amylin receptor, CGRP calcitonin gene-related peptide, CGRP-R CGRP receptor, CLR calcitonin-like receptor, mAb monoclonal antibody, RAMP receptor activity-modifying protein
Results from a postmarketing study in 78 patients with EM and CM demonstrated erenumab-aooe (70 mg) to be highly effective and well tolerated. In the EM patient group, a reduction of 3.8 MMD was reported at 4 weeks compared with baseline; in the CM patient group, reductions of 12.2 and 15.0 MMD from baseline were reported at 4 and 8 weeks, respectively [113]. Furthermore, in a real-world cohort of 65 individuals with severe migraine, a reduction in MMD of ≥ 50% was commonly observed in 40–50% of individuals within different patient subgroups when treated with erenumab-aooe. The cohort included patients who would have been excluded from randomized controlled trials (i.e., those with more chronic, frequent, severe, and refractory migraine). For example, subgroups included patients with CM of duration > 10 years, patients in whom > 3 prophylactic medications had failed, and patients scoring > 60 on the six-item Headache Impact Test [114].
Initial results (6 months) from a 1-year open-label extension trial in 312 patients treated with fremanezumab have also been reported. Patients with EM taking monthly doses of fremanezumab experienced reductions in MMD from 8.9 at baseline to 5.3 at 12 weeks and 4.0 at 6 months; patients on quarterly doses reported decreases in MMD from 9.2 at baseline to 5.3 at 12 weeks and 4.2 at 6 months. Patients with CM taking monthly doses of fremanezumab reported a decrease in MMD from 16.2 at baseline to 11.4 at 12 weeks and 8.3 at 6 months; patients on quarterly doses experienced reductions in MMD from 16.4 at baseline to 11.9 at 12 weeks and 9.9 at 6 months [115].
Overall, mAbs have long elimination half-lives, approximately 27–31 days for the mAbs approved for the treatment of migraine [116,117,118]. Physiological antibody concentrations are maintained, at least in part, by the neonatal fragment crystallizable receptor, which protects mAbs and other immunoglobulin G molecules from lysosomal degradation, providing a salvage pathway that returns approximately two-thirds of immunoglobulin antibodies to the cell surface, the remainder being degraded [119].
Although the immunogenic potential of therapeutic antibodies is reduced by humanization, erenumab-aooe is the only fully human antibody [26]. Further differentiating erenumab-aooe from the ligand-targeted mAbs is the fact that those are CGRP-release dependent, whereas receptor-targeted strategies are release independent. It may be that, for migraine prevention, receptor-targeted strategies will provide a more sustained modulation of CGRP-R signaling than ‘waiting’ for ligand release to drive a feed-forward cycle.
CGRP expression is increased in RAMP1-deficient mice [120]. Conversely, CGRP-R expression is elevated in α-CGRP knockout mice [121]. Thus, upregulation of CGRP-R or AMY1 expression might be anticipated in response to chronic ligand depletion or receptor blockade in humans. If this occurs and receptor density increases, the observed efficacy of CGRP-R-targeted mAbs may increase over time because receptors remain saturated. The binding of antibodies to CGRP or to CGRP-R may result in the formation of distinct receptor conformations that signal through unique pathways (novel signaling bias), although this requires further investigation [122].
CGRP Receptor Signaling Monoclonal Antibody Safety
Given the widespread role of CGRP, it might be expected that CGRP-R signaling blockade could result in safety concerns; however, the clinical trial data for anti-CGRP and anti-CGRP-R mAbs showed only mild drug-related adverse events, including injection-site reactions, hypersensitivity reactions (such as erythema or pruritus), and constipation (Table 1) [116,117,118]. Analysis of 5-year data from patients with EM participating in an open-label extension after a 12-week double-blind phase showed no new safety signals and no increase in the incidence of adverse events or of serious adverse events with longer-term erenumab-aooe use compared with the double-blind treatment phase [110]. A pooled analysis of four double-blind randomized trials and their extension studies in 2375 patients who received at least one dose of erenumab-aooe (70 mg or 140 mg; cumulative exposure, 2641 patient-years) found that erenumab-aooe had a favorable and stable adverse event profile for over 3 years in patients with EM or CM. Exposure-adjusted adverse event rates with erenumab-aooe were similar to placebo during the double-blind phase, except injection site-reactions (17.1 vs. 10.8 per 100 patient-years), constipation (7.0 vs. 3.8 per 100 patient-years), and muscle spasm (2.3 vs. 1.2 per 100 patient-years). Adverse events reported during the long-term extensions were similar to those in the double-blind treatment phase, and rates of injection-site reactions, constipation, and muscle spasm were lower [123].
Furthermore, studies have explored any potential cardiovascular impact of erenumab-aooe. No differences between erenumab-aooe and placebo were observed for any endpoint during treadmill testing or the 12-week safety follow-up in a clinical trial of patients with ischemic cardiovascular disease [124], nor were any differences in arterial blood pressure observed in healthy volunteers in a placebo-controlled study of concomitant administration of erenumab-aooe and sumatriptan [125]. The cardiovascular, cerebrovascular, and peripheral vascular safety of erenumab-aooe in patients with CM or EM was investigated in a pooled analysis of four double-blind, placebo-controlled studies and their open-label extensions. A total of 2443 patients were included; of these, 1043 received placebo, 893 erenumab-aooe 70 mg, and 507 erenumab-aooe 140 mg. The incidence of vascular adverse events was similar between placebo and erenumab-aooe over 12 weeks, and the emergence of adverse events did not increase over time (up to 256 weeks) [126]. The lack of cardiovascular adverse effects associated with CGRP-R blockade suggests redundancy in the CGRP-R system (both CGRP and amylin bind to AMY1 with high affinity) or compensatory vasodilatory mechanisms [124, 127, 128]. However, the potential for cardiovascular adverse effects following CGRP ligand blockade cannot be ruled out based on results from these studies alone. Development of hypertension and worsening of pre-existing hypertension have been reported following the use of erenumab-aooe in the postmarketing setting. Many of these patients had pre-existing hypertension or risk factors for hypertension. In most cases, the onset or worsening of hypertension was reported after the first dose and were more frequently reported within seven days of dose administration although they could occur at any time during treatment. There were cases of hypertension requiring pharmacological treatment and, in some cases, hospitalization. Per the Aimovig (erenumab-aooe) US Prescribing Information, patients treated with erenumab-aooe should be monitored for new-onset or worsening of pre-existing hypertension and treatment discontinuation considered if an alternative etiology cannot be established [116].
