
Abstract
Hereditary variant transthyretin amyloidosis (ATTRv) is a rare genetic defect that affects about 5000–10,000 people worldwide, causing amyloidosis secondary to misfolding of mutant transthyretin (TTR) protein fibrils. TTR mutations can cause protein deposits in many extracellular regions of organs, but those deposits in cardiac and axonal cells are the primary cause of this clinical syndrome. Treatment options are limited, but new drugs are being developed. Patisiran, a novel drug, is a liposomal siRNA against TTR that specifically targets this protein, reducing the accumulation of TTR in tissues, with subsequent improvement in both neuropathy and cardiac function. Patisiran is likely to serve as a prototype for the development of further intelligent drug solutions for use in targeted therapy. In this review we summarize the evidence currently available on the treatment of polyneuropathy in people with ATTRv with patisiran. We review the evidence on its efficacy, safety, and indications of use, citing novel and seminal papers on these subjects.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Hereditary variant transthyretin amyloidosis (ATTRv) is a rare genetic defect, affecting about 5000–10,000 people worldwide, that causes amyloidosis secondary to misfolding of mutant transthyretin (TTR) proteins. |
TTR mutations cause protein deposits, mainly in cardiac and axonal cells, leading to the syndromes of polyneuropathy. |
Patisiran, a novel drug, is a liposomal siRNA against TTR that allows targeted therapy against this specific protein and reduces the accumulation of TTR in tissues. |
Patisiran improves both neuropathy and cardiac function and is likely to serve as a prototype for the development of further intelligent drug solutions for targeted therapy. |
Introduction
Hereditary variant transthyretin amyloidosis (ATTRv) is a rare, progressive, and lethal autosomal dominant disease characterized by the deposition of misfolded mutant transthyretin (TTR) protein fibrils [1, 2]. TTR, a single polypeptide chain comprised of 127 amino acid residues, is a plasma transport protein for thyroid hormone and retinol-binding protein [3]. Produced by the liver, the wild-type mature structure of TTR is in a tetrameric configuration, which, when destabilized, has the ability to misfold into a monomeric configuration [2, 3]. The disease state of TTR amyloidosis occurs via the formation of insoluble deposits of misfolded TTR fibrils in various body tissues [2, 3]. Amyloidosis can be categorized in terms of deposition of misfolded TTR into two types: TTR amyloidosis wild-type (ATTRwt) and ATTRv [4]. A pathogenic mutation is not present in ATTRwt, and systemic amyloidosis develops due to misfolding of wild-type TTR fibrils and the subsequent deposition and build-up of these fibrils, which results in the disease and associated symptoms [4]. ATTRv is an inherited disease that is due to mutations in the TTR gene and is characterized by the misfolding of mutated TTR fibrils; it can be further categorized into disease with predominant symptoms of polyneuropathy or cardiomyopathy, respectively [4, 5]. The distinction differs in terms of the dominant symptom of the disease—cardiomyopathy or polyneuropathy—although the distinction is not absolute as the presentation may also be mixed [6]. In this review we focus on ATTRv with polyneuropathy. Polyneuropathy, in relation to ATTRv, is a disease condition of the peripheral nerve system, involving somatic, sensory, and autonomic neurons [5, 7]. The disease state and polyneuropathy occurs due to the extracellular deposition of mutated TTR fibrils, leading to the destruction of peripheral nerves [5].
This article is based on previously conducted studies and does not contain any studies with human participants or animals performed by any of the authors.
Epidemiology
The epidemiology of ATTRv is not well established. The disease is considered to be endemic to Portugal, Sweden, and Japan. Its global prevalence has been estimated to about 5000–10,000 (range 5526–38,468) persons, and it has been reported in a total of 36 countries worldwide, including the USA [8]. Prevalence is quite different in those countries where it is deemed endemic versus those where it is considered to be nonendemic, although cases in both endemic and nonendemic countries have been found to have similar genetic mutations [8]. The prevalence of the clinical disease in the USA is estimated to be around 3000–3500 persons [7]). It is important to note that the total global prevalence of ATTRv is not currently known [7]. Once diagnosed, ATTRv is associated with a mean survival time of 5–15 years. The mean age at onset of symptoms is 68.1 years [6, 7], although ATTRv may occur at any stage of life from early adulthood onwards [6].
Genetics of ATTRv Amyloidosis and Subsequent Polyneuropathy
As ATTRv is inherited in an autosomal dominant pattern, genetic inheritance is the greatest risk factor for disease development. Located on the long arm of chromosome 18, the TTR gene contains a total of four exons [9]. TTR gene mutations are primarily missense mutations, although one gene deletion has been discovered [9]. More than 130 TTR gene mutations have been discovered and characterized [10]. The most common TTR variant associated with ATTRv is the p.Val50Met mutation [10], which is responsible for 50% of the mutations associated with ATTRv worldwide [11]. However, although the p.Val50Met mutation dominates prevalence globally, in different regions across the world a higher prevalence of the disease may associated with a different TTR mutation [11]. For example, 89% of patients with ATTRv in Bulgaria present with the Glu89Gln mutation [11]. In particular, TTR gene mutations elicit distinct dominant symptoms and increase severity for ATTRv [9]. The p.Val50Met mutation is more often associated with polyneuropathy as the dominant symptom, while the most dominant symptom of ATTRv associated with the Val122Ile mutation is cardiomyopathy [4, 9, 11]. TTR variants Thr60Ala and Ile107Val are associated with more progressive and lethal disease compared to the p.Val50Met mutation [12]. Parental and geographic origin dictates penetrance of the disease [11]. For example, increased penetrance and earlier onset of symptoms are associated with the inheritance of the variant mutation from the mother rather than from the father [11]. Age of onset and gender are both important risk factors for ATTRv pathology, as male patients and patients with a later age of onset more often have disease that corresponds with cardiac symptoms compared to polyneuropathy [11, 13]. As such, patients with an earlier age of onset are more often associated with polyneuropathy symptoms [14]. The differences in inheritance and how these are related to risk of disease highlight the importance of distinct variants correlating with various levels of penetrance, disease severity, and dominant symptoms [9, 11].
