
Abstract
The chemistry of hoop-shaped π-conjugated molecules has increased dramatically in recent years. We present here a computational modeling of photoinduced electron transfer processes in a series of host–guest complexes of Twin1, Twin2, and Twin3 double nanohoops with C60 fullerene. According to our findings, charge transfer from cycloparaphenylene (CPP) fragments to C60 is energetically favorable and occurs on a sub-nanosecond time scale. The slow decay of the generated charge-separated state suggests that the complexes may be of interest for organic photovoltaics.
Graphical abstract

Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Molecular systems with unusual topology attract significant attention from scientists of different research fields. Their electronic and chemical properties, as well as esthetic beauty, encourage scientists to look for new distorted, strained, bend or interlocked molecules [1,2,3,4]. Highly expanded π-conjugated architectures hitherto unknown are of great interest for photovoltaic applications. An example of such systems with unusual topology are cycloparaphenylenes (CPPs)—radially π-conjugated nanohoops constructed of para-linked phenylene rings [5]. Over the past 10 years, significant developments in organic synthesis have allowed for the precise control of the amount of phenylene units in the target nanohoops, resulting in CPPs with a diameter range of 7–28 Å. The variability in the size of nanohoops has led to their wide application in supramolecular chemistry [5, 6]. A change in size dramatically affects their photophysical properties [7,8,9]. The first host–guest complex of CPP was reported by Iwamoto et al. in 2011 [10]. The authors found that [10]CPP with ten phenylene units has a nearly ideal diameter (13.8 Å) to accommodate C60 fullerene. Bigger CPPs can bind larger fullerenes and endohedral metallofullerenes [11,12,13]. The formation of fullerene-nanohoop host–guest complexes suppresses fullerene self-aggregation, which encourages their use as a component of organic solar cells. It has been reported that certain supramolecular donor–acceptor (DA) complexes of CPPs with different fullerenes are capable of photoinduced electron transfer (PET). CPPs and their π-extended analogs serve as an electron-donating species in many of them [14, 15].
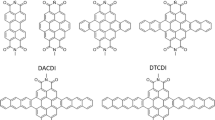
In 2021, Du and co-workers [16] presented an unusual double-nanohoop molecule (Twin1)—a highly strained all-phenylene bismacrocycle, termed conjoined (1,4)[10]cycloparaphenylenophane. It consists of two CPP units linked by a twisted benzene ring (Fig. 1). The absorption spectrum of Twin1 is similar to that of [10]CPP, while the extinction coefficient is twice as high as that of the parental CPP. However, the emission spectrum of Twin1 demonstrates a significant redshift (of more than 50 nm) compared to [10]CPP. The authors presume that the observed shift is associated with a high strain energy (greater than 110 kcal/mol), and the corresponding enhancement of vibrational couplings. The Twin1 bismacrocycle exhibits a suitable diameter to form inclusion complexes with C60. The binding constant was determined to be Ka1 = (7.46 ± 0.33) × 105 M−1 and Ka2 = (5.85 ± 0.25) × 104 M−1 for the mono (Twin1 ⊃ C60) and the bis (Twin1 ⊃ 2C60) adduct, respectively.

Structure of the double-nanohoop systems studied in this work
Also, Juríček and co-workers [17, 18] reported a new member of the double-nanohoop family (Twin2)—a giant π-conjugated framework with two CPP units linked by a peropyrene fragment (Fig. 1). The absorption spectrum of Twin2 was found to be a superposition of absorption spectra of CPPs and peropyrene, while the emission spectrum is nearly identical to that of the peropyrene unit. The authors demonstrated that such a system, with the bay region of central peropyrene core embedded in nanohoops, can form stable complexes with C60 fullerene. They managed to obtain Twin2 ⊃ C60 complex, while a bis adduct has never been observed in the solid state.
In 2022, Zhu, Cong, and co-workers [19] successfully synthesized a double nanohoop (Twin3), which combines two [10]CPP units and a flexible cyclooctatetrathiophene core. This combination was found to be essential for the formation of host–guest complexes with C60 and C70 fullerenes. The binding constant for the mono (Twin3 ⊃ C60) and the bis (Twin3 ⊃ 2C60) adducts was found to be Ka1 = (1.3 ± 0.1) × 105 M−1 and Ka2 = (1.9 ± 0.2) × 104 M−1, respectively.
In this paper, we present a computational investigation of the structural, electronic, and photoinduced electron transfer properties of host–guest complexes of the Twin1, Twin2, and Twin3 double nanohoops with C60 fullerene. We compare the rates of the formation and the decay of charge transfer (CT) states to find the best candidate for further experimental research. Our results shed light on the role of structure and electronic nature of the linker between two CPP units in electron transfer processes.
Computational methods
Geometry optimizations were performed using ORCA 4.2.1 program [20]. BLYP [21, 22] functional was used with def2-SVP basis set [23, 24]. Resolution of identity approximation [25, 26] and D3 dispersion correction by Grimme with Becke-Johnson damping [27, 28] were also employed. Vibrational frequencies were computed for all structures. The absence of imaginary frequencies confirmed that the obtained geometries correspond to the minima on the potential energy surface. The interaction energy of the complexes was computed at the BLYP-D3(BJ)/def2-TZVP level of theory. BLYP-D3(BJ) functional was chosen as the DFT functional with the best accuracy-to-cost ratio for non-covalent interactions [29, 30]. As previously demonstrated, using range-separated functionals is required for accurate prediction of charge transfer rates [31,32,33]. Consequently, vertical excitation energies were calculated using Tamm–Dankov approximation (TDA) formalism [34] with range-separated CAM-B3LYP [35] functional and def2-SVP basis set, [22, 23] using Gaussian 16 (rev. A03) [36]. The same program was used for population analysis and calculation of Mulliken [37, 38], Lowdin [39], Hirshfeld, [40] and CM5 [41] charges. Energy decomposition analysis (EDA) was performed using the Amsterdam Density Functional (ADF) program [42]. Topological analysis of the electron density distribution was conducted using the “Quantum Theory of Atoms in Molecules” (QTAIM). [43] AIMALL package [44] was applied to evaluate the bond critical point properties and associated bond descriptors. Excited states were analyzed by constructing natural transition orbitals (NTO) introduced by Luzanov et al. [45] and implemented within modern many-body codes by Head-Gordon et al. [46] Chemcraft 1.8 [47] was used to visualize chemical structures and frontier molecular orbitals.