Approved anti-CGRP-R signaling mAbs are administered by subcutaneous injection, via autoinjectors (erenumab-aooe and galcanezumab-gnlm), prefilled syringe (fremanezumab-vfrm), or intravenous infusion (eptinezumab-jjmr) [116,117,118, 129]. Injection-site reactions after subcutaneous administration are common, resulting in localized pain, pruritus, and erythema [130], and rates appear to be higher with fremanezumab-vfrm and galcanezumab-gnlm than with erenumab-aooe, with 26–30%, 6–21%, and 3–6% of patients, respectively, reporting injection-site pain in Phase 3 trials [116,117,118]. Fewer injection-site reactions with erenumab-aooe, together with the availability of an autoinjector, may have a positive effect on adherence, although this will need confirmation in dedicated studies. Reactions may be linked to the immunogenicity of the drug or excipients such as polysorbate-related activation of complement or reactive degradation products [131]. Additionally, drug volume, viscosity, sugar concentration, pH, and having L-histidine as an excipient may all contribute to the variability of injection-site reactions.
CGRP may have a role in regulating uteroplacental blood flow, other vascular adaptations during pregnancy, and relaxation of uterine smooth muscle, resulting in potential complications in pregnancy [132]. However, to date, no adverse effects have been observed on pregnancy in rats and rabbits (fremanezumab-vfrm, galcanezumab-gnlm, and eptinezumab-jjmr), or monkeys (erenumab-aooe) following anti-CGRP-R signaling mAb administration at doses higher than used clinically [116,117,118, 129].
The gastrointestinal tract is highly innervated by enteric neurons expressing CGRP, which is thought to affect intestinal blood flow, smooth muscle relaxation, and slow wave amplitude [133, 134]. Constipation was reported as an adverse event in erenumab-aooe clinical trials in 1–3% of patients [116, 135], and constipation rates did not increase with longer-term treatment (≤ 5 years) [110]. Suspected cases of constipation with serious complications have been observed in a postmarketing setting, although the incidence has not been reported [116]. For context, no cases of serious constipation were reported with erenumab-aooe during Phase 3 trials, which accumulated over 300 person-years [135, 136]. An incidence of serious constipation in people initiating migraine treatment of 0.63 per 100 person-years has been reported [137]. It is important to monitor and to manage patients receiving erenumab-aooe as clinically appropriate if cases of constipation with serious complications occur [116], although these cases are likely to be uncommon. Constipation is also now listed as a side effect of galcanezumab [138], implying that constipation may be a class effect. This side effect is consistent with preclinical studies showing that CGRP causes diarrhea in mice [139]. None of the approved CGRP-R and CGRP ligand mAbs are contraindicated in patients with impaired hepatic or renal function, and such impairment is not expected to alter the pharmacokinetics of the drugs [116,117,118]. The lack of any hepatotoxic effects in this drug class is in sharp contrast to those effects observed with repeated exposure to certain early gepants [23].
A known phenomenon of therapeutic antibodies is the development of neutralizing antibodies [140]. Pooled data from erenumab-aooe clinical trials showed that only a small proportion of patients developed anti-erenumab-aooe-binding antibodies (70 mg, 6.3% [56/884]; 140 mg, 2.6% [13/504]), only three of whom had neutralizing antibodies. By the end of the study, > 50% of patients who developed binding antibodies reverted to negative status and the mean change in MMD did not appreciably differ between patients who did or did not develop anti-erenumab-aooe-binding antibodies [141]. Furthermore, anti-erenumab-aooe-binding antibodies had no impact on safety, and the concentrations of erenumab-aooe overlapped with those of patients who were antibody negative [141]. In placebo-controlled studies of galcanezumab-gnlm, 4.8% of patients (33/688) developed anti-drug antibodies [118]; in a 12-month open-label trial, 12.5% of patients (16/128) developed anti-drug antibodies, most of which were neutralizing [118]. For fremanezumab-vfrm, placebo-controlled trials revealed that 0.4% of patients (6/1701) developed anti-drug antibodies, while in an ongoing open-label study, 1.6% of patients (30/1888) developed anti-drug antibodies [117]. In patients receiving eptinezumab 100 mg or 300 mg every 3 months in Phase 3 trials (PROMISE-1 and PROMISE-2), the incidence of anti-eptinezumab antibodies was 20.6% (92/447) in PROMISE-1 (up to 56 weeks) and 18.3% (129/706) in PROMISE-2 (up to 32 weeks) [129, 142, 143]. The impact of neutralizing antibody development on clinical efficacy has not been shown.
Drug-drug interactions are unlikely to occur between mAbs and other medications, because mAbs are degraded into peptides or amino acids, although the potential for issues arising with concomitant treatment with mAbs and other preventive therapies for migraine is not known. Additionally, it is unknown if there are issues with switching between receptor- and ligand-binding CGRP mAbs [140].
Future Directions for Monoclonal Antibodies for Migraine
Migraine presents with an abundance of non-pain signs and symptoms. Patients receiving anti-CGRP-R signaling treatments commonly experience improvements in other neurological abnormalities of migraine, including reduced sensory aura and improved cognition. However, no longitudinal data are available to indicate the long-term effects (e.g., rebound pain, disease modification) of anti-CGRP-R signaling mAbs [128]. Since the introduction of these agents, patients increasingly mention that the ‘brain fog’ associated with migraine has lifted, with many remarking that this change is more noticeable between than during attacks.
Anti-CGRP-R signaling mAbs are likely to be used effectively in combination with other migraine-preventive treatments, which have different molecular targets. Anti-CGRP-R signaling mAbs might be expected to decrease signaling between attacks, protecting against trigeminal nerve hypersensitivity, although longitudinal studies are required to demonstrate this. Additionally, although there is some evidence that the integration of anti-CGRP-R signaling mAbs may reduce the use of acute migraine treatments [144], further research is needed to evaluate the efficacy and safety of these mAbs in combination with other standard-of-care medications. Further to this, a post hoc analysis has been performed on erenumab-aooe Phase 2 trial data in patients with CM and medication overuse in whom preventive treatment had previously failed. Erenumab-aooe reduced the number of MMD and moderate-severe headache days, reduced the use of acute migraine medication, and increased the proportion of patients achieving nonmedication overuse status versus placebo [144]. Moreover, in a Phase 3 trial, fremanezumab-vfrm reduced the number of days of acute headache medication use over a 12-week period versus placebo in patients with medication overuse at baseline [145]. It will also be necessary to assess anti-CGRP-R signaling mAbs in children, adolescents, during pregnancy, and in similar headache conditions [146].
