
Abstract
Nanoclusters of nickel sulphide (NiS) n and iron sulphide (FeS) n for n = 3–5 are studied using B3LYP exchange correlation function with 6-31G as a basis set. The structural stability of different isomers of NiS and FeS is analysed with the optimized energy, binding energy and vibrational studies. The electronic properties of isomers are discussed in terms of HOMO–LUMO gap, ionization potential, electron affinity, and embedding energy of different clusters. Based on the calculated energy, planar ring, linear ladder and bipyramidal cube of NiS and FeS nanoclusters are found to be more stable. The increase in number of atoms in the clusters leads to increase in its stability. The dipole moment is high for planar rhombus and linear ladder structures of NiS and FeS nanoclusters. The ionization potential and electron affinity are high for planar ring structure of NiS and FeS clusters. The value of energy gap of linear ladder NiS nanocluster and cube and bipyramidal cube structures of FeS is found to be low. The binding energy for cube structure of NiS and FeS clusters is found to be high. Hexagonal ring structure of NiS and FeS clusters has low embedding energy value. The other parameters such as dipole moment and point symmetry are also discussed. The structural stability and electronic properties will provide an insight for experimentalist to tailor new materials that have potential importance in engineering applications.
Similar content being viewed by others
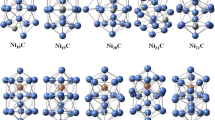

Background
Chalcogenides are an important class of materials which have significant electrical properties, optical properties and chemical characteristics. Chalcogen includes sulphur, selenium and tellurium elements. The sulphur compounds have a wide range of properties which attract the scientific community, predominantly in thin film technology and nanoparticle synthesis. The applications of chalcogenide materials include a variety of chalcogenide glasses, infrared sensors, solar energy conversion and window layer [1–6]. Various methods are utilized for the synthesis of binary, ternary and quaternary chalcogenides.
In the family of chalcogenide nanomaterials, nickel sulphide (NiS) is one of the important materials which find its potential application in photoconduction [7]. Han et al. [8, 9] reported NiS as an attractive cathode material for rechargeable lithium batteries and charge–discharge mechanism of NiS as a cathode material. Fernandez et al. [10] studied ZnS–NiS–CuS optical filters with wide-range solar control characteristics and as a catalyst [11]. Different morphologies of NiS can be prepared by various methods: Shen et al. [12] reported phase-controlled synthesis of nickel sulphide nanorods; Wang et al. [13] synthesized NiS nanotubes using self-assembly route in AAO templates; Chen et al. [14] prepared NiS nanotubes and nanoneedles derived from rolled nanosheets from microemulsion; and Huang et al. [15] reported NiS with a core–shell structure. NiS can be synthesized using solvothermal method [16] and hydrothermal process [17].
Iron sulphide (FeS) has importance in the function of electron-transfer groups in biological applications such as enzyme catalysis, protein folding and metabolic regulation, photosynthesis and respiratory chain. Esrafili et al. [18], studied electronic structure of small Fe m S n nanoclusters; Henderson et al. [19] reported FeS-based materials for groundwater remediation; Ritchie et al. [20] lithium-ion/iron sulphide rechargeable batteries; and Gomes et al. [21] synthesized FeS by electrodeposition method.
The motivation of the present work is to tailor proper structure of NiS and FeS with suitable method that will enhance its structural and electronic properties with improved electrical, optical and chemical properties. With this as motivation, the survey was conducted using Thomson Reuters “web of science” database, only less work has been reported in NiS and FeS. The reported work mainly concentrates on synthesis and characterization of nickel sulphide and iron sulphide. The quantum chemical studies on these chalcogenide materials will fine-tune the structural and electronic properties which will be suitable for its engineering application. Using the International Centre for Diffraction Data (ICDD) data as reference, the nanoclusters are constructed; NiS is taken from card number: 89-7141 and FeS is taken from card number: 89-9021. The density functional theory (DFT) is a better approach to study the structural and electronic properties for chalcogenide materials. The electron movement is confined in nanoclusters which resemble zero-dimensional material. The structural and electronic property mainly depends on the geometry of the clusters; DFT method effectively explores the electronic properties of nanostructures. With this as the objective, some of the possible six realistic structures for each group are studied using the NWChem package and the results are analysed and reported.