Interaction and deformation energies
Interaction energy, ΔEint, was calculated from the electronic energy of the complex and electronic energies of its subsystems using the BLYP-D3(BJ)/def2-TZVP//BLYP-D3(BJ)/def2-SVP scheme. For TwinX ⊃ C60 and TwinX ⊃ 2C60 complexes, the interaction energy can be calculated as follows:
The deformation energy, ΔEdef, is the amount of energy required to deform the individual fragments from their equilibrium structure to the geometry that they adopt in the complex:
Energy decomposition analysis
A quantitative energy decomposition analysis [48, 49] was used to investigate the interaction energy in the gas phase within the framework of the Kohn–Sham MO model. ΔEint was decomposed into electrostatic interactions, Pauli repulsion, and attractive orbital interactions, with the addition of the term ∆Edisp to account for dispersion correction:
The term ∆Velstat usually refers to the classical electrostatic interactions between the unaltered charge distributions of the prepared fragments. The Pauli repulsion, ∆EPauli, is responsible for any steric repulsion and consists of the destabilizing interactions between occupied orbitals. The orbital interactions, ∆Eoi, account for electron-pair bonding, charge transfer interactions, and polarization. The term ∆Edisp accounts for the dispersion corrections [50].
Analysis of excited states
Exciton delocalization and charge transfer in donor–acceptor complexes were quantitatively analyzed using the transition density [51,52,53]. The analysis was carried out in the more convenient Löwdin orthogonalized basis. The matrix λC of orthogonalized MO coefficients is obtained from the coefficients C in the original basis λC = S1/2 C, where S is the atomic orbital overlap matrix. The transition density matrix T0i for an excited state Φi constructed as a superposition of singly excited configurations (where an occupied MO ψi is replaced by a virtual MO ψa) is computed,
where Aij→a is the expansion coefficient of the j→a configuration in excited state Φi.
A key quantity \(\Omega\)(D,A) is given by
The weights of local excitations on donor (D) and acceptor (A) are Ω(D,D) and Ω(A,A). The weight of electron transfer configurations D → A and A → D is represented by Ω(D,A) and Ω(A,D), respectively. The index Δq, which describes charge separation and charge transfer between D and A, is calculated as
Solvent effects
A COSMO-like polarizable continuum model [54,55,56] in the monopole approximation was used to estimate the equilibrium solvation energy \(E_{{\text{S}}}^{{{\text{eq}}}}\) of a molecule (in the ground or excited state) in the medium with the dielectric constant ε:
where f(ε) is the dielectric scaling factor, \(f(\varepsilon ) = \frac{\varepsilon - 1}{\varepsilon }\), Q is the vector of n atomic charges in the molecular system, and D is the n × n symmetric matrix determined by the shape of the boundary surface between solute and solvent. D = B+A−1B, where the m × m matrix A describes electrostatic interaction between m surface charges and the m × n B matrix describes the interaction of the surface charges with n atomic charges of the solute [54]. The molecular boundary surface was built using the GEPOL93 scheme [57].
The charge on atom X in the excited state Φi, \(q_{X}^{i}\), is calculated as:
where \(q_{X}^{0}\) is the atomic charge on X in the ground state and \(\Delta_{X}^{i}\) is the change caused by the excitation ψ0 → ψi that causes the redistribution of electron density between the atoms X and the rest of the atoms Y.
The non-equilibrium solvation energy for excited state ψi can be calculated as [58]:
In Eq. (10), n2 is the refraction index squared, ε is the optical dielectric constant of the medium, and the vector Δ describes the change of atomic charges in the molecule by excitation in terms of atomic charges; see Eq. (9).
Electron transfer rates
The nonadiabatic electron transfer rate, kET, can be calculated using the electronic coupling squared, V2, and the Franck–Condon weighted density of states (FCWD):
that accounts for the overlap of donor and acceptor vibrational states, and can be approximated using the classical Marcus equation [59]:
where λ is the reorganization energy and ∆G0 is the standard Gibbs energy change of the process. The electronic couplings between the LE, CT, and GS states were calculated within TDA using the fragment charge difference (FCD) method [60, 61].
The Marcus expression is derived for the high-temperature condition, \(\mathrm{\hslash }{\omega }_{l}\ll kT\), for all vibrational modes l. The semi-classical description of ET [62, 63] includes the effect of the quantum vibrational modes in an effective way, the solvent (low frequency) modes are treated classically, while a single high-frequency intramolecular mode, \({\omega }_{i}, \mathrm{\hslash }{\omega }_{i}\gg kT\), is described quantum mechanically. The rate k can be written as the sum of all channels connecting the initial state with the vibrational quantum number n = 0 to various vibrational levels of the final state, because ET generally occurs from the lowest vibrational level of the initial state.
with
An effective value of the Huang–Rhys factor S is estimated from the internal reorganization energy λi as
\(S = \lambda_{i} /\hbar \omega_{i}\). As can be observed, the semi-classical expression includes an additional parameter (in comparison to the Marcus equation): the frequency ωi of a vibrational mode that accurately captures the nuclear intramolecular relaxation following the ET.
Electronic coupling
The fragment charge difference method (FCD) [60, 61] was used to derive the coupling between diabatic states of interest from related adiabatic excited states computed using TDA approach [64]. Within the two-state model, the D–A coupling reads:
where (Ei – Ej) is vertical excitation energy, Δqi and Δqj are the difference in the donor and acceptor charges in the adiabatic states Φi and Φj, respectively, and Δqij is the charge difference computed from the Φi → Φj transition density matrix.
Reorganization energy
The reorganization energy was computed at the BLYP-D3(BJ)/def2-SVP level. It is usually divided into two parts, λ = λi + λs, including the internal and solvent terms. The solvent reorganization energy is the amount of energy required to move solvent molecules from their initial-state location to their CT state location without charge transfer. The λs for a particular CT states was computed as a difference between the equilibrium (Eeq, see Eq. 8) and non-equilibrium (Eneq, see Eq. 10) solvation energies for states of interest. The internal reorganization energy λi is the energy of structural changes that occur when donor/acceptor fragments go from initial-state geometries to final-state geometries.