It has been shown that lower injection frequency and increased efficacy are key drivers of patient adherence [147]. Efficacious mAbs with relatively infrequent dosing may therefore be well received by some patients compared with small molecules or botulinum toxin type A, based on their frequency of administration or number of injections per dose (31–39 injections/dose for botulinum toxin type A) [148]. Further studies on adherence in a real-world setting are needed.
The involvement of other receptors and pathways (such as AMY1, the pituitary adenylate cyclase-activating peptide system, the serotonin-5-HT1F receptor, purinergic receptors, hyperpolarization-activated cyclic nucleotide-gated channels, adenosine triphosphate-sensitive potassium channels, and the glutaminergic system [149]) and the potential for treatments that work synergistically (such as concomitant small-molecule CGRP-R antagonists and CGRP-based mAbs [150]) could also be assessed. Approved anti-CGRP-R signaling mAbs are effective for approximately 50% of patients [116,117,118, 129] and may encourage future development of centrally and peripherally acting CGRP drugs. Combining medications that have peripheral effects with treatments that target central processes may be a good combination therapy approach for migraine control.
Conclusion
There is an unmet need for preventive migraine therapies, and targeting CGRP-R signaling offers much potential. Several mAbs that target the CGRP-R or ligand are approved by the FDA and have proven to be efficacious and well tolerated in pivotal clinical trials. These migraine-specific treatments may help patients who are hypervigilant and to reduce allostatic load. Further evidence is required to determine if there are any real-world differences between ligand- and receptor-targeted therapies and to optimize their use. It will be interesting to discover to what degree these therapies are disease-modifying and, if so, whether this results from an ability to modulate allostatic load.
Change history
02 July 2021
The original article has been revised to correct the term Alder BioPharmaceuticals under Disclosures section.
References
GBD 2016 Headache Collaborators. Global, regional, and national burden of migraine and tension-type headache, 1990–2016: a systematic analysis for the Global Burden of Disease Study 2016. Lancet Neurol. 2018;17(11):954–76.
Russo AF. CGRP-based migraine therapeutics: how might they work, why so safe, and what next? ACS Pharmacol Transl Sci. 2018;2(1):2–8.
Durham PL. Diverse physiological roles of calcitonin gene-related peptide in migraine pathology: modulation of neuronal-glial-immune cells to promote peripheral and central sensitization. Curr Pain Headache Rep. 2016;20(8):48.
Lipton RB, Stewart WF, Diamond S, Diamond ML, Reed M. Prevalence and burden of migraine in the United States: data from the American Migraine Study II. Headache. 2001;41(7):646–57.
Young WB, Park JE, Tian IX, Kempner J. The stigma of migraine. PLoS ONE. 2013;8(1):e54074.
Brandes JL. Migraine and functional impairment. CNS Drugs. 2009;23(12):1039–45.
Eftekhari S, Edvinsson L. Possible sites of action of the new calcitonin gene-related peptide receptor antagonists. Ther Adv Neurol Disord. 2010;3(6):369–78.
de Vries B, Anttila V, Freilinger T, Wessman M, Kaunisto MA, Kallela M, et al. Systematic re-evaluation of genes from candidate gene association studies in migraine using a large genome-wide association data set. Cephalalgia. 2016;36(7):604–14.
Goadsby PJ, Holland PR, Martins-Oliveira M, Hoffmann J, Schankin C, Akerman S. Pathophysiology of migraine: a disorder of sensory processing. Physiol Rev. 2017;97(2):553–622.
Silva-Neto RP, Peres MF, Valenca MM. Odorant substances that trigger headaches in migraine patients. Cephalalgia. 2014;34(1):14–21.
Okuma H, Okuma Y, Kitagawa Y. Examination of fluctuations in atmospheric pressure related to migraine. SpringerPlus. 2015;4:790.
Charles A. The evolution of a migraine attack—a review of recent evidence. Headache. 2013;53(2):413–9.
Headache Classification Committee of the International Headache Society (IHS). The international classification of headache disorders, 3rd edition (beta version). Cephalalgia. 2013;33(9):629–808.
Headache Classification Committee of the International Headache Society (IHS). The international classification of headache disorders, 3rd edition. Cephalalgia. 2018;38(1):1–211.
Miller S. The acute and preventative treatment of episodic migraine. Ann Indian Acad Neurol. 2012;15(Suppl 1):S33–9.
Silberstein SD. Practice parameter: evidence-based guidelines for migraine headache (an evidence-based review): report of the Quality Standards Subcommittee of the American Academy of Neurology. Neurology. 2000;55(6):754–62.
Reuter U, Goadsby PJ, Lanteri-Minet M, Wen S, Hours-Zesiger P, Ferrari MD, et al. Efficacy and tolerability of erenumab in patients with episodic migraine in whom two-to-four previous preventive treatments were unsuccessful: a randomised, double-blind, placebo-controlled, phase 3b study. Lancet. 2018;392:2280–7.
Hepp Z, Dodick DW, Varon SF, Chia J, Matthew N, Gillard P, et al. Persistence and switching patterns of oral migraine prophylactic medications among patients with chronic migraine: a retrospective claims analysis. Cephalalgia. 2017;37(5):470–85.
Reuter U. A review of monoclonal antibody therapies and other preventative treatments in migraine. Headache. 2018;58(Suppl 1):48–59.
Diamond S, Bigal ME, Silberstein S, Loder E, Reed M, Lipton RB. Patterns of diagnosis and acute and preventive treatment for migraine in the United States: results from the American Migraine Prevalence and Prevention study. Headache. 2007;47(3):355–63.
Noseda R, Burstein R. Migraine pathophysiology: anatomy of the trigeminovascular pathway and associated neurological symptoms, CSD, sensitization and modulation of pain. Pain. 2013;154(Suppl 1):S44–53.
Brain SD, Grant AD. Vascular actions of calcitonin gene-related peptide and adrenomedullin. Physiol Rev. 2004;84(3):903–34.
Edvinsson L. CGRP receptor antagonists and antibodies against CGRP and its receptor in migraine treatment. Br J Clin Pharmacol. 2015;80(2):193–9.
Messlinger K. The big CGRP flood—sources, sinks and signalling sites in the trigeminovascular system. J Headache Pain. 2018;19(1):22.
Goadsby PJ, Edvinsson L, Ekman R. Vasoactive peptide release in the extracerebral circulation of humans during migraine headache. Ann Neurol. 1990;28(2):183–7.