Methods
The realistic clusters of NiS and FeS of different structures were completely optimized using NWChem package [31]. The optimization in this work is based on B3LYP [32–35] exchange correlation which completely exploits sulphide clusters with 6-31G as a basis set. Since, the atomic number of nickel, iron and sulphur are 28, 26 and 16, respectively; 6-31G is the suitable basis set which will optimize these clusters [36–38] and this is the reason behind the selection of this basis set. In the quantum chemical methods, the pseudo potential approximation will replace the complex field of bound electrons in the atoms and the effective potential of the nucleus is a modified potential term. NWChem utilizes Fock matrix elements, which in the case of Hartree–Fock calculation with the local basis set called Fock build is adopted; a similar algorithm of Kohn–Sham matrix elements is used to evaluate the computation in DFT calculations [39]. The convergence study was carried out with 10−5 eV with respect to energy.
Results and discussion
The realistic possible structure of sulphide clusters of NiS and FeS is designed in six different types. The hexagonal structure resembles bees hive like structure. The planar rhombus structure has rhombus structure which is separated by square. The cube structure has a cube-like arrangement. Planar ring structure has five transition metal atoms and five sulphur atoms which form a closed ring-like loop. In the linear ladder arrangement, all the atoms form a ladder-like structure. In the bipyramidal structure, the cube is projected with the pyramid at two faces. These are the different types of nanoclusters which are studied in the present work. The energy, dipole moment, point symmetry, HOMO–LUMO gap, ionization potential (IP), electron affinity (EA), binding energy (BE), embedding energy (EE) and vibration analysis of different sulphide clusters are analysed and reported.
Structures of (NiS) n
The possible geometrically optimized structures of (NiS) n are shown in the Fig. 1. The hexagonal ring and planar rhombus have the energy of −5,718.76 and −5,718.44 Hartrees, respectively, with the dipole moment of 1.118 and 3.266 Debye, respectively. Due to closed structure of hexagonal ring the dipole moment is comparatively lower than planar rhombus. The cube structure has the energy of −7,623.87 Hartrees, cube structure has low value of 0.300 Debye which is due to the ordered structure. The planar ring, linear ladder and bipyramidal cube have the energy in the order of −9,530 Hartrees. In these structures with (NiS)5, the dipole moment is large for linear ladder structure whereas, the other two are almost symmetrical and have the low value of dipole moment. The obtained energy and dipole moment of geometrically optimized clusters are tabulated in Table 1.

Structures of NiS nanoclusters
The HOMO–LUMO gap gives the electronic properties of NiS nanoclusters [22, 23]. Usually a low value of gap implies that the electron can easily move from the occupied level to the virtual level. Among all the different structures of NiS clusters, a low value is seen for linear ladder which is more suitable for electronic applications. In contrast, a high value of gap is observed for planar rhombus. In the planar rhombus Ni and S atoms are arranged symmetrically that gives rise to the high value of HOMO–LUMO gap due to overlapping of unfilled shells in d and p orbitals of Ni and S atoms. The values of HOMO–LUMO gap of NiS nanoclusters are tabulated in the Table 2.
The high value of IP implies that it is difficult to remove the electron from the cluster which will be more chemically inert [24]. Among all the structures planar ring has the high value of IP due to the closed arrangement of atoms in the ring. EA refers the amount of energy change when the electron is added to the cluster [25, 26]; a low value indicates less chemically reactive nature of the cluster. Low value of EA seen for cube structure is 2.63 eV. The IP and EA values of different clusters are shown in Fig. 2.

Variation of IP and EA with cluster type for NiS clusters
Structures of (FeS) n
The structures of (FeS) n are shown in the Fig. 3. The calculated energies and dipole moment of various FeS nanoclusters are shown in Table 3. The hexagonal ring and planar rhombus structure has energy of −4,984 Hartrees which has three Fe atoms and three sulphur in its clusters. In these structures a moderate dipole moment of 1.581 Debye is observed for hexagonal ring, whereas a high dipole moment of 4.165 Debye is seen for planar rhombus. In the cases of planar ring, linear ladder and bipyramidal cubes, all have the energy in and around −8,308 Hartrees with different dipole moments due to the positions of atoms: planar ring has a low dipole moment of 0.971 Debye, bipyramidal cube structure has a moderate value of 1.448 Debye and linear ladder has a high value of 5.111 Debye.