Diagnosis and Clinical Presentation of Polyneuropathy in ATTRv
Due to the breadth of genotypes associated with ATTRv, the clinical presentation varies according to the mutation acquired, geographic location, age at onset, and gender [4, 9, 11]. Patients with polyneuropathy as the dominant symptom are characterized by sensory, motor, and autonomic dysfunction [15]. Given the non-specific symptoms of ATTRv, differentiating the clinical presentation of ATTRv from other polyneuropathy pathologies is a clinical challenge [6]. Above the waist, small fiber neuropathies may exist, presenting neuropathic pain that feels like a lightening bolt and impaired temperature sensation, which may be the initial symptoms [5, 15, 16]. Patients with ATTRv may also progress to present with ascending sensorimotor neuropathy symptoms, with the feet affected before the upper limbs [15, 16]. These sensorimotor symptoms may vary with different mutations, but muscle weakness, imbalance, and sensory deprivation can be seen in all cases [15]. In addition to the progressive sensorimotor neuropathy symptoms, autonomic dysregulation may also present [5, 15]. Possible autonomic symptoms include orthostatic hypotension, sexual dysfunction, and gastrointestinal dysfunction (constipation, diarrhea, vomiting) [5, 15]. Bilateral carpal tunnel syndrome in conjunction with sensorimotor and autonomic dysfunction may hint toward a ATTRv presentation rather than more common types of neuropathies [15, 16]. Together with the polyneuropathy symptoms, the patient may also present cardiac, renal, and eye (dry eye, glaucoma, vitreous amyloidosis) symptoms [5]. Due to this variety of clinical presentations, the correct diagnosis can be extremely difficult to obtain; consequently, misdiagnosis is common [12, 17]. The diagnostic process starts with a comprehensive patient interview and detailed family history [16]. A combination of polyneuropathy symptoms, such as sensorimotor, small fiber, and autonomic neuropathies, together with other systemic organ involvement (e.g., renal, cardiac, gastrointestinal, and eye pathologies) may hint toward a diagnosis [6, 15, 16, 18]. If ATTRv is suspected, tissue biopsy (from regions such as the fat pad or salivary gland) is the standard diagnostic method for amyloidosis confirmation [6, 14, 19]. The presence of amyloid can be confirmed in biopsies stained with Congo Red by the presence of a yellow–green birefringence under polarized light, while immuno-histochemistry may help determine amyloid etiology [6, 15, 19]. Upon confirmation of TTR amyloid deposition, genetic testing determines the genotype of the TTR variant and distinguishes ATTRv from wild-type or acquired TTR amyloidosis [14, 15].
Pathophysiology of Polyneuropathy in ATTRv
Hereditary variant transthyretin amyloidosis is a progressively fatal disease that is caused by the accumulation of amyloid in many different organs and tissues [1]. As mentioned in the Introduction, current thinking separates ATTR into two main categories: a hereditary syndrome (ATTRv) in which misfolded mutated amyloid TTR accumulates in tissues, frequently caused by an autosomal dominant mutation in any of > 130 TTR genes [2], and a nonhereditary syndrome (ATTRwt) in which there is no pathogenic mutation but rather misfolded normal (wild-type) TTP is accumulated in tissues, resulting in systemic organ damage. The important point to be made here is that both disease states are characterized by the deposition of TTR in tissues.
TTR is a 55-kDa transporter protein that is made in the liver. It is involved in transporting thyroid hormone, retinol (vitamin A), and thyroxine [3]. TTR can be mutated via the genetic pathway described in the section Genetics of ATTRv Amyloidosis and Subsequent Polyneuropathy, resulting in the normal tetrameric complex becoming denatured and splitting into monomeric compounds. These monomeric versions of the TTR proteins can accumulate in tissues as amyloid fibrils, thus causing the amyloidosis.
ATTRv can classically result in cardiomyopathy, sensorimotor neuropathy, autonomic neuropathy, and nephropathy [4], but it is especially known for its association with peripheral sensorimotor neuropathy, and this symptom is one of the most classic of the disease [4]. The peripheral sensorimotor neuropathy starts in the feet and upper limbs and progresses proximally, possibly leading to muscle weakness that can cause walking difficulty, lack of balance, and loss of sensation in the upper and lower limbs [5]. Differentiating ATTRv-induced peripheral sensorimotor neuropathy from other kinds of polyneuropathy can be challenging, but some specific symptoms of ATTRv-induced peripheral sensorimotor neuropathy include neuropathic pain that is described as similar to being struck by a bolt of lightning, absence of ataxia, and loss of sensation above the wrist [6]. Any patient with these symptoms that appear to be progressing to additional autonomic dysfunction should be tested for ATTRv. This peripheral sensorimotor neuropathy is largely attributed to the accumulation of amyloid in the nerves. Nerve biopsy therefore shows the loss of axons with some remyelination and demyelination [6].
The specific mechanisms of ATTRv-induced peripheral sensorimotor neuropathy are currrently not completely understood, but there are a couple of important pathophysiological factors that contribute to this neuropathy. The aggregation of amyloid TTR monomers has toxic effects on the neighboring tissue and also disrupts the blood–nerve barrier, leading to TTR leakage into the endoneurial space, thereby promoting further nerve damage [7]. This accumulation of TTR in the endoneurial space can also damage Schwann cells, which furthers the progressive demyelination, resulting in neuropathy.
Current Treatment
Amyloid deposition in ATTRv clearly causes extensive tissue damage, strongly indicating that therapeutic strategies should focus on this pathophysiology. Research is concentrating on three main targets to address the tissue damage; thus, therapies to treat ATTRv could be a mixture of reducing the amount of TTR that is produced [8], preventing the misfolding of TTR by stabilizing it [9], or removing any TTR that has already been deposited [10]. There is also some evidence that a liver transplant for an early-onset patient could simply halt the production of variant TTR and thus completely prevent further disease progression [10]. However, late-onset patients cannot benefit from a liver transplant, and therefore this therapeutic strategy cannot address all people affected by ATTRv.
Tafamidis and diflunisal are two TTR stabilizers that can help manage ATTRv amyloidosis [11, 12]. These two drugs are based on addressing one of the therapeutic targets mentioned above and have had success in treating patients with ATTRv. Unfortunately, many patients who are treated with these drugs continue to have symptoms due to disease progression [13]. Both tafamidis and diflunisal are taken orally. Patisiran, in contrast, is a drug given intravenously that interferes with short interfering RNA (siRNA) that has been shown to be effective in treating ATTRv [14]. Patisiran reduces the production of both variant and wild-type amyloid transthyretin. This reduction in TTR is dose-dependent and is seen in both healthy patients and patients with ATTRv [15]. This reduction in TTR levels has been found to significantly improve the neuropathy induced by the TTR levels, as evidenced in a randomized control trial with 225 patients receiving patisiran or a placebo [16]. In this trial, patients receiving the placebo were more likely to report sensorimotor and autonomic impairment leading to a decrease in their functional abilities. There is even some evidence that patisiran may undo the effects of the polyneuropathy induced by ATTRv as the polyneuropathy disability score increased in 8% of patients in the patisiran group [16].