Edvinsson L. The CGRP pathway in migraine as a viable target for therapies. Headache. 2018;58(Suppl 1):33–47.
Cernuda-Morollon E, Larrosa D, Ramon C, Vega J, Martinez-Camblor P, Pascual J. Interictal increase of CGRP levels in peripheral blood as a biomarker for chronic migraine. Neurology. 2013;81(14):1191–6.
Sarchielli P, Pini LA, Zanchin G, Alberti A, Maggioni F, Rossi C, et al. Clinical-biochemical correlates of migraine attacks in rizatriptan responders and non-responders. Cephalalgia. 2006;26(3):257–65.
Avona A, Burgos-Vega C, Burton MD, Akopian AN, Price TJ, Dussor G. Dural calcitonin gene-related peptide produces female-specific responses in rodent migraine models. J Neurosci. 2019;39(22):4323–31.
Hansen JM, Hauge AW, Olesen J, Ashina M. Calcitonin gene-related peptide triggers migraine-like attacks in patients with migraine with aura. Cephalalgia. 2010;30(10):1179–86.
Juhasz G, Zsombok T, Jakab B, Nemeth J, Szolcsanyi J, Bagdy G. Sumatriptan causes parallel decrease in plasma calcitonin gene-related peptide (CGRP) concentration and migraine headache during nitroglycerin induced migraine attack. Cephalalgia. 2005;25(3):179–83.
Lassen LH, Haderslev PA, Jacobsen VB, Iversen HK, Sperling B, Olesen J. CGRP may play a causative role in migraine. Cephalalgia. 2002;22(1):54–61.
Russo AF. CGRP as a neuropeptide in migraine: lessons from mice. Br J Clin Pharmacol. 2015;80(3):403–14.
Goadsby PJ, Edvinsson L. The trigeminovascular system and migraine: studies characterizing cerebrovascular and neuropeptide changes seen in humans and cats. Ann Neurol. 1993;33(1):48–56.
Guo S, Vollesen AL, Olesen J, Ashina M. Premonitory and nonheadache symptoms induced by CGRP and PACAP38 in patients with migraine. Pain. 2016;157(12):2773–81.
McEwen BS. Stressed or stressed out: what is the difference? J Psychiatry Neurosci. 2005;30(5):315–8.
Bohm PE, Stancampiano FF, Rozen TD. Migraine headache: updates and future developments. Mayo Clin Proc. 2018;93(11):1648–53.
Dodick DW. A phase-by-phase review of migraine pathophysiology. Headache. 2018;58(Suppl 1):4–16.
Close LN, Eftekhari S, Wang M, Charles AC, Russo AF. Cortical spreading depression as a site of origin for migraine: role of CGRP. Cephalalgia. 2018;2:428–34.
Burstein R, Noseda R, Borsook D. Migraine: multiple processes, complex pathophysiology. J Neurosci. 2015;35(17):6619–29.
Costa C, Tozzi A, Rainero I, Cupini LM, Calabresi P, Ayata C, et al. Cortical spreading depression as a target for anti-migraine agents. J Headache Pain. 2013;14:62.
Melo-Carrillo A, Strassman AM, Nir RR, Schain AJ, Noseda R, Stratton J, et al. Fremanezumab—a humanized monoclonal anti-CGRP antibody—inhibits thinly myelinated (Aδ) but not unmyelinated (C) meningeal nociceptors. J Neurosci. 2017;37(44):10587–96.
Hay DL, Garelja ML, Poyner DR, Walker CS. Update on the pharmacology of calcitonin/CGRP family of peptides: IUPHAR Review 25. Br J Pharmacol. 2018;175(1):3–17.
Hendrikse ER, Bower RL, Hay DL, Walker CS. Molecular studies of CGRP and the CGRP family of peptides in the central nervous system. Cephalalgia. 2018;2:2.
Russell FA, King R, Smillie SJ, Kodji X, Brain SD. Calcitonin gene-related peptide: physiology and pathophysiology. Physiol Rev. 2014;94(4):1099–142.
van den Pol AN. Neuropeptide transmission in brain circuits. Neuron. 2012;76(1):98–115.
Schlereth T, Schukraft J, Kramer-Best HH, Geber C, Ackermann T, Birklein F. Interaction of calcitonin gene related peptide (CGRP) and substance P (SP) in human skin. Neuropeptides. 2016;59:57–62.
Iyengar S, Ossipov MH, Johnson KW. The role of calcitonin gene-related peptide in peripheral and central pain mechanisms including migraine. Pain. 2017;158(4):543–59.
Evans BN, Rosenblatt MI, Mnayer LO, Oliver KR, Dickerson IM. CGRP-RCP, a novel protein required for signal transduction at calcitonin gene-related peptide and adrenomedullin receptors. J Biol Chem. 2000;275(40):31438–43.
Walker CS, Eftekhari S, Bower RL, Wilderman A, Insel PA, Edvinsson L, et al. A second trigeminal CGRP receptor: function and expression of the AMY1 receptor. Ann Clin Transl Neurol. 2015;2(6):595–608.
Walker CS, Hay DL. CGRP in the trigeminovascular system: a role for CGRP, adrenomedullin and amylin receptors? Br J Pharmacol. 2013;170(7):1293–307.
Hay DL. Amylin. Headache. 2017;57(Suppl 2):89–96.
Walker CS, Raddant AC, Woolley MJ, Russo AF, Hay DL. CGRP receptor antagonist activity of olcegepant depends on the signalling pathway measured. Cephalalgia. 2018;38(3):437–51.
Walker CS, Conner AC, Poyner DR, Hay DL. Regulation of signal transduction by calcitonin gene-related peptide receptors. Trends Pharmacol Sci. 2010;31(10):476–83.
Gingell JJ, Hendrikse ER, Hay DL. New insights into the regulation of CGRP-family receptors. Trends Pharmacol Sci. 2019;40:71–83.
Manoukian R, Sun H, Miller S, Shi D, Chan B, Xu C. Effects of monoclonal antagonist antibodies on calcitonin gene-related peptide receptor function and trafficking. J Headache Pain. 2019;20(1):44.
Yarwood RE, Imlach WL, Lieu T, Veldhuis NA, Jensen DD, Klein Herenbrink C, et al. Endosomal signaling of the receptor for calcitonin gene-related peptide mediates pain transmission. Proc Natl Acad Sci USA. 2017;114(46):12309–14.
Bower RL, Yule L, Rees TA, Deganutti G, Hendrikse ER, Harris PWR, et al. Molecular signature for receptor engagement in the metabolic peptide hormone amylin. ACS Pharmacol Transl Sci. 2018;1(1):32–49.