Results and discussions
Ground state structure and properties
A unique peanut-like topology of the studied molecules as well as their ability to form inclusion complexes with fullerenes both in solution and in the solid state encouraged us to study in detail the binary and ternary complexes of the TwinX with one or two C60 fullerene molecules. To estimate the stability of the complexes, the interaction energy (∆Eint) between the twin nanorings and fullerene was calculated. The obtained values for mono (Twin1 ⊃ C60, Twin2 ⊃ C60, and Twin3 ⊃ C60) and bis (Twin1 ⊃ 2C60, Twin2 ⊃ 2C60, and Twin3 ⊃ 2C60) adducts are shown in Table 1. As previously demonstrated for similar systems, dispersion interactions play a significant role in the stability of such complexes [65, 66]. To confirm this, we used a Morokuma-type method of energy decomposition analysis (EDA) [67,68,69].
As can be seen in Table 1, the destabilizing term (Pauli repulsion) decreases when moving from Twin1 ⊃ 2C60 to Twin3 ⊃ 2C60 complex. The dispersion term dominates the binding forces (electrostatic, orbital, and dispersion interactions) and accounts for approximately 61% of the total. It is followed by the electrostatic (about 26%) and orbital (about 13%) interactions. Similar behavior has been observed in other van der Waals complexes of C60 with cycloparaphenylenes [66].
We compared the geometries of the complexes to explain the observed differences. The guest molecule (C60) is the same in all complexes. Its effective radius Reff (the mean distance from center to each atom) is 5.150 Å. The size of the host molecules, however, differs significantly. Let us first compare the mono adduct complexes Twin1 ⊃ C60, Twin2 ⊃ C60, and Twin3 ⊃ C60. As seen in Fig. 1, both wings of Twin1 are in the shape of a circle, whereas Twin2 and Twin3 have a distinct ellipsoid shape with wings extending away from the linker. The structures of the double nanohoops and their geometrical parameters are provided in Fig. S1, SI. In Twin1 ⊃ C60, the C60 fullerene is surrounded by ten phenylene units at a distance of 6.879 Å. The comparison of Twin1 ⊃ C60 with the [10]CPP ⊃ C60 complex, in which both units are perfectly size matched, demonstrates a high similarity of the effective radius (Reff = 6.867 Å in [10]CPP ⊃ C60), and the interaction energy (ΔEint = − 56.8 kcal/mol for [10]CPP ⊃ C60). In both Twin2 ⊃ C60 and Twin3 ⊃ C60, C60 interacts with only eight phenylene units at the effective distance of about 6.9 Å (Fig. S2, SI). Thus, the weaker interactions in Twin2 ⊃ C60 and Twin3 ⊃ C60 can be explained by the smaller number of phenylene units available for interaction with C60. The higher ΔEint values in Twin1 compared to Twin3 are consistent with the larger association constants of the former.
The relationship between nanohoop size and the interaction energy found for the mono adduct is also valid for Twin1 ⊃ 2C60, Twin2 ⊃ 2C60, and Twin3 ⊃ 2C60. The formation energy of the ternary complexes can be expected to be superadditive due to the additional interactions between two fullerenes, similar to that observed in the complexes of fullerenes with other curved carbon nanostructures [70, 71]. However, the distance between fullerenes in these complexes is too large (13.590 Å, 27.047 Å, and 26.694 Å for Twin1 ⊃ 2C60, Twin2 ⊃ 2C60, and Twin3 ⊃ 2C60) for such interactions (Fig. S3, SI). This is confirmed by the calculated ∆Eint (Table 1), which is less than − 0.5 kcal/mol.
The non-covalent interaction index (NCI) was used to describe the topology of the host–guest interactions [72]. The shape of NCI isosurfaces clearly demonstrates the difference between the intermolecular interactions in Twin1 ⊃ C60, Twin2 ⊃ C60, and Twin3 ⊃ C60. In the case of Twin1 ⊃ C60, the isosurface is distributed fairly uniformly between C60 and one of the nanohoops. In turn, in Twin2 ⊃ C60 and Twin3 ⊃ C60, the isosurface surrounds the fullerene only partially, indicating interactions with eight out of ten phenyl rings. Figures S4–S7 in SI provide the NCI isosurfaces and reduced density gradient (RDG) plots for mono and bis adducts.
The topological study based on Bader's Atoms in Molecules theory [43] provides more details regarding the host–guest interactions in the complexes. Table S1 in SI show the electron density, its Laplacian, and other topological parameters at bond critical points (BCPs). The analysis confirmed that the interactions between the fragments in both binary and ternary complexes correspond mostly to π···π interactions. When moving from Twin1 to Twin3-based complexes, the number of BCPs notably decreases. For example, the number of BCPs between the nanohoops and C60 in Twin1 ⊃ C60, Twin2 ⊃ C60, and Twin3 ⊃ C60 complexes is 22, 16, and 11, respectively. Only a few of them correspond to CH···π interactions. The number of BCPs and characteristics of the electron density at these points are in agreement with the interaction energy values of the complexes. The nature of non-covalent interactions in ternary Twin1 ⊃ 2C60, Twin2 ⊃ 2C60, and Twin3 ⊃ 2C60 complexes is similar to the nature in binary complexes. Figures S8 and S9 in SI show the QTAIM molecular graphs for each complex studied.
Let us compare the orbital energies of the host and guest molecules and their complexes. All complexes have similar HOMO and LUMO energies, as shown in Fig. 2. The HOMO is localized on the nanohoop, while the LUMO is localized on the fullerene unit (Fig. 3). The HOMO energies of nanohoops remain almost unchanged (within 0.1 eV) upon complex formation. A shift in the LUMO energy of the fullerene is less than 0.2 eV. These small differences in orbital energies when going from the individual molecules to their complex indicate the absence of notable changes in the fragment structures as well as the lack of charge separation between the units in the ground state (GS), in accordance with the low contribution of the ΔEoi term to the bonding interaction (vide supra).