Unfortunately, polyneuropathy is not the only important symptom in ATTRv; cardiac involvement is common and a frequent cause of death in patients with the disease. Therefore, any drug appropriate for treating ATTRv should address this cardiac involvement and ideally should reduce these cardiovascular events. Echocardiography has shown that patisiran improves the cardiac adverse effects seen in patients with ATTRv by reducing N-terminal pro-B-type naturetic peptide (NT-proBNP) levels and significantly decreasing cardiac arrhythmias. NT-proBNP is a natriuretic peptide that is elevated in conditions that result in high intracardiac pressure. A classic example of this in the setting of ATTRv is heart failure with preserved ejection fraction. However, one study found no significant difference in cardiac adverse effects and cardiac failure between the patisiran and placebo treatment arms [16], indicating that while patisiran is a useful drug it is not a silver bullet in the treatment of ATTRv.
Inotersen is a drug that is similar to patisiran that is used in the treatment of ATTRv. Inotersen decreases TTR levels by inhibiting its transcription. In a phase III randomized, double-blind, placebo-controlled study, inotersen was shown to stabilize or improve neuropathy symptoms and reduce TTR levels [17]. However, inotersen did not change any of the echocardiographic variables when compared to placebo and, therefore, may not have the same efficacious effects that patisiran has in terms of cardiac outcomes.
An emerging treatment for ATTRv may be the use of monoclonal antibodies to clear up the amyloid deposits. Serum amyloid P component (SAP) is a glycoprotein that is very commonly seen in amyloid deposits. The monoclonal antibody dezamizumab has been shown to decrease the levels of SAP in the bodies of patients with systemic amyloidosis [18], but dezamizumab’s development was discontinued after this phase I trial. However, the results do demonstrate how monoclonal antibodies and emerging therapies may hold the key to fully addressing the constellation of symptoms seen in ATTRv.
Patisiran (ONPATTRO®)
Patisiran (ONPATTRO®; Alnylam Pharmaceuticals Inc.) is an oligonucleotide that was approved by the United States Food and Drug Administration (FDA) and European Commission (EC) in August 2018 for the treatment of polyneuropathy associated with ATTRv in adults [20,21,22,23]. It is a double-stranded siRNA that can reduce the synthesis of TTR, which is misfolded and accumulates as insoluble amyloid fibrils in ATTRv [21]. The drug is formulated as a lipid nanoparticle for targeted delivery to hepatocytes, the primary site of TTR synthesis [20, 21]. It is administrated over approximately 80 min via intravenous infusion of 0.3 mg/kg once every 3 weeks if the patient weighs < 100 kg or 30 mg once every 3 weeks if the patient weighs ≥ 100 kg [20]. Risks associated with patisiran administration include possible infusion-related reactions, which can be minimized with premedication, and/or reduced serum vitamin A levels, which can be improved with vitamin A supplementation [20]. At least 60 min prior to the administration of patisiran, the patients must be premedicated with a corticosteroid and antihistamine, both given intravenously, and acetaminophen via the oral route to prevent infusion-related reactions [20]. The most common adverse reactions associated with patisiran are upper respiratory tract infections and infusion-related reactions, including musculoskeletal pain, flushing, abdominal pain, nausea, respiratory symptoms, headache, rash, tachycardia, and/or changes in blood pressure [20]. There are no contraindications to giving patisiran, but the safety and efficacy of the drug in pediatric patients, pregnant women, or adult patients with moderate/severe hepatic or severe renal impairments have not been established [20].
Mechanism of Action
In ATTRv, the TTR gene is mutated, leading to the substitution of Val for Met at position 30 [21]. This mutation destabilizes the protein’s quaternary structure, making it more vulnerable to dissociation compared to the wild-type structure [21, 24]. This susceptibility to dissociation leads to amyloidosis in multiple organs and peripheral nerves, resulting in clinical manifestations, such as polyneuropathy [21, 24, 25].
Patisiran (ONPATTRO®) is the first RNA interference therapy to be approved at the global scale [26,27,28]. It is a liposome formulation containing a slightly modified siRNA encapsulated with lipid excipients [26, 28, 29]. The siRNA was slightly modified with eleven 2′-methoxy-modified sugar residues and four 2′-deoxythymidine residues to improve its stability and to avoid off-target effects [25, 26, 29]. The drug is delivered using a lipid nanoparticle delivery system, which is composed of ionizable cationic lipids (DLin-MC3-DMA), phospholipid (DSPC), cholesterol, and polyethylene glycol-modified lipids (PEG2000-C-DMG), combined via rapid mixing under acidic conditions, followed by a pH-dependent fusion process [23, 26, 27, 29, 30]. With pH neutralization, smaller pH-sensitive liposomes fuse into a larger lipid nanoparticle with an electron-dense core [27]. Once patisiran is administered into the systemic circulation, the polyethylene glycol-modified lipids are lost and replaced by serum proteins, mainly apolipoprotein E (ApoE), which interacts with the cholesterol components of the lipid complex and targets the drug to the liver [29]. Hepatocytes take up ApoE-covered lipid nanoparticles and direct them to the endosome [29]. DLin-MC3-DMA is a pH-sensitive ionizable lipid that becomes cationic in the low pH environment of an endosome, triggering osmosis into the endosome until it ruptures [26, 29]. Once released into the cytoplasm, siRNA is incorporated into an inactive RNA-induced silencing complex (RISC), which then separates the siRNA duplex into single-stranded RNAs [24, 26, 28]. RISC matures once it releases the sense strand of siRNA, leaving the antisense siRNA to bind to its complementary mRNA [24, 26, 28]. The active pharmaceutical ingredient in ONPATTRO® is patisiran sodium (C412 H480 N148 Na40 O290 P40), and the molecular weight is 14,304 Da [20]. As a siRNA, patisiran binds to a genetically conserved sequence in the 3′ untranslated region (3′-UTR) of both wild-type and mutant TTR mRNA [20, 25, 26, 29]. This binding triggers activation of the Argonaute slicer protein (Ago2) in RISC, which then degrades the homologous mRNA, thereby inhibiting the synthesis of TTR protein, which subsequently decreases serum levels and tissue deposits of the protein [24, 26, 29].