Gingell JJ, Rees TA, Hendrikse ER, Siow A, Rennison D, Scotter J, et al. Distinct patterns of internalization of different calcitonin gene-related peptide receptors. ACS Pharmacol Transl Sci. 2020;3(2):296–304.
Kenakin T. Functional selectivity and biased receptor signaling. J Pharmacol Exp Ther. 2011;336(2):296–302.
Thalakoti S, Patil VV, Damodaram S, Vause CV, Langford LE, Freeman SE, et al. Neuron-glia signaling in trigeminal ganglion: implications for migraine pathology. Headache. 2007;47(7):1008–23.
Cady RJ, Glenn JR, Smith KM, Durham PL. Calcitonin gene-related peptide promotes cellular changes in trigeminal neurons and glia implicated in peripheral and central sensitization. Mol Pain. 2011;7:94.
Koyuncu Irmak D, Kilinc E, Tore F. Shared fate of meningeal mast cells and sensory neurons in migraine. Front Cell Neurosci. 2019;13:136.
Cornelison LE, Hawkins JL, Durham PL. Elevated levels of calcitonin gene-related peptide in upper spinal cord promotes sensitization of primary trigeminal nociceptive neurons. Neuroscience. 2016;339:491–501.
Schwedt TJ, Chiang CC, Chong CD, Dodick DW. Functional MRI of migraine. Lancet Neurol. 2015;14(1):81–91.
Minen MT, Begasse De Dhaem O, Kroon Van Diest A, Powers S, Schwedt TJ, Lipton R, et al. Migraine and its psychiatric comorbidities. J Neurol Neurosurg Psychiatry. 2016;87(7):741–9.
Hawkins JL, Moore NJ, Miley D, Durham PL. Secondary traumatic stress increases expression of proteins implicated in peripheral and central sensitization of trigeminal neurons. Brain Res. 2018;1687:162–72.
Lapp HS, Sabatowski R, Weidner K, Croy I, Gossrau G. C-tactile touch perception in migraineurs—a case–control study. Cephalalgia. 2020;40(5):478–92.
Borsook D, Maleki N, Becerra L, McEwen B. Understanding migraine through the lens of maladaptive stress responses: a model disease of allostatic load. Neuron. 2012;73(2):219–34.
Sink KS, Walker DL, Yang Y, Davis M. Calcitonin gene-related peptide in the bed nucleus of the stria terminalis produces an anxiety-like pattern of behavior and increases neural activation in anxiety-related structures. J Neurosci. 2011;31(5):1802–10.
Kimble M, Boxwala M, Bean W, Maletsky K, Halper J, Spollen K, et al. The impact of hypervigilance: evidence for a forward feedback loop. J Anxiety Disord. 2014;28(2):241–5.
Crestani CC, Alves FH, Gomes FV, Resstel LB, Correa FM, Herman JP. Mechanisms in the bed nucleus of the stria terminalis involved in control of autonomic and neuroendocrine functions: a review. Curr Neuropharmacol. 2013;11(2):141–59.
Han S, Soleiman MT, Soden ME, Zweifel LS, Palmiter RD. Elucidating an affective pain circuit that creates a threat memory. Cell. 2015;162(2):363–74.
Filiz A, Tepe N, Eftekhari S, Boran HE, Dilekoz E, Edvinsson L, et al. CGRP receptor antagonist MK-8825 attenuates cortical spreading depression induced pain behavior. Cephalalgia. 2019;39(3):354–65.
Di Tillo E, Cevoli S, Zenesini C, Fontana C, Grandi S, Tossani E, et al. Allostatic load in migraine patients: a pilot study using an integrated psychosomatic and biochemical approach. Cephalalgia. 2019;39:322.
Chapman CR. Painful Multi-Symptom Disorders: A Systems Perspective. In: Kruger L, Light AR, editors. Translational Pain Research: From Mouse to Man. Frontiers in Neuroscience. Boca Raton (FL) 2010.
Borsook D, Dodick DW. Taking the headache out of migraine. Neurol Clin Pract. 2015;5(4):317–25.
Silberstein SD. Preventive migraine treatment. Continuum (Minneap Minn). 2015;21(4 Headache):973–89.
Ramachandran R, Yaksh TL. Therapeutic use of botulinum toxin in migraine: mechanisms of action. Br J Pharmacol. 2014;171(18):4177–92.
Negro A, Koverech A, Martelletti P. Serotonin receptor agonists in the acute treatment of migraine: a review on their therapeutic potential. J Pain Res. 2018;11:515–26.
Ahn AH, Basbaum AI. Where do triptans act in the treatment of migraine? Pain. 2005;115(1–2):1–4.
Edvinsson L. Aspects on the pathophysiology of migraine and cluster headache. Pharmacol Toxicol. 2001;89(2):65–73.
Durham PL, Masterson CG. Two mechanisms involved in trigeminal CGRP release: implications for migraine treatment. Headache. 2013;53(1):67–80.
Holland PR, Goadsby PJ. Targeted CGRP small molecule antagonists for acute migraine therapy. Neurotherapeutics. 2018;15(2):304–12.
Negro A, Martelletti P. Gepants for the treatment of migraine. Expert Opin Investig Drugs. 2019;28(6):555–67.
Edvinsson L, Chan KY, Eftekhari S, Nilsson E, de Vries R, Saveland H, et al. Effect of the calcitonin gene-related peptide (CGRP) receptor antagonist telcagepant in human cranial arteries. Cephalalgia. 2010;30(10):1233–40.
Gupta S, Mehrotra S, Avezaat CJ, Villalon CM, Saxena PR, Maassen Van Den Brink A. Characterisation of CGRP receptors in the human isolated middle meningeal artery. Life Sci. 2006;79(3):265–71.
Ho TW, Ferrari MD, Dodick DW, Galet V, Kost J, Fan X, et al. Efficacy and tolerability of MK-0974 (telcagepant), a new oral antagonist of calcitonin gene-related peptide receptor, compared with zolmitriptan for acute migraine: a randomised, placebo-controlled, parallel-treatment trial. Lancet. 2008;372(9656):2115–23.
Ho TW, Mannix LK, Fan X, Assaid C, Furtek C, Jones CJ, et al. Randomized controlled trial of an oral CGRP receptor antagonist, MK-0974, in acute treatment of migraine. Neurology. 2008;70(16):1304–12.