Structures of FeS nanoclusters
The high value of HOMO–LUMO gap is noticed for planar rhombus; the electron requires more energy to move from HOMO to LUMO level. The high value of gap in planar rhombus structures arise due to metal–metal bonding of Fe atoms which has unoccupied d shells. In the case of FeS clusters, all the clusters have a low value of gap which seems to have more metallic-like property. The HOMO–LUMO gap of FeS clusters are tabulated in the Table 4.
The high value of IP around 5.58 eV is noticed for planar ring due to the closed loop. Comparing with the IP value of NiS structures, the FeS clusters have low values of IP. A low value of EA is observed for cube and bipyramidal cube structures of FeS. The IP and EA variations for different clusters are shown in the Fig. 4.

Variation of IP and EA with cluster type for FeS clusters
Binding energies and embedding energies of (NiS) n and (FeS) n clusters
Binding energy is also one of the important parameter to analyse the structural stability of the nanoclusters [27]. The BE per atom of the nanoclusters can be calculated from the following Eq. (1).
where E[TM] is the energy of the transition metal, E[S] is the energy of sulphur, E[TMS] is the energy of transition metal sulphide and n is the number of transition metal and sulphur atom in the cluster. The energy of individual atoms (TM and S) is calculated separately in the same basis set and the resultant energy values are taken into account to calculate E[TM] and E[S]. BE plays a vital role in determining the stability of the nanoclusters.
Among all the clusters of NiS, BE for cube and bipyramidal cube was high which can be said to be more stable than that of other clusters; low BE is observed for hexagonal ring and are tabulated in Table 5. Interestingly, in the case of FeS clusters also the cube and bipyramidal cube have the high BE. The BE values of FeS nanoclusters are tabulated in Table 6. The high value of BE for cube and bipyramidal cube ascends due to the similar kind of geometrical symmetry. Comparing the BE of all the clusters of NiS and FeS, high value is seen for FeS clusters. In NiS and FeS clusters, cube and bipyramidal cube have the high value of BE. The reason may be due to the perfect symmetry of structure between the cube and bipyramidal cube.
The other parameter which can be explored is the EE; it refers to the gain in energy to embed a foreign atom during the growth process in the cluster [28]. The EE of the cluster is given by relation as given in the Eq. 2,
where TM represents transition metals such as Ni, Fe, S for sulphur and n represents the number of transition metal atoms and sulphur atom present in the clusters. For the calculation of EE, the individual energy of TM and S atoms is calculated in the same basis set and is incorporated to find E(TM) and E(S).
The variation in the EE with the cluster size of NiS is shown in the Table 7. For NiS clusters, the bipyramidal cube structure of NiS cluster has a high value of EE which is difficult to add a foreign atom in this cluster. In FeS cluster, the EE is maximum for bipyramidal cube structure of FeS. The EE of FeS is tabulated in the Table 8. Among all the clusters, the EE of bipyramidal cube has the high value.
Vibrational analysis of (NiS) n and (FeS) n clusters for n = 3–5
The vibrational analysis provides the insight for the stability of the nanoclusters [29, 30]. A particular cluster with a real positive frequency is said to be more stable. More IR intensity in the positive frequency leads to an increase in cluster stability. Tables 9 and 10 show the vibrational spectrum and mode assignment of NiS nanoclusters. Hexagonal ring has the significant vibrational frequency at 491.34 and 688.54 cm−1 with the intensity of 59.75 and 27.36 km/mole both assigned to molecular stretching. In the case of planar rhombus, high IR intensity of 31.29 and 89.59 km/mole is observed at the vibrational frequency of 346.90 and 1,108.89 cm−1 assigned to molecular stretching and S–S stretching, respectively. Looking at the vibrational frequency of cube structure, the maximum intensity of 7.35 and 21.12 km/mole for this structure is observed at the frequency of 951.55 and 1,047.37 cm−1 respectively mode assigned to molecular stretching. The prominent IR intensity of 26.18 and 42.05 km/mole is observed for planar ring at the frequency of 28.61 and 738.04 cm−1 respectively assigned to molecular stretching and S–Ni–S stretching. Linear ladder has the intensity of 12.09, 15.36 and 20.27 km/mole at the frequency of 636.19, 655.15 and 991.08 cm−1 which is mode assigned to molecular twist, molecular stretching and Ni–S–Ni stretching, respectively. Bipyramidal cube has the intensity of 20.16 and 21.32 km/mole for the frequency of 634.23 and 1,070.82 cm−1 assigned to molecular stretching and Ni–S–Ni stretching, respectively.