Pharmacokinetics and Pharmacodynamics
Through RNA interference, patisiran degrades TTR mRNA, inhibiting TTR protein synthesis, and thus decreasing serum TTR levels [20, 25, 26, 28, 29]. Results from studies showed that after a single intravenous administration of 0.3 mg/kg of patisiran, the mean serum TTR was reduced by approximately 80% within 10–14 days and was similar across genotype, sex, age, or race of the patient [20, 25]. With continued dosing at 0.3 mg/kg once every 3 weeks, mean reductions in serum TTR after 9 and 18 months of treatment were 83 and 84%, respectively, with an 88% maximum reduction in serum TTR levels over 18 months of treatment [20]. Patisiran decreases serum TTR levels, which helps improve polyneuropathy in patients with ATTRv, but serum TTR is also a carrier of retinol-binding protein, which helps transport vitamin A in the blood [20]. Continued use of patisiran for longer than 18 months was found to result in a 45% mean reduction in serum retinol-binding protein and a 62% mean reduction in serum vitamin A [20].
According to the ONPATTRO full prescribing information (FDA), after a single administration of patisiran over the range of 0.01–0.5 mg/kg, the mean steady-state concentrations and areas under the curve (AUC) increase linearly with increasing dose in a dose-dependent manner [20]. At the recommended dosing regimen of 0.3 mg/kg once every 3 weeks, steady-state is reached within 24 weeks of treatment, and at steady state, the area under the curve was found to be 3.2-fold of what it was at the first dose [20]. At this dosing regimen, the estimated mean (± SD) steady-state peak concentrations, trough concentrations, AUC, and volume of distribution were 7.15 ± 2.14 µg/mL, 0.021 ± 0.044 µg/mL, 184 ± 159 µg/mL, and 0.26 ± 0.20 L/kg, respectively [20]. Over 95% of patisiran in circulation is associated with the lipid nanoparticle, and plasma binding of the drug is low, with ≤ 2.1% binding in vitro [20]. Patisiran distributes primarily to the liver, the primary site of TTR synthesis [20]. The mean (± SD) terminal elimination half-life of patisiran is 3.2 ± 1.8 days [20]. It is metabolized by nucleases into nucleotides of various lengths, and the mean (± SD) total body clearance at steady-state is 3.0 ± 2.5 mL/h/kg [20]. Most of the drug is cleared through metabolism, so < 1% of the administered dose is excreted unchanged into urine [20]. Age, race, and sex were found to have no effect on the steady-state pharmacokinetics of patisiran or TTR reduction [20]. Mild/moderate renal impairment (estimated glomerular filtration rate ≥ 30 to < 90 mL/min/1.73 m2) or mild hepatic impairment (bilirubin ≤ 1 × upper limit of normal [ULN]; aspartate aminotransferase > 1 × ULN; bilirubin > 1.0–1.5 × ULN) has no effect on patisiran exposure or TTR reduction [20]. Patisiran has not been studied in people with severe renal impairment, moderate/severe hepatic impairment, or in those with a history of liver transplant [20]. Patisiran is not an inhibitor, inducer, or substrate of cytochrome P450 (CYP) enzymes, so it is not expected to cause any drug–drug interactions. In a population pharmacokinetic analysis, concomitant use of CYP3A inducers/inhibitors had no effect on the pharmacokinetic properties of patisiran [20].
Clinical Trials
APOLLO Phase 1 Trial
In the APOLLO phase 1 trial, an anti-TTR siRNA encapsulated in the first-and second-generation formulations of lipid nanoparticles, ALN-TTR01 and ALN-TTR02, respectively, were given in single doses in a randomized, placebo-controlled, single-blind, multicenter, dose-ranging trial with the aim to assess their respective safety and efficacy in reducing TTR levels. The trial on the first-generation formulation, ALN-TTR01 enrolled patients (n = 32) and ran from July 2010 through to September 2011; the trial on the second-generation formulation, ALN-TTR02, enrolled patients (n = 17) and ran from March 2012 through to May 2012. Patients in the ALN-TTR01 trial received a randomly assigned dose of between 0.01 and 1.0 mg/kg or 0.9% saline placebo, while those in the ALN-TTR02 trial received a dose between 0.01 and 0.05 mg/kg or 0.9% saline placebo. ALN-TTR01 was infused over 15 min, and ALN-TTR02 was infused over 60 min. In both trials, patients received premedication with dexamethasone, acetaminophen, ranitidine, and diphenhydramine or cetirizine to reduce the risk of infusion-related reactions. Pharmacodynamic activity of the drugs was monitored through measuring serum TTR levels using enzyme-linked immunosorbent assay (ELISA), and pharmacokinetics were monitored by ELISA-based hybridization [31].
Neither ALN-TTR01 or ALN-TTR02 use resulted in any significant changes in thyroid, renal, liver, or hematologic function measurements, nor were there any serious adverse events related to the drugs. There were mild to moderate infusion-related reactions in 20.8% of subjects receiving ALN-TTR01 at doses of ≥ 0.4 mg/kg, and there was one moderate infusion-related reaction in a subject who received 0.5 mg/kg of ALN-TTR02. All mild reactions resolved without intervention, and moderate reactions resolved by temporarily stopping administration of the formulation and then continuing infusion at a slower rate. No reactions were associated with proinflammatory cytokines. There was a transient elevation in complement C3a in the subject who had a moderate reaction to the 0.5 mg/kg dose of ALN-TTR02 [31]. The pharmacokinetic values of the mean AUC and the maximal plasma concentration each increased in a dose-proportionate manner for ALN-TTR01 and ALN-TTR02. For ALN-TTR01, there was no significant change from baseline in serum TTR levels for doses 0.01–0.7 mg/kg when compared to the placebo group; however, for the group which received 1.0 mg/kg, there was a significant decrease in serum TTR from baseline on day 1 and a mean reduction of 38% of serum TTR on day 7 (p = 0.01). There was considerable variability in knockdown levels for patients with the p.Val50Met (n = 4) and Ser77Tyr (n = 1) mutations, ranging from 16 to 41%. For ALN-TTR02, there was no significant change in serum TTR levels for the two lowest doses when compared with the placebo. However, significant knockdown rates were observed through to day 28 compared to placebo for subjects receiving doses of 0.15–0.5 mg/kg (p < 0.001) [31].
APOLLO Phase 2 Trial
In the APOLLO Phase 2 trial, nine cohorts of patients received two intravenous doses of patisiran ranging from 0.01 to 0.3 mg/kg every 3 or 4 weeks (dosing and interval varying by cohort). The primary objective of this trial was to assess the safety and tolerability of ascending dose regimens of patisiran. Secondary objectives included assessing the pharmacodynamics of patisiran on total serum TTR protein levels with ELISA, separately measuring wild-type and mutant TTR protein with a proprietary mass spectrometry method, and measuring plasma and urine pharmacokinetics of patisiran in blood and urine samples [32].