Structure and HOMO/LUMO energy levels of the studied binary and ternary complexes

Localization of Kohn–Sham HOMO and LUMO in the TwinX ⊃ C60 complexes
The deformation energy (ΔEdef) was calculated to quantify structural changes. The greatest ΔEdef = 6.5 kcal/mol was found for Twin1 ⊃ C60, with a dominant contribution from the nanohoop. In other complexes, both fullerene and nanohoop fragments make similar contribution (Table S2, SI). As seen in Fig. S10, the deformations in Twin1 ⊃ C60 are mainly associated with changes in the twist angle of the phenylene units of the nanohoops. In Twin3 ⊃ C60, the changes in the dimensions of the nanohoop wings are caused by the interaction with fullerene. Twin2 ⊃ C60 has the smallest ΔEdef, which correlates with the smallest root-mean-square deviation (RMSD) of atomic positions. Population analysis performed with several charge schemes shows no significant charge transfer between the host and guest molecules in the complexes (Table S3, SI).
Singlet excited states
PET properties of the nanohoop are dependent on both their structural (size of wing and type of linker) and electronic (HOMO energy) characteristics. Each system was divided into two (for TwinX ⊃ C60) or three (for TwinX ⊃ 2C60) fragments: nanohoop, acting as an electron donor; and C60 fullerene, acting as an electron acceptor. The lowest 80 excited states were investigated for their contribution to charge transfer and exciton delocalization (Table 2).
Three types of the excited states were identified: (1) locally excited (LE) states are those in which excitation is localized on either the fullerene (LEGuest) or the nanohoop (LEHost) and charge transfer between the units is less than 0.1 eV (CT < 0.1 e); (2) charge transfer (CT) states with high charge separation (CT > 0.8 e); and (3) mixed states, which combine the contributions of both LE and CT states (0.1 e < CT < 0.8 e).
The lowest 80 vertical singlet excitation energies of the binary complexes in the gas phase range from 2.50 to 4.37 eV. In all complexes, the lowest-lying excited states are localized on the fullerene unit (LEGuest). Excited states localized on the twin nanohoops (LEHost) are higher in energy compared to LEGuest. The lowest LEHost state was found in Twin2 ⊃ C60, with a small energy difference between LEGuest and LEHost of 0.36 eV. For Twin1 ⊃ C60 and Twin3 ⊃ C60, such a difference is 0.73 and 0.92 eV correspondingly (Table 2). Among the excited states studied, two types of CT states have been found. Both are generated by electron transfer from TwinX to C60 and can be denoted as TwinX+ ⊃ C60−. For the first CT type, the dipole moment difference between GS and CT states ranges between 4 and 14 D, while the second type of CT is characterized by a significantly stronger dipole moment difference (56.7 and 39.9 D in Twin2 ⊃ C60 and Twin3 ⊃ C60, respectively). It is important to note that the CT2 state was not found in Twin1 ⊃ C60. Since Twin1 has the benzene ring linker, unlike the other complexes with extended π-conjugated linkers, the formation of the CT2 state could be related to electron transfer from the linker to the fullerene. The low-lying CT2 state is not observed in Twin1 ⊃ C60 complex, because the HOMO energy of the benzene linker is significantly lower compared to the HOMOs of peropyrene and cyclooctatetrathiophene. Moreover, the higher energy of CT2 state in Twin3 ⊃ C60 can be explained by the lower HOMO energy of cyclooctatetrathiophene fragment compared to CPP fragment (see Fig. 2 and Table S4, SI). We repeated the investigation of the excited states dividing the TwinX ⊃ C60 complexes into three fragments (fullerene, linker, and CPP wings) to gain further insight. We have found that in the Twin2 ⊃ C60 complex, electron transfer in CT2 occurs exclusively from the linker to C60. The two types of CT states in Twin2 ⊃ C60 are shown in Fig. 4.

NTO orbitals for two types of CT states in Twin2 ⊃ C60
The nature of CT1 and CT2 states in Twin3 ⊃ C60 is similar to that in Twin2 ⊃ C60. Thus, CT1 is generated by electron transfer from the CPP wing of TwinX to the fullerene unit, while the case of CT2 a dominant involvement of the linker in electron transfer is observed. As expected, no CT2 state was found in Twin1 ⊃ C60. Figures S11–S13 in SI depict natural transition orbitals (NTOs) for the LE and CT states.
Behavior of the ternary complexes TwinX ⊃ 2C60 is similar to the binary ones. Due to a higher density of states, the energies of the lowest 80 excited singlet states in the systems range from 2.48 to 3.80 eV. The energies of both LEGuest and LEHost states in the ternary and binary complexes are very similar. A qualitative difference between Twin1 ⊃ C60 and Twin1 ⊃ 2C60 was found for the LEHost states (see Figs. S11 and S14, SI). In Twin1 ⊃ C60, LEHost is only on one CPP wing, while in Twin1 ⊃ 2C60 the LEHost is delocalized over the entire Twin1 molecule. In the binary system, two CPP wings are not structurally similar—one contains fullerene and the other does not. Interaction with C60 increases the energy of the orbital located at this CPP, resulting in LEHost being located only on this wing. In Twin1 ⊃ 2C60, both CPP wings are nearly equivalent and the state is delocalized over them.
In Twin2 ⊃ 2C60 and Twin3 ⊃ 2C60, two types of CT states can also be distinguished. In Twin2 ⊃ 2C60 and Twin2 ⊃ C60 their characteristics are very similar, whereas in Twin3 ⊃ 2C60 the energy of the CT2 state is 0.2 eV lower compared to Twin3 ⊃ C60. Analysis of the CT2 states in Twin3 ⊃ C60 and Twin3 ⊃ 2C60 complexes revealed that its lower energy in Twin3 ⊃ 2C60 is associated with greater delocalization of the occupied NTO along the phenyl rings of CPP (compare Figs. S13 and S16, SI). The NTOs describing LE and CT states in TwinX ⊃ 2C60 complexes are given in Figs. S14–S16 in SI.
Solvent effects on generation of CT states
It is widely accepted that solvation has a great influence on both the ground and excited states. While CT states can be highly stabilized or destabilized by the solvent, the influence of solvation on LE states is often minimal. The equilibrium COSMO-like solvation model [66, 73, 74] with dichloromethane (DCM) as the solvent was used to evaluate the effect of the solvent on the excited states. All of the investigated complexes in GS have rather small dipole moments. For binary systems, the dipole moments vary from 0.25 to 0.87 D, whereas for more symmetrical ternary complexes, these values decrease to 0.06–0.13 D. For Twin1 ⊃ C60, Twin2 ⊃ C60, and Twin3 ⊃ C60, the GS solvation energies are −0.77, −0.92, and −1.00 eV, correspondingly. The similarity in the dipole moments results in similar solvation energies. The solvation energies of the ternary analogs differ by no more than 0.05 eV (Table S5, SI).