Diener HC, Barbanti P, Dahlof C, Reuter U, Habeck J, Podhorna J. BI 44370 TA, an oral CGRP antagonist for the treatment of acute migraine attacks: results from a phase II study. Cephalalgia. 2011;31(5):573–84.
Hewitt DJ, Aurora SK, Dodick DW, Goadsby PJ, Ge YJ, Bachman R, et al. Randomized controlled trial of the CGRP receptor antagonist MK-3207 in the acute treatment of migraine. Cephalalgia. 2011;31(6):712–22.
Olesen J, Diener HC, Husstedt IW, Goadsby PJ, Hall D, Meier U, et al. Calcitonin gene-related peptide receptor antagonist BIBN 4096 BS for the acute treatment of migraine. N Engl J Med. 2004;350(11):1104–10.
Kish T. Emerging therapies for patients with difficult-to-treat migraine. PT. 2018;43(10):616–21.
Prescribing information: Ubrelvy (ubrogepant). Madison, NJ: Allergan Inc, 2020 (package insert). Available at: https://media.allergan.com/products/Ubrelvy_pi.pdf. Accessed 9 Feb 2021.
Dodick DW, Lipton RB, Ailani J, Lu K, Finnegan M, Trugman JM, et al. Ubrogepant for the treatment of migraine. N Engl J Med. 2019;381(23):2230–41.
Tepper SJ. Anti-calcitonin gene-related peptide (CGRP) therapies: update on a previous review after the American Headache Society 60th Scientific Meeting, San Francisco, June 2018. Headache. 2018;58(Suppl 3):276–90.
Croop R, Goadsby PJ, Stock DA, Conway CM, Forshaw M, Stock EG, et al. Efficacy, safety, and tolerability of rimegepant orally disintegrating tablet for the acute treatment of migraine: a randomised, phase 3, double-blind, placebo-controlled trial. Lancet. 2019;394(10200):737–45.
Biohaven Pharmaceuticals. Nurtec ODT (rimegepant) Prescribing information. Available at: https://www.accessdata.fda.gov/drugsatfda_docs/label/2020/212728s000lbl.pdf. Accessed 9 Feb 2021.
Goadsby PJ, Dodick DW, Ailani J. et al. Orally administered atogepant was efficacious, safe, and tolerable for the prevention of migraine: results from a phase 2b/3 study. Presented at the 62nd Annual Scientific Meeting of the American Headache Society, July 11–14, 2019, Philadelphia, PA, USA.
Allergan. Allergan’s Oral CGRP Receptor antagonist atogepant demonstrates robust efficacy and safety in episodic migraine prevention in a Phase 2b/3 Clinical Trial. https://www.prnewswire.com/news-releases/allergans-oral-cgrp-receptor-antagonist-atogepant-demonstrates-robust-efficacy-and-safety-in-episodic-migraine-prevention-in-a-phase-2b3-clinical-trial-300663770.html. Accessed 9 Feb 2021.
Hostetler ED, Joshi AD, Sanabria-Bohorquez S, Fan H, Zeng Z, Purcell M, et al. In vivo quantification of calcitonin gene-related peptide receptor occupancy by telcagepant in rhesus monkey and human brain using the positron emission tomography tracer [11C]MK-4232. J Pharmacol Exp Ther. 2013;347(2):478–86.
Lipman NS, Jackson LR, Trudel LJ, Weis-Garcia F. Monoclonal versus polyclonal antibodies: distinguishing characteristics, applications, and information resources. ILAR J. 2005;46(3):258–68.
Shepard HM, Phillips GL, Thanos CD, Feldmann M. Developments in therapy with monoclonal antibodies and related proteins. Clin Med (Lond). 2017;17(3):220–32.
Suzuki M, Kato C, Kato A. Therapeutic antibodies: their mechanisms of action and the pathological findings they induce in toxicity studies. J Toxicol Pathol. 2015;28(3):133–9.
Yu YJ, Watts RJ. Developing therapeutic antibodies for neurodegenerative disease. Neurotherapeutics. 2013;10(3):459–72.
Johnson KW, Morin SM, Wroblewski VJ, Johnson MP. Peripheral and central nervous system distribution of the CGRP neutralizing antibody [125I] galcanezumab in male rats. Cephalalgia. 2019;39(10):1241–8.
Garces F, Mohr C, Zhang L, Huang CS, Chen Q, King C, et al. Molecular insight into recognition of the CGRPR complex by migraine prevention therapy Aimovig (Erenumab). Cell Rep. 2020;30(6):1714–23.
Silberstein S, Lenz R, Xu C. Therapeutic monoclonal antibodies: what headache specialists need to know. Headache. 2015;55(8):1171–82.
Tepper SJ, Ashina M, Reuter U, Brandes JL, Dolezil D, Silberstein SD, et al. Long-term safety and efficacy of erenumab in patients with chronic migraine: results from a 52-week, open-label extension study. Cephalalgia. 2020;40(6):543–53.
Ashina M, Goadsby PJ, Reuter U, Silberstein S, Dodick DW, Xue F, et al. Long-term efficacy and safety of erenumab in migraine prevention: results from a 5-year, open-label treatment phase of a randomized clinical trial. Eur J Neurol. 2021;28(5):1716–25.
Ashina M, Tepper S, Brandes JL, Reuter U, Boudreau G, Dolezil D, et al. Efficacy and safety of erenumab (AMG334) in chronic migraine patients with prior preventive treatment failure: a subgroup analysis of a randomized, double-blind, placebo-controlled study. Cephalalgia. 2018;38(10):1611–21.
Goadsby PJ, Paemeleire K, Broessner G, Brandes J, Klatt J, Zhang F, et al. Efficacy and safety of erenumab (AMG334) in episodic migraine patients with prior preventive treatment failure: a subgroup analysis of a randomized, double-blind, placebo-controlled study. Cephalalgia. 2019;39(7):817–26.
Barbanti P, Aurilia C, Egeo G, Fofi L. Erenumab: from scientific evidence to clinical practice-the first Italian real-life data. Neurol Sci. 2019;40(Suppl 1):177–9.
Jenkins B, Cheng S, Hutton E. Will refractory patients respond to erenumab in the real world? J Neurol Neurosurg Psychiatry. 2019;90:A10.
Robblee J, VanderPluym J. Fremanezumab in the treatment of migraines: evidence to date. J Pain Res. 2019;12:2589–95.
Prescribing information: Aimovig (erenumab-aooe). Thousand Oaks, CA: Amgen Inc. and Novartis Pharmaceuticals Corporation, 2020 (package insert). Available at: https://www.pi.amgen.com/~/media/amgen/repositorysites/pi-amgen-com/aimovig/aimovig_pi_hcp_english.ashx. Accessed 9 Feb 2021.