Of the 29 patients enrolled, 26 completed the study; however, all 29 received treatment and were included in the intent-to-treat population [32]. In general, multiple patisiran doses were shown to be safe in this study, with no dose-limiting toxicities observed. The most common treatment-emergent adverse event was a mild to moderate infusion-related reaction in three of the 29 patients, including decreased oxygen saturation, abdominal pain, dyspnea, tachypnea, tachycardia, bronchospasm, chills, pyrexia, pallor, and erythema. No other treatment-emergent adverse events were observed for more than one patient in a dosage group. One patient withdrew due to nausea and vomiting; this patient also experienced urinary tract infection and sepsis (all four symptoms listed as serious adverse effects). Another serious adverse effect was extravasation-related cellulitis, which occurred in one patient. Nausea, vomiting, and cellulitis were all attributed to patisiran specifically. No clinically significant changes in hematologic parameters, liver function tests, or renal function were observed. Transient increases in white blood cell count recorded roughly 24 h after each infusion were attributed to the use of dexamethasone as premedication. Transient increases in complement factor Bb in patients receiving doses of 0.05–0.3 mg/kg were not associated with any adverse effects [32].
At baseline, serum TTR protein levels were similar across all cohorts. There was a statistically significant, dose-dependent knockdown of serum TTR protein levels across cohorts, which is consistent with the knockdown rates in the phase 1 placebo-controlled study [31, 32]. The rate of TTR protein knockdown with patisiran was unchanged with concurrent use of tafamidis or diflunisal, which suggests that the pharmacologic activity of patisiran is not affected by these TTR stabilizers. Patisiran use in those patients with the p.Val50Met mutation resulted in suppression of both wild-type and mutated TTR [32]. Pharmacokinetic analysis showed a decrease in mean drug concentration after infusion and no observed increase in siRNA following the second dose. After both doses, the mean AUC and the maximal plasma concentration both increased proportionally to dose. The median terminal half-life of the drug was 39–59 h at doses > 0.01 mg/kg and remained relatively constant between the two doses in each cohort [32].
APOLLO Phase 3 Trial
The APOLLO phase 3 trial was an 18-month-long randomized, double-blind, placebo-controlled study conducted from December 2013 to January 2016 to assess the efficacy and safety of patisiran in 225 patients from 46 locations in 19 countries. Patients were aged 18–85 years and had a diagnosis of ATTRv amyloidosis with an anticipated survival ≥ 2 years, a neuropathy impairment score (NIS) of 5–130, a polyneuropathy disability (PND) score ≤ IIIb, and satisfactory renal and liver function. Exclusion criteria included, among other factors, prior or planned liver transplant; autonomic or sensorimotor neuropathy unrelated to ATTRv amyloidosis; type 1 diabetes mellitus (DM); type 2 DM ≥ 5 years; active hepatitis B or C infection; human immunodeficiency virus; and current use of tafamidis, diflunisal, doxycycline, or tauroursodeoxycholic acid without a washout period. Cardiac parameters defined within the exclusion criteria included New York Heart Association (NYHA) classification > 2; acute coronary syndrome within the last 3 months; unstable angina; and uncontrolled arrhythmia. Patients were randomized 2:1 to receive 0.3 mg/kg patisiran or 0.9% saline as the placebo, as an intravenous infusion over 70 min once every 3 weeks for 18 months. Premedication was used 60 min before the injection of patisiran or placebo to decrease the risk of infusion reactions, as in the APOLLO phase 1 and 2 trials. The groups were matched in terms of NIS, early-onset ATTRv due to Val30Met (V30M) mutation versus other mutations, and prior use of TTR stabilizer at the start of the study [25, 33]. In total, 93% of patients in the patisiran group (n = 138) and 71% of patients in the placebo group (n = 55) completed the trial. Causes of attrition included adverse event, death, withdrawal by a physician, or withdrawal due to disease progression [25].
Neurologic Outcomes
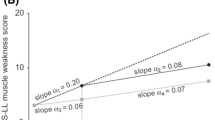
The primary outcome in the APPOLO phase 3 trial was the 18-month difference between trial groups in terms of modified NIS + 7 (mNIS + 7) score, a 304-point scale of neurologic impairment, including neurologic examination of the cranial nerves, upper limbs, and lower limbs; electrophysiologic conduction measurment in small and large nerve fibers; smart somatotopic quantitative sensory testing (including touch pressure and heat pain); and autonomic function, including postural hypotension [25]. The mean mNIS + 7 scores at baseline for the patisiran group and placebo group were 80.9 ± 41.5 and 74.6 ± 37.0, respectively. The benefit of patisiran was indicated by the significantly lower change from baseline at 18 months in the patisiran group than in the placebo group (least squares [LS] mean ± standard error [SE] − 6.0 ± 1.7 [patisiran] vs. 28.0 ± 2.6 [placebo; 95% confidence interval [CI] − 39.9 to − 28.1, p < 0.001) [25]. After 18 months, 56.1% of patients who received patisiran, compared to 3.9% of patients who received a placebo, showed an improvement in their mNIS + 7 score from baseline [34]. The positive effect of patisiran was significant in all components of the scoring scale. There was a correlation between mNIS + 7 score improvement and reduction of serum TTR level [25].
A secondary measure of medication efficacy is the Norfolk Quality of Life–Diabetic Neuropathy (Norfolk QOL-DN) questionnaire, with a scale from − 4 to 136 in which a lower score indicates improved polyneuropathy symptoms, including nerve and autonomic function. The trial groups were initially matched for Norfolk QOL-DN score. The benefit of patisiran was indicated by the improved score at 18 months compared to placebo in all components of the scoring scale (LS mean ± SE: − 6.7 ± 1.8 [patisiran] and 14.4 ± 2.7 [placebo]; 95% CI − 27.2 to − 15.0; p < 0.001). At the study endpoint, 51.4% of patients who received patisiran and 10.4% of those who received placebo had an improved Norfolk QOL-DN score from baseline [25, 34].
The COMPASS-31 scale, which assesses autonomic neuropathy on a scale from 0 to 100, with a lower score indicating improved symptoms, was another secondary endpoint of the efficacy of patisiran in this trial. The LS mean change during the study duration was − 5.3 for the patisiran group and 2.2 in the placebo group, correlating with a LS mean difference of − 7.5 (p = 0.0008). There was a significant benefit in all components of the COMPASS-31 score, including secretomotor, bladder, vasomotor, and pupillomotor function, while the greatest improvement compared to placebo was seen in the orthostatic intolerance and gastrointestinal components [34].