For the LEGuest and LEHost states, the overall picture is very similar to the GS state. A comparison of excitation energies computed in the gas phase and in DCM solution shows that the LE transition energies remain almost unchanged, which in turn correlates perfectly with negligibly small changes in the dipole moment when going from GS to LE states. A difference (∆μ) of the dipole moment vector in the LE and GS states in the complexes does not exceed 1 D. The changes in the dipole moment associated with CT states are significantly larger compared to the LE states. Despite the fact that both types of CT have a similar electronic structure, their response to solvation can be different. Indeed, the solvation energies calculated for the CT states are in good agreement with the corresponding ∆μ values. For CT1 states characterized by a moderate ∆μ, the difference in solvation energies of the GS and CT states ranges from 0.18 to 0.30 eV. The CT2 states in Twin2 and Twin3 based complexes show significantly larger ∆μ values and, as a result, larger solvation energies. Figure 5 displays the energies of the GS, LE, and CT states in the gas phase and DCM. Detailed data for all complexes is collected in Table S5 in SI.

Energies of the LE and CT states (in eV) computed for the binary (TwinX ⊃ C60) and ternary (TwinX ⊃ 2C60) complexes in vacuum (VAC) and dichloromethane (DCM). In the figure, LE1 stands for LEGuest
In all cases, the solvent stabilization of the CT1 state is enough to balance the energies of the LEGuest and CT1 states. The solvation of the Twin2-based complexes was found to lower the energy of the CT2 state making it by almost 1 eV smaller than that of the LEGuest. In the case of Twin3-based complexes, the higher energy of the CT2 state in vacuum and its smaller solvation energy do not allow this state to become sufficiently low. Simulated absorption spectra for the complexes are given in Fig. S17 in SI.
Rates of charge separation and charge recombination
Because all CT states in the complexes have very weak oscillator strengths, they cannot be populated directly by light absorption. On the other hand, locally excited states populated by strongly absorbing transitions dissipates to the lowest-lying LE state by a non-radiative decay channel. In turn, the last state can decay to a lower CT states by electron transfer between the donor and acceptor sites.
Semi-classical approach proposed by Ulstrup and Jortner [62, 63] was used to estimate the rates of charge separation (kCS) and charge recombination (kCR) processes. According to this method, an effective vibrational mode is used to describe the intramolecular relaxation associated with ET. In addition, the rate is controlled by the electronic coupling between the initial and final states, Vij, solvation reorganization energy, λs, Gibbs free energy, ∆G0, and effective Huang-Rhys factor, Seff. We have previously shown that changing the effective frequency from 1400 to 1800 cm−1 does not lead to a significant change in the charge separation rate [75, 76]. Thus, the effective frequency of 1600 cm−1, which corresponds to the stretching of C=C bonds, was used to estimate the rates. Table 3 includes the computed parameters for charge separation processes in DCM solvent for the complexes of interest.
As can be seen in Table 3, the reorganization energy for the ET reactions varies significantly. The LEGuest → CT1 charge separation process in all complexes is characterized by moderate internal reorganization energies (from 0.13 to 0.15 eV) and solvation reorganization energies (from 0.16 to 0.25 eV). This process occurs in the normal Marcus regime (|∆G0|< λ) [59, 77] on the sub-nanosecond timescale. In contrast, ET parameters that control generation of the CT2 states are quite different. The LEGuest → CT2 charge separation process shows significantly higher reorganization energies. In the Twin2- and Twin3-based complexes, the solvent reorganization energy for this reaction is more than twice as high as for LEGuest → CT1 process. In Twin2 ⊃ C60 and Twin2 ⊃ 2C60 complexes, LEGuest → CT2 reaction is nearly barrierless. The characteristic time (τ) was found to be 0.05 and 0.09 ns, respectively. The CT2 charge separation in Twin3 ⊃ C60 and Twin3 ⊃ 2C60 is characterized by a strong positive Gibbs energy and thus this reaction is unlikely to occur.
Typically, excited CT states decay to the ground state by charge recombination. The effect of internal geometry reorganization on ∆G0 is rather small for large π-conjugated systems, such as the complexes studied, and can be safely ignored [65, 78]. The computed charge recombination rates (kCR) are provided in Table 4. Charge recombination rates of CT2 for Twin3 ⊃ C60 and Twin3 ⊃ 2C60 complexes were not considered because of their low probability.
In contrast to charge separation, the charge recombination reactions take place in the deep inverted Marcus region (|∆G0 |≫ λ) [59, 77]. The decay of CT1 states is significantly slower than their generation. However, the charge recombination of CT2 states in Twin2 ⊃ C60 and Twin2 ⊃ 2C60 complexes is an order of magnitude faster than the charge separation reaction. The fast decay of CT2 states prevents their experimental observation. Note that the charge recombination rates assessments are rather qualitative and should be treated with caution.
Conclusions
Using the TD-DFT approach, we investigated the ground and excited-state properties of the host–guest complexes based on three experimentally obtained double nanohoops and C60 fullerene. The energy decomposition analysis revealed that the stability of the complexes is determined by dispersion interactions between the host and guest molecules. Two types of CT states were found: CT1, generated by electron transfer from CPP to C60, and CT2, in which an electron is transferred from the linker to C60. Efficient population of the CT2 state is possible only for Twin2-based complexes due to the peropyrene linker. However, the rapid deactivation of this state is a significant disadvantage that prevents its experimental observation. We demonstrated that the efficient photoinduced electron transfer from the CPP fragment to C60 occurs on sub-nanosecond timescale. Sufficiently slow charge recombination processes found for the CT1 states lead to an efficient separation of electrons and holes, making the complexes attractive candidates for use in organic photovoltaics.