The vibrational analysis of FeS is shown in Tables 11 and 12. In the case of hexagonal ring, prominent IR intensity of 75.95 and 46.15 km/mole is observed at the frequency of 443.12 and 504.46 cm−1 for Fe–S–Fe stretching and Fe–S–Fe bending mode respectively. The planar rhombus structure has the vibrational frequency at 741.44 and 1,118.84 cm−1 that has the IR intensity of 12.57 and 61.50 km/mole for molecular stretching and S–S stretching respectively. The cube structure has the IR intensity of 17.24, 11.39 and 25.64 km/mole for frequency of 985.88, 1,110.02 and 1,137.97 cm−1 for the mode of molecular bending, molecular twisting and molecular twisting, respectively. Planar ring structure has the IR intensity of 39.33, 74.09 and 104.59 km/mole for the frequency of 570.5, 795.6 and 906.13 cm−1 assigned to molecular stretching, S–Fe–S stretching and Fe–S–Fe stretching, respectively. In the case of linear ladder, the vibrational frequency occurs at 671.1, 714.71 and 752.79 cm−1 with IR intensity of 38.13, 330.66 and 42.85 km/mole which is assigned to molecular stretching, molecular bending and Fe–S stretching, respectively. The bipyramidal cube has the vibrational frequency of 440.66, 656.47 and 1,160.71 cm−1 with IR intensity of molecular twist, molecular stretching and Fe–S stretching.
Conclusions
Nanoclusters of (NiS) n and (FeS) n for n = 3–5 are completely optimized with B3LYP/6-31G as a basis set. The stability of the cluster increases when the cluster size increases. The dipole moment and point symmetry for all different types of isomers depend on the positioning of atoms in the cluster. The electronic properties are studied in terms of HOMO–LUMO gap, IP and EA of different isomers, among these clusters chemically reactive and inert clusters are identified and reported. The BE of different clusters are studied and reported. The embedding energy of transition metal sulphide clusters provides the insight for the inclusion of the foreign atom in the cluster. Vibrational analysis also reveals the stability of the clusters, the particular cluster with no imaginary frequency can be concluded as more stable. The structural and electronic properties of different isomers provide insight for tailoring new materials which find its potential importance in functional nanomaterials.
References
Tikhomirov, V.K., Asatryan, K., Galstian, T.V., Vallee, R., Seddon, A.B.: Photoinduced volume changes related to photoinduced anisotropy in chalcogenide glasses. Philos. Mag. Lett. 83, 117–124 (2003)
Jain, P.K., Deepika, K.S., Saxena, N.S.: Glass transition, thermal stability and glass-forming ability of Se90In10−xSbx (x = 0, 2, 4, 6, 8, 10) chalcogenide glasses. Philos. Mag. 89, 641–650 (2009)
Zogg, H., Arnold, M.: Narrow spectral band monolithic lead-chalcogenide-on-Si mid-IR photodetectors. Opto Electron. Rev. 14, 33–36 (2006)
Ruxandra, V.: Slow transient photovoltage in thin film CuxS/CdS solar cells. J. Mater. Sci. Lett. 16, 1833–1835 (1997)
Li, K., Wee, A.T.S., Lin, J., Tan, K.L., Zhou, L., Li, S.F.Y., Feng, Z.C., Chou, H.C., Kamra, S., Rohatgi, A.: A microstructural study on the surface and interface of CdTe/CdS solar cells. J. Mater. Sci. Mater. Electron. 8, 125–132 (1997)
Hartley, A., Irvine, S.J.C.: The effects of the growth parameters of the CdS window layer on the photovoltaic properties of MOCVD-grown CdS/CdTe solar cells. J. Mater. Sci. Mater. Electron. 11, 569–573 (2000)
Mane, R.S., Lokhande, C.D.: Chemical deposition method for metal chalcogenide thin films. Mater. Chem. Phys. 65, 1–31 (2000)