Prior Tafamidis Use
In the APPOLO phase 3 trial, patients who reported prior tafamidis use were required to discontinue tafamidis at least 14 days prior to starting patisiran. In this trial, 74 patients (32.9%) were in the prior tafamidis use subgroup; 46 patients discontinued tafamidis to enroll in APOLLO; and 25 discontinued tafamidis due to progression of ATTRv amyloidosis. Theimprovement in the mNIS + 7 score in the patisiran group over the placebo group with prior tafamidis use at 18 months was a LS mean difference of − 27.0, versus − 34.0 in the overall APOLLO population. The improvement in the Norfolk QOL-DN in the patisiran group over the placebo group with prior tafamidis use at 18 months was a LS mean difference of − 15.9 compared to placebo, versus − 21.1 in the overall APOLLO population. Patients in the treatment groups had a similar rate of adverse events as the general APOLLO population, indicating that overall, patients with prior use of tafamidis experienced similar benefits with patisiran compared to placebo as the general APOLLO population [35]. A review article analyzed NIS, Norfolk Qol-DN, and modified BMI score in randomized trials comparing patisiran and tafamidis to placebo, respectively. Patisiran was found to have a larger treatment benefit than tafamidis for all measures, suggesting patisiran has a more positive treatment effect than tafamidis for patients with hereditary ATTRv [18].
Cardiac Effects
The APOLLO phase 3 study also studied several cardiac parameters within a prespecified cardiac subpopulation consisting of patients without a history of hypertension or aortic valve disease with a baseline left ventricular (LV) wall thickness ≥ 13 mm. Of the 126 patients in the cardiac subpopulation (median age 61 years; 78% male), 90 (71.4%) received patisiran and 36 (28.6%) received placebo. Cardiac function was measured at baseline and at months 9 and 18 using 2-dimensional echocardiography measures and levels of troponin 1 and NT-proBNP [36].
LV mean wall thickness, as well as corresponding interventricular septal wall, posterior wall, and relative wall thicknesses, decreased in the patisiran group compared to the placebo group (LS mean difference ± standard error of the mean 0.9 ± 0.4 mm; p = 0.017). There was significant improvement across all echocardiographic measures in the patisiran group, including increased cardiac output (0.38 ± 0.19 L/min; p = 0.044) and increased LV end-diastolic volume (8.31 ± 3.91 mL; p = 0.036). Analysis of troponin-1 outcomes could not be conducted because 90.2% of the values were below the detection limit of 0.1 μg/L. In the cardiac subgroup, the patients in the patisiran group had significantly improved their 10-m walking test speed at 9 and 18 months compared to those receiving placebo. The level of NT-proBNP was significantly reduced by 55% in the patisiran group at 18 months compared to the placebo group [36].
The cardiac subpopulation was also analyzed to assess the effect of patisiran on regional LV myocardial strain. Cardiac amyloidosis causes a universal deficiency in LV global longitudinal strain (GLS), often with sparing of the cardiac apex, which is an independent predictor of mortality in this patient population. Baseline LV GLS was reduced with worse regional longitudinal strain deficit in the basal section with apical sparing. At 18 months, patisiran was found to improve absolute GLS versus placebo, with the most improvement observed in the basal section and no significant improvement in the middle and apex (LS mean difference ± SE 1.4 ± 0.6%; 95% CI 0.3–2.5%; p = 0.02). There was approximately a 21% change from baseline in the basal section versus a 6% change at the apex. These changes were not correlated with improvement in NT-proBNP levels. In noncardiac population patients, there were no significant changes in regional strain [37].
The exposure-adjusted rate of cardiac hospitalization and all-cause death was 10.1 per 100 patient-years in the patisiran group, compared to 18.7 in the placebo group. Of the other APOLLO Phase 3 patients not included in the cardiac subpopulation, no significant echocardiogram changes were seen, but NT-proBNP levels decreased in comparison to placebo for all non-cardiac subgroup patients, with an average reduction of 51% [36].
Orthostatic Hypotension
A common manifestation of ATTRv is orthostatic intolerance (OI), with the deposition of amyloid in the autonomic nervous system leading to an increased risk of syncope and falling. In the phase 3 APOLLO study, patients were asked, “In the past year, have you ever felt faint, dizzy, ‘goofy,’ or had difficulty thinking soon after standing up from a sitting or lying position?” Changes in the symptoms of OI were then evaluated. At baseline, approximately two-thirds of patients from both groups reported symptoms of OI (mild to severe). After 18 months, 30% of the patisiran group reported improvement in their OI symptoms compared to 10% of the placebo group. In addition, only 14% of the patisiran group reported that OI symptoms worsened after 18 months of treatment compared to 23% of the placebo group [38].
Overall Disability and Health Status
In the phase 3 APOLLO study, the Rasch-built Overall Disability Scale (R-ODS) was administered to all patients at baseline to study the impact of hereditary ATTRv amyloidosis disability on daily living and work. The scale ranges from 0 to 48, with higher scores indicating improved outcomes. At baseline, 69% of patients reported being unable to work, and 24% received government compensation due to their illness. Patients with worse neuropathy required greater government compensation. Patients reported mild difficulty performing low-intensity tasks, including reading a newspaper, eating, and brushing their teeth. Most patients were unable to perform high-intensity tasks, including dancing, standing for an extended period, or running. The change in R-ODS score at 18 months in patients receiving patisiran or placebo has not been published to date [39].
When compared to the placebo group, a larger percentage of the patisiran group had maintenance or improvement of baseline scores for each of the following measured domains: self-care (66 vs. 21%), pain/discomfort (73 vs. 31%), mobility (70 vs. 22%), anxiety/depression (81 vs. 45%), and usual activities (72 vs. 22%). The EQ visual analogue scale, a measure of overall health status assessment, showed an increase of 2.4 points for patients on patisiran and a decrease of 7.1 points for patients on placebo, with a total difference in points of 9.5 (p value = 0.0004) [40].