References
Rickhaus, M., Mayor, M., Juríček, M.: Chirality in curved polyaromatic systems. Chem. Soc. Rev. 46, 1643–1660 (2017)
Majewski, M.A., Stępień, M.: Bowls, hoops, and saddles: synthetic approaches to curved aromatic molecules. Angew. Chem. Int. Ed. 58, 86–116 (2019)
Cheung, K.Y., Segawa, Y., Itami, K.: Synthetic strategies of carbon nanobelts and related belt-shaped polycyclic aromatic hydrocarbons. Chem. Eur. J. 26, 14791–14801 (2020)
Xu, Y., von Delius, M.: The supramolecular chemistry of strained carbon nanohoops. Angew. Chem. Int. Ed. 59, 559–573 (2020)
Lewis, S.E.: Cycloparaphenylenes and related nanohoops. Chem. Soc. Rev. 44, 2221–2304 (2015)
Hermann, M., Wassy, D., Esser, B.: Conjugated nanohoops incorporating donor, acceptor, hetero- or polycyclic aromatics. Angew. Chem. Int. Ed. 60, 15743–15766 (2021)
Leonhardt, E.J., Jasti, R.: Emerging applications of carbon nanohoops. Nat. Rev. Chem. 3, 672–686 (2019)
Lv, Y., Lin, J., Song, K., Song, X., Zang, H., Zang, Y., Zhu, D.: Single cycloparaphenylene molecule devices: achieving large conductance modulation via tuning radial π-conjugation. Sci. Adv. 7, eabk3095 (2021)
Maust, R.L., Li, P., Shao, B., Zeitler, S.M., Sun, P.B., Reid, H.W., Zakharov, L.N., Golder, M.R., Jasti, R.: Controlled polymerization of norbornene cycloparaphenylenes expands carbon nanomaterials design space. ACS Cent. Sci. 7, 1056–1065 (2021)
Iwamoto, T., Watanabe, Y., Sadahiro, T., Haino, T., Yamago, S.: Size-selective encapsulation of C60 by [10]Cycloparaphenylene: formation of the shortest fullerene-peapod. Angew. Chem. Int. Ed. 50, 8342–8344 (2011)
Nakanishi, Y., Omachi, H., Matsuura, S., Miyata, Y., Kitaura, R., Segawa, Y., Itami, K., Shinohara, H.: Size-selective complexation and extraction of endohedral metallofullerenes with cycloparaphenylene. Angew. Chem. Int. Ed. 53, 3102–3106 (2014)
Minameyer, M.B., Xu, Y., Frühwald, S., Görling, A., von Delius, M., Drewello, T.: Investigation of cycloparaphenylenes (CPPs) and their noncovalent ring-in-ring and fullerene-in-ring complexes by (matrix-assisted) laser desorption/ionization and density functional theory. Chem. Eur. J. 26, 8729–8741 (2020)
González-Veloso, I., Cabaleiro-Lago, E.M., Rodríguez-Otero, J.: Fullerene size controls the selective complexation of [11]CPP with pristine and endohedral fullerenes. Phys. Chem. Chem. Phys. 20, 11347–11358 (2018)
Lu, D., Huang, Q., Wang, S., Wang, J., Huang, P., Du, P.: The supramolecular chemistry of cycloparaphenylenes and their analogs. Front. Chem. 7, 668 (2019).
Wang, J., Zhang, X., Jia, H., Wang, S., Du, P.: Large π-extended and curved carbon nanorings as carbon nanotube segments. Acc. Chem. Res. 54, 4178–4190 (2021)
Zhang, X., Shi, H., Zhuang, G., Wang, S., Wang, J., Yang, S., Shao, X., Du, P.: A highly strained all-phenylene conjoined bismacrocycle. Angew. Chem. Int. Ed. 60, 17368–17372 (2021)
Yang, Y., Blacque, O., Sato, S., Juríček, M.: Cycloparaphenylene-phenalenyl radical and its dimeric double nanohoop. Angew. Chem. Int. Ed. 60, 13529–13535 (2021)
Yang, Y., Huangfu, S., Sato, S., Juríček, M.: Cycloparaphenylene double nanohoop: structure, lamellar packing, and encapsulation of C60 in the solid state. Org. Lett. 23, 7943–7948 (2021)
Zhan, L., Dai, C., Zhang, G., Zhu, J., Zhang, S., Wang, H., Zeng, Y., Tung, C.-H., Wu, L.-Z., Cong, H.: A conjugated figure-of-eight oligoparaphenylene nanohoop with adaptive cavities derived from cyclooctatetrathiophene core. Angew. Chem. Int. Ed. 61, e202113334 (2022)
Neese, F.: Software update: the ORCA program system, version 4.0. Wiley Interdiscip. Rev. Comput. Mol. Sci. 8, e1327 (2018)
Becke, A.D.: Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A 38, 3098–3100 (1988)
Lee, C., Yang, W., Parr, R.G.: Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 37, 785–789 (1988)
Weigend, F., Ahlrichs, R.: Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 7, 3297–3305 (2005)
Weigend, F.: Accurate Coulomb-fitting basis sets for H to Rn. Phys. Chem. Chem. Phys. 8, 1057–1065 (2006)
Eichkorn, K., Treutler, O., Öhm, H., Häser, M., Ahlrichs, R.: Auxiliary basis sets to approximate Coulomb potentials. Chem. Phys. Lett. 240, 283–290 (1995)
Eichkorn, K., Weigend, F., Treutler, O., Ahlrichs, R.: Auxiliary basis sets for main row atoms and transition metals and their use to approximate Coulomb potentials. Theor. Chem. Acc. 97, 119–124 (1997)
Grimme, S., Antony, J., Ehrlich, S., Krieg, H.: A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 132, 154104 (2010)
Grimme, S., Ehrlich, S., Goerigk, L.: Effect of the damping function in dispersion corrected density functional theory. J. Comp. Chem. 32, 1456–1465 (2011)
Brauer, B., Kesharwani, M.K., Kozuch, S., Martin, J.M.L.: The S66x8 benchmark for noncovalent interactions revisited: explicitly correlated ab initio methods and density functional theory. Phys. Chem. Chem. Phys. 18, 20905–20925 (2016)
Sedlak, R., Janowski, T., Pitoňák, M., Řezáč, J., Pulay, P., Hobza, P.: Accuracy of quantum chemical methods for large noncovalent complexes. J. Chem. Theory Comput. 9, 3364–3374 (2013)
Wong, B.M., Hsieh, T.H.: Optoelectronic and excitonic properties of oligoacenes: substantial improvements from range-separated time-dependent density functional theory. J. Chem. Theory Comput. 6, 3704–3712 (2010)