Prescribing information: Ajovy (fremanezumab-vfrm). North Wales, PA: Teva Pharmaceuticals USA, Inc., 2020 (package insert). Available at: https://www.ajovy.com/globalassets/ajovy/ajovy-pi.pdf. Accessed 9 Feb 2021.
Prescribing information: Emgality (galcanezumab-gnlm). Indianapolis, IN: Eli Lilly and Company, 2019 (package insert). Available at: https://uspl.lilly.com/emgality/emgality.html#pi. Accessed 9 Feb 2021.
Ryman JT, Meibohm B. Pharmacokinetics of monoclonal antibodies. CPT Pharmacometr Syst Pharmacol. 2017;6(9):576–88.
Mishima T, Ito Y, Nishizawa N, Amano H, Tsujikawa K, Miyaji K, et al. RAMP1 signaling improves lymphedema and promotes lymphangiogenesis in mice. J Surg Res. 2017;219:50–60.
Wan Y, Ceci L, Wu N, Zhou T, Chen L, Venter J, et al. Knockout of α-calcitonin gene-related peptide attenuates cholestatic liver injury by differentially regulating cellular senescence of hepatic stellate cells and cholangiocytes. Lab Invest. 2019;99(6):764–76.
Wootten D, Christopoulos A, Marti-Solano M, Babu MM, Sexton PM. Mechanisms of signalling and biased agonism in G protein-coupled receptors. Nat Rev Mol Cell Biol. 2018;19(10):638–53.
Ashina M, Kudrow D, Reuter U, Dolezil D, Silberstein S, Tepper SJ, et al. Long-term tolerability and nonvascular safety of erenumab, a novel calcitonin gene-related peptide receptor antagonist for prevention of migraine: a pooled analysis of four placebo-controlled trials with long-term extensions. Cephalalgia. 2019;39(14):1798–808.
Depre C, Antalik L, Starling A, Koren M, Eisele O, Lenz RA, et al. A randomized, double-blind, placebo-controlled study to evaluate the effect of erenumab on exercise time during a treadmill test in patients with stable angina. Headache. 2018;58(5):715–23.
de Hoon J, Van Hecken A, Vandermeulen C, Herbots M, Kubo Y, Lee E, et al. Phase 1, randomized, parallel-group, double-blind, placebo-controlled trial to evaluate the effects of erenumab (AMG 334) and concomitant sumatriptan on blood pressure in healthy volunteers. Cephalalgia. 2019;39(1):100–10.
Kudrow D, Pascual J, Winner PK, Dodick DW, Tepper SJ, Reuter U, et al. Vascular safety of erenumab for migraine prevention. Neurology. 2020;94(5):e497–510.
Maassen Van Den Brink A, Meijer J, Villalon CM, Ferrari MD. Wiping out CGRP: potential cardiovascular risks. Trends Pharmacol Sci. 2016;37(9):779–88.
Dodick DW. CGRP ligand and receptor monoclonal antibodies for migraine prevention: evidence review and clinical implications. Cephalalgia. 2019;39(3):445–58.
Lundbeck Seattle BioPharmaceuticals. Vyepti (eptinezumab-jjmr) Prescribing Information, 2020. Available at: https://www.accessdata.fda.gov/drugsatfda_docs/label/2020/761119s000lbl.pdf. Accessed 9 Feb 2021.
Mitsikostas DD, Reuter U. Calcitonin gene-related peptide monoclonal antibodies for migraine prevention: comparisons across randomized controlled studies. Curr Opin Neurol. 2017;30(3):272–80.
Singh SK, Mahler HC, Hartman C, Stark CA. Are injection site reactions in monoclonal antibody therapies caused by polysorbate excipient degradants? J Pharm Sci. 2018;107(11):2735–41.
Yallampalli C, Chauhan M, Thota CS, Kondapaka S, Wimalawansa SJ. Calcitonin gene-related peptide in pregnancy and its emerging receptor heterogeneity. Trends Endocrinol Metab. 2002;13(6):263–9.
Deen M, Correnti E, Kamm K, Kelderman T, Papetti L, Rubio-Beltran E, et al. Blocking CGRP in migraine patients—a review of pros and cons. J Headache Pain. 2017;18(1):96.
Pawlik WW, Obuchowicz R, Biernat J, Sendur R, Jaworek J. Role of calcitonin gene related peptide in the modulation of intestinal circulatory, metabolic, and myoelectric activity during ischemia/reperfusion. J Physiol Pharmacol. 2000;51(4 Pt 2):933–42.
Goadsby PJ, Reuter U, Hallstrom Y, Broessner G, Bonner JH, Zhang F, et al. A controlled trial of erenumab for episodic migraine. N Engl J Med. 2017;377(22):2123–32.
Dodick DW, Ashina M, Brandes JL, Kudrow D, Lanteri-Minet M, Osipova V, et al. ARISE: a Phase 3 randomized trial of erenumab for episodic migraine. Cephalalgia. 2018;38(6):1026–37.
Chia V, Park A, Goli V, Win N, Navetta NS, Xue F. Incidence of constipation in patients treated with commonly used migraine medications. Presented at the 19th Congress of the International Headache Society (ICH); September 5–8, 2019; Dublin, Ireland.
Eli Lilly. Emgality - summary of product characteristics. Available at: https://www.ema.europa.eu/en/documents/product-information/emgality-epar-product-information_en.pdf. Accessed 9 Feb 2021.
Kaiser EA, Rea BJ, Kuburas A, Kovacevich BR, Garcia-Martinez LF, Recober A, et al. Anti-CGRP antibodies block CGRP-induced diarrhea in mice. Neuropeptides. 2017;64:95–9.
Zhou H, Mascelli MA. Mechanisms of monoclonal antibody-drug interactions. A Rev Pharmacol Toxicol. 2011;51:359–72.
Vargas B, Starling A, Silberstein S, Zhou Y, Trotman M-L, Xue F, et al. Erenumab immunogenicity: a pooled analysis of phase 2 and phase 3 migraine prevention clinical trials (P4.098). Neurology. 2018;90(15):98.
Ashina M, Saper J, Cady R, Schaeffler BA, Biondi DM, Hirman J, et al. Eptinezumab in episodic migraine: a randomized, double-blind, placebo-controlled study (PROMISE-1). Cephalalgia. 2020;40(3):241–54.