Safety and Adverse Events
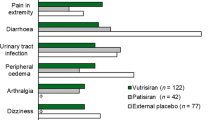
In the phase 3 APOLLO study, adverse events were reported by 97% of patients in both the patisiran and placebo groups. While these were most often mild or moderate in severity, severe events were reported in 28% of patients who received patisiran and 36% of patients who received a placebo, and serious events were reported in 36 and 40% of patients, respectively. Common adverse events in the patisiran and placebo group respectively were diarrhea (37 vs. 38%), peripheral edema (30 vs. 22%), falls (17 vs. 29%), nausea (15 vs. 21%), and infusion-related reaction (19 vs. 9%). Infusion reaction symptoms were mild or moderate, with no severe or serious events reported, and they did not cause withdrawal from the study; symptoms included back pain, abdominal pain, flushing, and nausea, and their incidence declined over time. In the patisiran group 5% of patients experienced an adverse event that led to discontinuation of the trial regimen, compared to 14% in the placebo group. The patisiran and placebo groups experienced similar rates of cardiac adverse events (28 vs. 36%, respectively), cardiac serious adverse events (14 vs. 13%), and cardiac failure (9 vs. 10%), although the patisiran group had a lower incidence of cardiac arrhythmia (19 vs. 29%). There were no clinically significant laboratory value abnormalities with patisiran, including platelet count, liver function, and renal function. Death, chiefly due to cardiovascular causes consistent with the natural course of ATTRv, occurred in seven patients receiving patisiran and six patients on placebo; these deaths were considered unlikely to be related to patisiran [25, 36]. Post hoc analysis indicated that the rates of all-cause hospitalization or death in the patisiran and placebo groups were 34.7 and 71.8 per 100 patient-years, respectively [36].
Budget Impact
The economic impact of reimbursing patisiran (including monitoring, pre-medications, and potentially serious adverse event costs) was assessed using a model that estimated the per-member-per-month budget impact in 2018 US dollars on a hypothetical US health plan covering one million lives over 3 years. The incremental budget impact was $0.008 per-member-per-month. A Medicare Advantage perspective yielded a similar budget impact ($0.009 per-member-per-month). The home infusion had a predicted savings of $0.002 per-member-per-month. These costs were considered to have a minimal impact on health plans from both perspectives [41].
Summary and Conclusion
Hereditary variant transthyretin amyloidosis is an autosomal dominant systemic disease caused by mutations in the TTR gene, which in turn lead to amyloid deposits in the peripheral nerves and multiple organs [21, 24, 25]. Treatment options for ATTRv were limited to liver transplantation and TTR tetramer stabilizers until 2018, when patisiran, the first RNA interference therapeutic, was approved to treat adults with polyneuropathy associated with ATTRv [20, 21, 25]. The approval of patisiran (ONPATTRO®) was based on promising findings from the APOLLO trial, a large global phase 3 randomized controlled trial [25]. The trial demonstrated that patisiran significantly improves polyneuropathy, quality of life, walking, nutritional status, and activities of daily living in patients with ATTRv [25]. Patisiran is a siRNA encapsulated within a lipid nanoparticle delivery system for targeted delivery to the liver, the primary source of serum TTR [20, 21, 23, 24, 26, 28, 30]. Once in the liver, siRNA binds to the 3′-untranslated region of TTR mRNA and degrades it, inhibiting TTR synthesis. With the exception of possible infusion-related reactions and reduced serum vitamin A levels, both of which can be prevented or alleviated, the drug has been shown to be safe and effective in adults with ATTRv with polyneuropathy [20, 25].
Change history
12 January 2021
A Correction to this paper has been published: https://doi.org/10.1007/s40120-020-00228-x
References
Vita G, Vita GL, Stancanelli C, Gentile L, Russo M, Mazzeo A. Genetic neuromuscular disorders: living the era of a therapeutic revolution. Part 1: peripheral neuropathies. Neurol Sci. 2019;40(4):661–9.
Lamb YN, Deeks ED. Tafamidis: a review in transthyretin amyloidosis with polyneuropathy. Drugs. 2019;79(8):863–74.
Benson MD, Kincaid JC. The molecular biology and clinical features of amyloid neuropathy. Muscle Nerve. 2007;36(4):411–23.
Frederick RL, Berk JL. Transthyretin (TTR) cardiac amyloidosis. Circulation. 2012;126(10):1286–300.
Adams D, Koike H, Slama M, Coelho T. Hereditary transthyretin amyloidosis: a model of medical progress for a fatal disease. Nat Rev Neurol. 2019;15(7):387–404.
Hawkins PN, Ando Y, Dispenzeri A, Gonzalez-Duarte A, Adams D, Suhr OB. Evolving landscape in the management of transthyretin amyloidosis. Ann Med. 2015;47(8):625–38.
Mickle K, Lasser KE, Hoch JS, Cipriano LE, Dreitlein WB, Pearson SD. The effectiveness and value of patisiran and inotersen for hereditary transthyretin amyloidosis. J Manag Care Spec Pharm. 2019;25(1):10–5.
Schmidt HH, Waddington-Cruz M, Botteman MF, et al. Estimating the global prevalence of transthyretin familial amyloid polyneuropathy. Muscle Nerve. 2018;57(5):829–37.
Rizk M, Tüzmen Ş. Update on the clinical utility of an RNA interference-based treatment: focus on Patisiran. Pharmgenom Pers Med. 2017;10:267–78.
Coelho T, Ines M, Conceicaõ I, Soares M, De Carvalho M, Costa J. Natural history and survival in stage 1 Val30Met transthyretin familial amyloid polyneuropathy. Neurology. 2018;91(21):E1999–2009.
Parman Y, Adams D, Obici L, et al. Sixty years of transthyretin familial amyloid polyneuropathy (TTR-FAP) in Europe: where are we now? A European network approach to defining the epidemiology and management patterns for TTR-FAP. Curr Opin Neurol. 2016;29:S3–13.
Plante-Bordeneuve V. Transthyretin familial amyloid polyneuropathy: an update. J Neurol. 2018;265(4):976–83.
Waddington-Cruz M, Ackermann EJ, Polydefkis M, et al. Hereditary transthyretin amyloidosis: baseline characteristics of patients in the NEURO-TTR trial. Amyloid. 2018;25(3):180–8.
Coelho T, Maurer MS, Suhr OB. THAOS—The Transthyretin Amyloidosis Outcomes Survey: initial report on clinical manifestations in patients with hereditary and wild-type transthyretin amyloidosis. Curr Med Res Opin. 2013;29(1):63–76.
Gertz MA, Mauermann ML, Grogan M, Coelho T. Advances in the treatment of hereditary transthyretin amyloidosis: a review. Brain Behav. 2019;9(9):1–12.
Conceição I, González-Duarte A, Obici L, et al. Red-flag symptom clusters in transthyretin familial amyloid polyneuropathy. J Peripher Nerv Syst. 2016;21(1):5–9.
Adams D, Cauquil C, Labeyrie C. Familial amyloid polyneuropathy. Curr Opin Neurol. 2017;30(5):481–9.