Imamura, Y., Kobayashi, R., Nakai, H.: Construction of orbital-specific hybrid functional by imposing the linearity condition for orbital energies in density functional theory. Procedia Comput. Sci. 4, 1151–1156 (2011)
Besalú-Sala, P., Voityuk, A.A., Luis, J.M., Solà, M.: Evaluation of charge-transfer rates in fullerene-based donor–acceptor dyads with different density functional approximations. Phys. Chem. Chem. Phys. 23, 5376–5384 (2021)
Hirata, S., Head-Gordon, M.: Time-dependent density functional theory within the Tamm-Dancoff approximation. Chem. Phys. Lett. 314, 291–299 (1999)
Yanai, T., Tew, D.P., Handy, N.C.: A new hybrid exchange–correlation functional using the Coulomb-attenuating method (CAM-B3LYP). Chem. Phys. Lett. 393, 51–57 (2004)
Frisch, M.J., Trucks, G.W., Schlegel, H.B., Scuseria, G.E., Robb, M.A., Cheeseman, J.R., Scalmani, G., Barone, V., Petersson, G.A., Nakatsuji, H., Li, X., Caricato, M., Marenich, A.V., Bloino, J., Janesko, B.G., Gomperts, R., Mennucci, B., Hratchian, H.P., Ortiz, J.V., Izmaylov, A.F., Sonnenberg, J.L., Williams, Ding, F., Lipparini, F., Egidi, F., Goings, J., Peng, B., Petrone, A., Henderson, T., Ranasinghe, D., Zakrzewski, V.G., Gao, J., Rega, N., Zheng, G., Liang, W., Hada, M., Ehara, M., Toyota, K., Fukuda, R., Hasegawa, J., Ishida, M., Nakajima, T., Honda, Y., Kitao, O., Nakai, H., Vreven, T., Throssell, K., Montgomery Jr., J.A., Peralta, J.E., Ogliaro, F., Bearpark, M.J., Heyd, J.J., Brothers, E.N., Kudin, K.N., Staroverov, V.N., Keith, T.A., Kobayashi, R., Normand, J., Raghavachari, K., Rendell, A.P., Burant, J.C., Iyengar, S.S., Tomasi, J., Cossi, M., Millam, J.M., Klene, M., Adamo, C., Cammi, R., Ochterski, J.W., Martin, R.L., Morokuma, K., Farkas, O., Foresman, J.B., Fox, D.J.: Gaussian 16 Rev. A.03, Wallingford (2016)
Mulliken, R.S.: Electronic population analysis on LCAO–MO molecular wave functions. I. J. Chem. Phys. 23, 1833–1840 (1955)
Mulliken, R.S.: Electronic population analysis on LCAO–MO molecular wave functions. II. Overlap populations, bond orders, and covalent bond energies. J. Chem. Phys. 23, 1841–1846 (1955)
Löwdin, P.O.: On the non-orthogonality problem connected with the use of atomic wave functions in the theory of molecules and crystals. J. Chem. Phys. 18, 365–375 (1950).
Hirshfeld, F.L.: Bonded-atom fragments for describing molecular charge densities. Theor. Chim. Acta 44, 129–138 (1977)
Marenich, A.V., Jerome, S.V., Cramer, C.J., Truhlar, D.G.: Charge model 5: an extension of Hirshfeld population analysis for the accurate description of molecular interactions in gaseous and condensed phases. J. Chem. Theory Comput. 8, 527–541 (2012)
Baerends, E.J., et al. ADF 2018, SCM, Theoretical Chemistry, Vrije Universiteit, Amsterdam, The Netherlands. http://www.scm.com
Bader, R.F.W.: A quantum theory of molecular structure and its applications. Chem. Rev. 91, 893–928 (1991)
AIMAll (Version 14.06.21), T. A. K., TK Gristmill Software, Overland Park (2019) (aim.tkgristmill.com)
Luzanov, A.V., Sukhorukov, A.A., Umanskii, V.É.: Application of transition density matrix for analysis of excited states. Theor. Exp. Chem. 10, 354–361 (1976)
Head-Gordon, M., Grana, A.M., Maurice, D., White, C.A.: Analysis of electronic transitions as the difference of electron attachment and detachment densities. J. Phys. Chem. 99, 14261–14270 (1995)
Zhurko, G.A.: Chemcraft 1.80 (build 523b) - graphical program for visualization of quantum chemistry computations. https://chemcraftprog.com
Ziegler, T., Rauk, A.: On the calculation of bonding energies by the Hartree Fock Slater method. Theor. Chim. Acta 46, 1–10 (1977)
Ziegler, T., Rauk, A.: Carbon monoxide, carbon monosulfide, molecular nitrogen, phosphorus trifluoride, and methyl isocyanide as σ donors and π acceptors. A theoretical study by the Hartree-Fock-Slater transition-state method. Inorg. Chem. 18, 1755–1759 (1979)
Grimme, S.: Accurate description of van der Waals complexes by density functional theory including empirical corrections. J. Comput. Chem. 25, 1463–1473 (2004)
Plasser, F., Lischka, H.: Analysis of excitonic and charge transfer interactions from quantum chemical calculations. J. Chem. Theory Comput. 8, 2777–2789 (2012)
Plasser, F., Bäppler, S.A., Wormit, M., Dreuw, A.: New tools for the systematic analysis and visualization of electronic excitations. II. Applications. J. Chem. Phys. 141, 024107 (2014)
Luzanov, A.V., Zhikol, O.A.: Electron invariants and excited state structural analysis for electronic transitions within CIS, RPA, and TDDFT models. Int. J. Quantum Chem. 110, 902–924 (2010)
Klamt, A., Schüürmann, G.: COSMO: a new approach to dielectric screening in solvents with explicit expressions for the screening energy and its gradient. J. Chem. Soc., Perkin Trans. 2, 799–805 (1993)
Tomasi, J., Mennucci, B., Cammi, R.: Quantum mechanical continuum solvation models. Chem. Rev. 105, 2999–3094 (2005)
Voityuk, A.A., Vyboishchikov, S.F.: A simple COSMO-based method for calculation of hydration energies of neutral molecules. Phys. Chem. Chem. Phys. 21, 18706–18713 (2019)