Han, S.C., Kim, H.S., Song, M.S., Kim, J.H., Ahn, H.J., Lee, J.Y.: Nickel sulfide synthesized by ball milling as an attractive cathode material for rechargeable lithium batteries. J. Alloys Compd. 351, 273–278 (2003)
Han, S.C., Kim, K.W., Ahn, H.J., Ahn, J.H., Lee, J.Y.: Charge–discharge mechanism of mechanically alloyed NiS used as a cathode in rechargeable lithium batteries. J. Alloys Compd. 361, 247–251 (2003)
Fernandez, A.M., Nair, M.T.S., Nair, P.K.: Chemically deposited ZnS–NiS–CuS optical filters with Wide range solar control characteristics. Mater. Manuf. Process. 8, 535–548 (1993)
Vandenborre, H., Vermeiren, Ph, Leysen, R.: Hydrogen evolution at nickel sulphide cathodes in alkaline medium. Electrochim. Acta 29, 297–301 (1984)
Shen, G., Chen, D., Tang, K., An, C., Yang, Q., Qian, Y.: Phase-controlled synthesis and characterization of nickel sulfides nanorods. J. Solid State Chem. 173, 227–231 (2003)
Wang, W., Wang, S.Y., Gao, Y.L., Wang, K.Y., Liu, M.: Nickel sulfide nanotubes formed by a directional infiltration self-assembly route in AAO templates. Mater. Sci. Eng. B 133, 167–171 (2006)
Chen, D., Gao, L.: Novel morphologies of nickel sulfides: nanotubes and nanoneedles derived from rolled nanosheets in a w/o microemulsion. J. Cryst. Growth 262, 554–560 (2004)
Huang, S., Harris, K.D.M., Capel, E.L., Manning, D.A.C., Rickard, D.: “Amorphous Nickel Sulfide” is hydrated nanocrystalline NiS with a core–shell structure. Inorg. Chem. 48, 11486–11488 (2009)
Meng, Z., Peng, Y., Xu, L., Qian, Y.: Solvothermal-reduction route to nanocrystalline α- and β-NiS. Mater. Lett. 53, 165–167 (2002)
Wang, L., Zhu, Y., Li, H., Li, Q., Qian, Y.: Hydrothermal synthesis of NiS nanobelts and NiS2 microspheres constructed of cuboids architectures. J. Solid State Chem. 183, 223–227 (2010)
Esrafili, M.D., Rezaei, S., Eftekhari, E.: A theoretical investigation on geometry and electronic structure of small FemSn nanoclusters (1 ≤ m, n ≤ 4). Theochem 1001, 1–6 (2012)
Henderson, A.D., Demond, A.H.: Permeability of iron sulfide (FeS)-based materials for groundwater remediation. Water Res. 47, 1267–1276 (2013)
Ritchie, A.G., Bowles, P.G., Scattergood, D.P.: Lithium-ion/iron sulphide rechargeable batteries. J. Power Sources 136, 276–280 (2004)
Gomes, A., da Silva Pereira, M.I., Mendonça, M.H., Costa, F.M.: Effect of the substrate on the electrodeposition of iron sulphides. Solid State Sci. 4, 1083–1088 (2002)
Sundaraganesan, N., Elango, G., Meganathan, C., Karthikeyan, B., Kurt, M.: Molecular structure, vibrational spectra and HOMO, LUMO analysis of 4-piperidone by density functional theory and ab initio Hartree–Fock calculations. Mol Simulat 35, 705–713 (2009)
Siddiqui, S.A.: First principle study of CrFn (n = 1–7) nano clusters: an investigation of superhalogen properties. Struct. Chem. 23, 267–274 (2012)
Schwenn, P.E., Burn, P.L., Powell, B.J.: Calculation of solid state molecular ionisation energies and electron affinities for organic semiconductors. Org. Electron. 12, 394–403 (2011)
Li, P., Wang, W., Sun, H., Bi, S.: A DFT study on the electron affinity of tetrachloro-p-benzoquinone: toward to understanding its electron-accepting ability in solution. THEOCHEM 1006, 127–132 (2013)
Zhan, C.G., Nichols, J.A., Dixon, D.A.: Ionization potential, electron affinity, electronegativity, hardness, and electron excitation energy: molecular properties from density functional theory orbital energies. J. Phys. Chem. A 107, 4184–4195 (2003)