Planté-Bordeneuve V, Lin H, Gollob J, et al. An indirect treatment comparison of the efficacy of patisiran and tafamidis for the treatment of hereditary transthyretin-mediated amyloidosis with polyneuropathy. Expert Opin Pharmacother. 2019;20(4):473–81.
Freeman R, Barroso F. Recent advances in familial amyloid polyneuropathy. Curr Opin Neurol. 2015;28(5):494–9.
US Food and Drug Administration (FDA). ONPATTRO full prescribing information. FDA, Washington DC. 2018. https://www.accessdata.fda.gov/drugsatfda_docs/nda/2018/210922Orig1s000TOC.cfm.
Al Shaer D, Al Musaimi O, Albericio F, de la Torre BG. 2018 FDA tides harvest. Pharmaceuticals (Basel) 2019;12(2):52. https://doi.org/10.3390/ph12020052.
Yang J. Patisiran for the treatment of hereditary transthyretin-mediated amyloidosis. Expert Rev Clin Pharmacol. 2019;12(2):95–9.
Wahlich J, Desai A, Greco F, et al. Nanomedicines for the delivery of biologics. Pharmaceutics (Basel) 2019;11(5):210. https://doi.org/10.3390/pharmaceutics11050210.
Saw PE, Song EW. siRNA therapeutics: a clinical reality. Beijing: Science in China Press; 2019.
Adams D, Gonzalez-Duarte A, O’Riordan WD, et al. Patisiran, an RNAi therapeutic, for hereditary transthyretin amyloidosis. N Engl J Med. 2018;379(1):11–21.
Weng Y, Xiao H, Zhang J, Liang XJ, Huang Y. RNAi therapeutic and its innovative biotechnological evolution. Biotechnol Adv. 2019;37:801–25.
Kulkarni JA, Witzigmann D, Leung J, et al. Fusion-dependent formation of lipid nanoparticles containing macromolecular payloads. Nanoscale. 2019;11(18):9023–31.
Hu B, Weng Y, Xia XH, Liang XJ, Huang Y. Clinical advances of siRNA therapeutics. J Gene Med. 2019;21:e3097.
Setten RL, Rossi JJ, Han SP. The current state and future directions of RNAi-based therapeutics. Nat Rev Drug Discov. 2019;18:421–46.
Dong Y, Siegwart DJ, Anderson DG. Strategies, design, and chemistry in siRNA delivery systems. Adv Drug Deliv Rev. 2019;144:133–47.
Coelho T, Adams D, Silva A, et al. Safety and efficacy of RNAi therapy for transthyretin amyloidosis. N Engl J Med. 2013;369(9):819–29.
Suhr OB, Coelho T, Buades J, et al. Efficacy and safety of patisiran for familial amyloidotic polyneuropathy: a phase II multi-dose study. Orphanet J Rare Dis. 2015;10(1):1–9.
Adams D, Suhr OB, Dyck PJ, et al. Trial design and rationale for APOLLO, a phase 3, placebo-controlled study of patisiran in patients with hereditary ATTR amyloidosis with polyneuropathy. BMC Neurol. 2017;17(1):1–12.
Mauermann M, Adams D, Gonzalez-Duarte A, et al. Impact of Patisiran on autonomic neuropathy in hereditary transthyretin-mediated amyloidosis patients. J Neuromusculoskelet Dis. 2018;5(1):S220–1.
Lin H, Obici L, Planté-Bordeneuve V, White MT, Riese R, Suhr OB. Efficacy of Patisiran in patients with hATTR amyloidosis and prior tafamidis use: analysis of APOLLO. Peripher Nerve Soc Annu Meet, 24 June 2019, Genoa. 2019;210922. https://www.alnylam.com/wp-content/uploads/2019/06/PNS-2019_Pati-APOLLO-Prior-Taf_FINAL.pdf
Solomon SD, Adams D, Kristen A, et al. Effects of PATISIRAN, an RNA interference therapeutic, on cardiac parameters in patients with hereditary transthyretin-mediated amyloidosis: analysis of the APOLLO study. Circulation. 2019;139(4):431–43.
Minamisawa M, Claggett B, Adams D, et al. Association of Patisiran, an RNA interference therapeutic, with regional left ventricular myocardial strain in hereditary transthyretin amyloidosis: the APOLLO study. JAMA Cardiol. 2019;4(5):466–72.
Gonzalez-Duarte A, Judge DP, Dispenzieri A, et al. Impact of Patisiran, an RNAi therapuetic, on orthostatic intolerance in patients with hereditary transthyretin-mediated amyloidosis. Neurology. 2020;94(Supp 15):766.
Berk J, Lin H, Agarwal S, et al. Impact of hereditary transthyretin-mediated amyloidosis on daily living and work productivity: baseline results from APOLLO. Amyloidosis. 2018;21(1):1.
Ajroud-Driss S, Adams D, Coelho T, et al. Impact of Patisiran on overall health status in hATTR amyloidosis: results from the APOLLO trial (S27.007). Neurology. 2019;92(Suppl 15).
Lin H, Merkel M, Pelligra C, Shah A. Pbi18 evaluating the budget impact of Patisiran, the first approved RNAi therapeutic, for treating the polyneuropathy of hATTR amyloidosis. Value Health. 2019;22(May):S50. https://doi.org/10.1016/j.jval.2019.04.101.
Acknowledgements
Funding
No funding or sponsorship was received for this study or publication of this article.
Authorship
All named authors meet the International Committee of Medical Journal Editors (ICMJE) criteria for authorship for this article, take responsibility for the integrity of the work as a whole, and have given their approval for this version to be published.
Disclosures
All authors (I Urits, D Swanson, MC Swett, A Patel, K Berardino, A Amgalan, AA Berger, H Kassem, AD Kaye, O Viswanath) have nothing to disclose.
Compliance with Ethics Guidelines
This article is based on previously conducted studies and does not contain any studies with human participants or animals performed by any of the authors.
Data Availability
Data sharing is not applicable to this article as no datasets were generated or analyzed during the current study.
Author information
Authors and Affiliations
Corresponding author
Additional information
The original article has been revised to include the author's mid initial - Alan D. Kaye
Digital Features
To view digital features for this article go to https://doi.org/10.6084/m9.figshare.12732299.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/ .
About this article

Cite this article
Urits, I., Swanson, D., Swett, M.C. et al. A Review of Patisiran (ONPATTRO®) for the Treatment of Polyneuropathy in People with Hereditary Transthyretin Amyloidosis. Neurol Ther 9, 301–315 (2020). https://doi.org/10.1007/s40120-020-00208-1
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40120-020-00208-1