Pascual-ahuir, J.L., Silla, E., Tuñon, I.: GEPOL: an improved description of molecular surfaces. III. A new algorithm for the computation of a solvent-excluding surface. J. Comput. Chem. 15, 1127–1138 (1994)
Klamt, A.: Calculation of UV/Vis spectra in solution. J. Phys. Chem. 100, 3349–3353 (1996)
Marcus, R.A., Sutin, N.: Electron transfers in chemistry and biology. Biochim. Biophys. Acta Rev. Bioenerg. 811, 265–322 (1985)
Voityuk, A.A., Rösch, N.: Fragment charge difference method for estimating donor–acceptor electronic coupling: application to DNA π-stacks. J. Chem. Phys. 117, 5607–5616 (2002)
Voityuk, A.A.: Electronic coupling for charge transfer in donor–bridge–acceptor systems. Performance of the two-state FCD model. Phys. Chem. Chem. Phys. 14, 13789–13793 (2012)
Ulstrup, J., Jortner, J.: The effect of intramolecular quantum modes on free energy relationships for electron transfer reactions. J. Chem. Phys. 63, 4358–4368 (1975)
Jortner, J.: Temperature dependent activation energy for electron transfer between biological molecules. J. Chem. Phys. 64, 4860–4867 (1976)
Voityuk, A.A.: Estimation of electronic coupling for photoinduced charge separation and charge recombination using the fragment charge difference method. J. Phys. Chem. C 117, 2670–2675 (2013)
Stasyuk, A.J., Stasyuk, O.A., Solà, M., Voityuk, A.A.: Photoinduced electron transfer in nanotube⊃C70 inclusion complexes: phenine vs. nanographene nanotubes. Chem. Commun. 56, 12624–12627 (2020)
Stasyuk, O.A., Stasyuk, A.J., Solà, M., Voityuk, A.A.: [10]CPP-based inclusion complexes of charged fulleropyrrolidines. Effect of the charge location on the photoinduced electron transfer. Chem. Eur. J. 27, 8737–8744 (2021)
Morokuma, K.: Molecular orbital studies of hydrogen bonds. III. C=O···H–O Hydrogen Bond in H2CO···H2O and H2CO···2H2O. J. Chem. Phys. 55, 1236–1244 (1971).
Fernández, I., Bickelhaupt, F.M.: The activation strain model and molecular orbital theory: understanding and designing chemical reactions. Chem. Soc. Rev. 43, 4953–4967 (2014)
Zhao, L., von Hopffgarten, M., Andrada, D.M., Frenking, G.: Energy decomposition analysis. Wiley Interdiscip. Rev. Comput. Mol. Sci. 8, e1345 (2018)
Stasyuk, A.J., Stasyuk, O.A., Solà, M., Voityuk, A.A.: Photoinduced charge shift in Li+-doped giant nested fullerenes. J. Phys. Chem. C 123, 16525–16532 (2019)
Zank, S., Fernández-García, J.M., Stasyuk, A.J., Voityuk, A.A., Krug, M., Solà, M., Guldi, D.M., Martín, N.: Initiating electron transfer in doubly curved nanographene upon supramolecular complexation of C60. Angew. Chem. Int. Ed. 61, e202112834 (2022)
Johnson, E.R., Keinan, S., Mori-Sánchez, P., Contreras-García, J., Cohen, A.J., Yang, W.: Revealing noncovalent interactions. J. Am. Chem. Soc. 132, 6498–6506 (2010)
Stasyuk, A.J., Stasyuk, O.A., Solà, M., Voityuk, A.A.: Hypsochromic solvent shift of the charge separation band in ionic donor–acceptor Li+@C60⊂[10]CPP. Chem. Commun. 55, 11195–11198 (2019)
Izquierdo, M., Platzer, B., Stasyuk, A.J., Stasyuk, O.A., Voityuk, A.A., Cuesta, S., Solà, M., Guldi, D.M., Martín, N.: All-fullerene electron donor-acceptor conjugates. Angew. Chem. Int. Ed. 58, 6932–6937 (2019)
Stasyuk, A.J., Stasyuk, O.A., Solà, M., Voityuk, A.A.: Triquinoline-versus fullerene-based cycloparaphenylene ionic complexes: comparison of photoinduced charge-shift reactions. Chem. Eur. J. 26, 10896–10902 (2020)
Stasyuk, O.A., Stasyuk, A.J., Solà, M., Voityuk, A.A.: Photoinduced electron transfer in nano-Saturn complexes of fullerene. Phys. Chem. Chem. Phys. 23, 2126–2133 (2021)
Suppan, P.: The Marcus inverted region. In: Mattay, J. (ed.) Photoinduced Electron Transfer IV, pp. 95–130. Springer Berlin Heidelberg, Berlin, Heidelberg (1992)
Stasyuk, A.J., Stasyuk, O.A., Solà, M., Voityuk, A.A.: Photoinduced electron transfer in mechanically interlocked suit[3]ane systems. J. Mat. Chem. C 9, 9436–9445 (2021)
Acknowledgements
We acknowledge financial support from the Spanish MINECO (Network RED2018-102815-T, project PID2020-113711GB-I00, and Juan de la Cierva contract IJC2019-039846-I to A.J.S.), the Catalan DIUE (2017SGR39), and the University of Girona (POSTDOC-UdG 2021/31 to O.A.S.). This research was supported in part by PLGrid Infrastructure.
Funding
Open Access funding provided thanks to the CRUE-CSIC agreement with Springer Nature. Ministerio de Ciencia e Innovación (Grant nos: RED2018-102815-T, PID2020-113711 GB-I00, IJC2019-039846-I); Departament d'Innovació, Universitats i Empresa, Generalitat de Catalunya (Grant no. 2017SGR39); Universitat de Girona (Grant no. POSTDOC-UdG 2021/31).
Author information
Authors and Affiliations
Contributions
OAS and AJS: investigation, formal analysis, writing—original draft, writing—review and editing. MS: supervision, writing—review and editing, funding acquisition. AAV: supervision, writing—review and editing.
Corresponding authors
Ethics declarations
Conflict of interest
There are no conflicts to declare.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Stasyuk, O.A., Stasyuk, A.J., Solà, M. et al. Photoinduced electron transfer in host–guest complexes of double nanohoops. J Nanostruct Chem 14, 293–306 (2024). https://doi.org/10.1007/s40097-022-00518-w
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40097-022-00518-w