Lipton RB, Goadsby PJ, Smith J, Schaeffler BA, Biondi DM, Hirman J, et al. Efficacy and safety of eptinezumab in patients with chronic migraine: PROMISE-2. Neurology. 2020;94(13):e1365–77.
Tepper SJ, Diener HC, Ashina M, Brandes JL, Friedman DI, Reuter U, et al. Erenumab in chronic migraine with medication overuse: Subgroup analysis of a randomized trial. Neurology. 2019;92(20):e2309–20.
Silberstein SD, Cohen JM, Seminerio MJ, Yang R, Ashina S, Katsarava Z. The impact of fremanezumab on medication overuse in patients with chronic migraine. J Headache Pain. 2020;21:114.
Szperka CL, VanderPluym J, Orr SL, Oakley CB, Qubty W, Patniyot I, et al. Recommendations on the use of anti-CGRP monoclonal antibodies in children and adolescents. Headache. 2018;58(10):1658–69.
Poulos C, Kinter E, Yang JC, Bridges JF, Posner J, Gleissner E, et al. A discrete-choice experiment to determine patient preferences for injectable multiple sclerosis treatments in Germany. Ther Adv Neurol Disord. 2016;9(2):95–104.
Allergan. Botulinum toxin type A - summary of product characteristics. Available at: https://www.medicines.org.uk/emc/product/859/smpc. Accessed 9 Feb 2021.
Haanes KA, Edvinsson L. Pathophysiological mechanisms in migraine and the identification of new therapeutic targets. CNS Drugs. 2019;33(6):525–37.
Mullin K, Kudrow D, Croop R, Lovegren M, Conway CM, Coric V, et al. Potential for treatment benefit of small molecule CGRP receptor antagonist plus monoclonal antibody in migraine therapy. Neurology. 2020;94(20):e2121–5.
Loder E. What is the evolutionary advantage of migraine? Cephalalgia. 2002;22(8):624–32.
Corrigendum: CGRP ligand and receptor monoclonal antibodies for migraine prevention: Evidence review and clinical implications. Cephalalgia. 2019;39(8):1069.
Sun H, Dodick DW, Silberstein S, Goadsby PJ, Reuter U, Ashina M, et al. Safety and efficacy of AMG 334 for prevention of episodic migraine: a randomised, double-blind, placebo-controlled, phase 2 trial. Lancet Neurol. 2016;15(4):382–90.
Tepper S, Ashina M, Reuter U, Brandes JL, Dolezil D, Silberstein S, et al. Safety and efficacy of erenumab for preventive treatment of chronic migraine: a randomised, double-blind, placebo-controlled phase 2 trial. Lancet Neurol. 2017;16(6):425–34.
Dodick DW, Silberstein SD, Bigal ME, Yeung PP, Goadsby PJ, Blankenbiller T, et al. Effect of fremanezumab compared with placebo for prevention of episodic migraine: a randomized clinical trial. JAMA. 2018;319(19):1999–2008.
Silberstein SD, Dodick DW, Bigal ME, Yeung PP, Goadsby PJ, Blankenbiller T, et al. Fremanezumab for the preventive treatment of chronic migraine. N Engl J Med. 2017;377(22):2113–22.
Stauffer VL, Dodick DW, Zhang Q, Carter JN, Ailani J, Conley RR. Evaluation of galcanezumab for the prevention of episodic migraine: the EVOLVE-1 randomized clinical trial. JAMA Neurol. 2018;75(9):1080–8.
Skljarevski V, Matharu M, Millen BA, Ossipov MH, Kim BK, Yang JY. Efficacy and safety of galcanezumab for the prevention of episodic migraine: results of the EVOLVE-2 Phase 3 randomized controlled clinical trial. Cephalalgia. 2018;38(8):1442–54.
Detke HC, Goadsby PJ, Wang S, Friedman DI, Selzler KJ, Aurora SK. Galcanezumab in chronic migraine: the randomized, double-blind, placebo-controlled REGAIN study. Neurology. 2018;91(24):e2211–21.
Acknowledgements
We are grateful to Herman Ng for his helpful review and additions to the final manuscript draft.
Funding
This publication, including the journal’s Rapid Service Fee, was supported by Novartis Pharmaceuticals Corporation, East Hanover, NJ, USA, as part of a collaboration between Novartis and Amgen Inc.
Medical Writing and/or Editorial Assistance
The authors gratefully acknowledge Kim Wager and Angela Pozo Ramajo (Oxford PharmaGenesis Ltd, Oxford, UK) for providing medical writing and medical editing support. Medical writing assistance was supported by Novartis Pharmaceuticals Corporation, East Hanover, NJ, USA.
Authorship
All authors were equally responsible for the design/conceptualization of the study; acquisition, analysis/interpretation of data; and drafting/revising the manuscript for intellectual content.
Disclosures
Andrew Blumenfeld serves as a consultant and/or a promotional speaker for Alder, Allergan, Amgen, Biohaven, electroCore, Lilly, Novartis, Promius, Supernus, Teva, and Theranica. Paul L. Durham serves as a consultant for Novartis and has received grant support from Alder BioPharmaceuticals, CBD Life Sciences, electroCore, and Teva. Alexander Feoktistov serves on speaker bureaus for Allergan, Amgen, electroCore, Promius Pharma, Supernus Pharmaceuticals, and Teva. He also serves on advisory boards for Amgen and Lilly. Debbie L. Hay serves as a consultant for Intarcia Therapeutics and Merck, and has received grant support from Alder BioPharmaceuticals and Living Cell Technologies. Andrew F. Russo serves as a consultant for Alder BioPharmaceuticals, Amgen, Lilly, Novartis, PharmNovo, and Schedule 1 Therapeutics, and receives grant support from Alder BioPharmaceuticals. Ira Turner serves as a consultant (advisory boards/speaker bureaus) for Allergan, Amgen, Biohaven, Depomed, electroCore, Impax, Lilly, Novartis, Promius, Supernus, and Teva. He also receives research support from Alder BioPharmaceuticals, Allergan, Amgen, ATI, Biohaven, electroCore, Lilly, and Teva.
Compliance with Ethics Guidelines
This article is based on previously conducted studies and does not contain any studies with human participants or animals performed by any of the authors.
Data Availability
Data sharing is not applicable to this article as no data sets were generated or analyzed for this review.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third-party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Blumenfeld, A., Durham, P.L., Feoktistov, A. et al. Hypervigilance, Allostatic Load, and Migraine Prevention: Antibodies to CGRP or Receptor. Neurol Ther 10, 469–497 (2021). https://doi.org/10.1007/s40120-021-00250-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40120-021-00250-7