Chandiramouli, R., Sriram, S., Balamurugan, D.: Quantum chemical studies on (ZnO)n/(NiO)n heterostructured nanoclusters. Mol. Phys. 112. doi:10.1080/00268976.2013.805846 (2014)
Bandyopadhyay, D.: Chemisorptions effect of oxygen on the geometries, electronic and magnetic properties of small size Nin (n = 1–6) clusters. J. Mol. Model. 18, 737–749 (2012)
Yadav, P.S., Pandey, D.K., Agrawal, S., Agrawal, B.K.: Ab initio study of structural, electronic, optical, and vibrational properties of ZnxSy (x + y = 2 to 5) nanoclusters. J. Nanopart. Res. 12, 737–757 (2012)
Sriram, S, Chandiramouli, R, Jeyaprakash, B.G: Influence of fluorine substitution on the properties of CdO nanocluster: a DFT approach. Struct. Chem. doi:10.1007/s11224-013-0302-5
Valiev, M., Bylask, E.J., Govind, N., Kowalski, K., Straatsma, T.P., van Dam, H.J.J., Wang Nieplocha, D.J., Apra, E., Windus, T.L., de Jong, W.A.: NWChem: A comprehensive and scalable open-source solution for large scale molecular simulations. Comput. Phys. Commun. 181, 1477–1489 (2010)
Osuna, S., Morera, J., Cases, M., Morokuma, K., Sola, M.: Diels Alder reaction between Cyclopentadiene and C60: an analysis of the performance of the ONIOM method for the study of chemical reactivity in fullerenes and nanotubes. J. Phys. Chem. A 113, 9721–9726 (2009)
Hayer, A., Regemorter, T., Hofer, B., Mak, C.S.K., Beljonne, D., Hler, A.K.: On the formation mechanism for electrically generated exciplexes in a carbazole-pyridine copolymer. J. Polym. Sci. Part B Polym. Phys. 50, 361–369 (2012)
Huang, W., Bulusu, S., Pal, R., Zeng, X.C., Wang, L.S.: CO chemisorption on the surfaces of the golden cages. J. Chem. Phys. 131, 234305 (2009)
Gomes, D.E.B., Lins, R.D., Pascutti, P.G., Lei, C., Thereza Soares, A.: Conformational variability of organophosphorous hydrolase upon soman and paraoxon binding. J. Phys. Chem. B. 115, 15389–15398 (2011)
Ermakov, A.I., Belousov, V.V.: Relaxation Of STO-3G and 6–31G* basis set functions in the series of LiF isoelectronic molecules of second row elements. J. Struct. Chem. 48, 6–15 (2007)
Sriram, S, Chandiramouli, R: DFT studies on the stability of linear, ring, and 3D structures in CdTe nanoclusters. Res Chem Intermed. doi:10.1007/s11164-013-1334-6
Sriram, S., Chandiramouli, R., Balamurugan, D., Thayumanvan, A.: A DFT study on the structural and electronic properties of ZnTe nanoclusters. Eur. Phys. J. Appl. Phys. 62, 30101 (2013)
Harrison, R.J., Guest, M.F., Kendall, R.A., Bernholdt, D.E., Wong, A.T., Stave, M., Anchell, J.L., Hess, A.C., Littlefield, R.J., Fann, G.L., Nieplocha, J., Thomas, G.S., Elwood, D., Tilson, J.L., Shepard, R.L., Wagner, A.F., Foster, I.T., Lusk, E., Stevens, R.: Toward high-performance computational chemistry: II. A scalable self-consistent field program. J. Comput. Chem. 17, 124–132 (1996)
Acknowledgments
The authors are thankful to the Vice Chancellor, SASTRA University for his constant support and encouragement.
Conflict of interest
The authors declare that they have no competing interests.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
This article is published under license to BioMed Central Ltd. Open Access This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
About this article
Cite this article
Nagarajan, V., Chandiramouli, R., Sriram, S. et al. Quantum chemical studies on the structural and electronic properties of nickel sulphide and iron sulphide nanoclusters. J Nanostruct Chem 4, 87 (2014). https://doi.org/10.1007/s40097-014-0087-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s40097-014-0087-0