
Abstract
Forty isolates of Ascochyta spp. infecting pea collected from different commercial pea growing areas of Himachal Pradesh were characterized into three species—Ascochyta pinodes, A. pisi and A. pinodella—based on symptoms, morphological and cultural traits. The identity of the isolates was confirmed by studying the rDNA sequence patterns. Based on various cultural and morphological trait combinations, 36 isolates were identified as A. pinodes, one isolate as A. pisi and one isolate as A. pinodella. Whereas two isolates, As3 and As14, could not be placed in any of the species as equal number of characters matched with A. pisi and A. pinodes. However, sequencing of ITS region of these isolates resolved the ambiguity and placed them in A. pinodes. This necessitates ITS sequencing as one of the criteria to establish the taxonomic status of Ascochyta species infecting pea. RAPD, RAMS, and Rep-PCR analysis with 10, 4 and universal ERIC, and BOX-AIR primers categorized different isolates according to their geographical locations. This study established the predominance of A. pinodes in Himachal Pradesh where edible pea is one of the remunerative off-season vegetable crops. This also constitutes first report of genetic variability of Ascochyta species infecting pea in India.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Pea (Pisum sativum L.), an edible legume is grown throughout the world. As a winter season crop in India, pea enjoys the status of an important off-season crop in temperate Himachal Pradesh regions like Kinnaur, Lahaul and Spiti and mountainous locations of Kullu, Mandi, Shimla and Sirmaur districts. The unique geographical diversity of Himachal Pradesh offers an opportunity to produce pea both as a winter crop in low and mid hills, and as summer crop in high hills. It is grown in an area of 16,348 ha with annual production of 1,77,036 metric tonnes [3] and during off-season it sells at a premium in the plains of India. Consequently, pea has emerged as the major off-season vegetable crop of the state.
Full genetic potential of the crop is not realized owing to a variety of biological constraints, and Ascochyta blight is one besides others such as powdery mildew. Ascochyta blight complex is a severe disease of peas throughout the world and causes huge losses to growers every year [8, 14, 26, 42]. The disease complex is caused by three Ascochyta species: A. pisi (teleomorph Didymella pisi) [12], A. pinodes (teleomorph Mycosphaerella pinodes) and Phoma medicaginis var. pinodella [25]. However, recently the fourth species Phoma koolunga was reported to occur in the south Australia [13]. The principal characteristics used to distinguish the former three species were: (i) the presence of pseudothecia in M. pinodes, (ii) size of the conidia (conidia of P. pinodella are smaller than those of A. pisi or M. pinodes), and (iii) carrot red spore masses of A. pisi compared with the light buff of M. pinodes and P. pinodella [25]. The taxonomic position of Ascochyta species on the morphological and pathological basis has been controversial for long.
Morphological criteria being influenced by environmental conditions, makes disease diagnosis a cumbersome process [18]. The symptom expression and morpho-cultural methods are highly unstable being environment dependent. Secondly, lack of resistance in pea to Ascochyta spp. restricts the detection of variability in this pathogen. Use of molecular markers offers an opportunity to establish the taxonomic status and variability in such pathogens. Recently, molecular approaches using RAPD, AFLP, RFLP, and sequence analysis have been applied in systematic studies of Ascochyta species. RAPD fingerprinting methods have been criticized for poor reproducibility [6, 16, 23, 37]. RFLP analysis of rDNA has been applied in population studies of plant pathogens by southern hybridization or restriction digestion of amplified DNA [1, 5, 38]. However, without information on the position of restriction sites, the potential for character homoplasy must be considered when using this type of data for phylogenetic reconstruction [45, 50]. Thus, direct sequence determination provides the most robust data from which phylogenetic reconstruction can proceed with confidence. The most contemporary phylogenetic studies have been based on ITS, mtSSU, mtLSU, β-tubulin, glyceraldehyde-3-phosphate dehydrogenase gene (G3PD), chitin synthase 1 gene (CHS), translation elongation factor 1 alpha gene (EF) and MAT locus sequence analysis, and have supported the organization of Ascochyta species into distinct species groups [7, 12, 13, 20, 36].
Information about the dominance of a particular Ascochyta species in a particular pea growing region of Himachal Pradesh is lacking. Work on such aspects of the disease has not been undertaken in India. Accurate pathogen identification is most important for diagnosis of the disease and understanding the mechanism of host-pathogen relationship for sustainable disease management strategies. Hence, the objective of this study was to establish the identity of Ascochyta specie(s) associated with blight complex of pea using morpho-cultural traits, their predominance in the region and elucidation of molecular diversity among the different species.
Materials and Methods
Collection of Fungus Isolates, Their Purification and Maintenance
Extensive survey of pea growing areas were conducted during June–August 2007–2009 when pea is cultivated as an off-season crop, and 40 isolates of Ascochyta spp. causing blight complex were collected to study the population structure and genetic analysis. Each isolate was cultured on potato dextrose agar medium, purified by single spore isolation and maintained for further studies. The symptom development of each isolate was studied on susceptible variety Azad P1 and the characteristic symptoms produced by each isolate were compared according to description given by Lawyer [28] and Sagar [41] (Supplementary Table 1).
Cultural and Morphological Characteristics
The cultural characteristics of the isolates were studied on oat meal agar dispensed (20 ml) into 9 cm diameter Petri plates. Mycelial plugs of 5 mm diameter were cut with the help of cork borer from the margin of an actively growing colony and placed in the center of media plates and incubated at 22 ± 1 °C. Observations were recorded on colony color, zonation and sporulation after 20–25 days of inoculation. Various morphological characters like pycnidiospores (length and breadth), septation of conidia and formation of chlamydospores were recorded. Conidial suspensions prepared from 20 day old sporulating cultures were stained with cotton blue lactophenol followed by microscopic examination of conidial length, breadth, number of septa (range), percent septa, and constriction at septa at 400× magnification using at least 10 pycnidia/plate.
The formation of pseudoperithecia was also studied on oat meal agar slants incubated at 15 ± 1 °C for 25–30 days for formation of perfect stage. Diameter of pseudoperithecia, length and breadth of asci and ascospores were measured as per standard procedure. In order to assign Ascochyta isolates into a particular species, consensus speciation was done with traits showing maximum homology.
Molecular Characterization
DNA Fingerprinting
The genetic diversity of Ascochyta isolates associated with pea blight was studied by employing random amplified polymorphic DNA (RAPD), rep-PCR and randomly amplified microsatellites (RAMS). Total genomic DNA of each isolate was extracted using CTAB method [44]. Out of 140 RAPD oligonucleotide primers (Operon and Life Technologies) screened with 5 randomly selected isolates, eight most polymorphic primers (OPA-02, OPA-09, OPA-13, OPD-11, OPQ-13, OPQ-18, S-1466, and S-144) showing consistent banding pattern with two DNA samples of same isolate were used for fingerprinting following the procedure of Padder et al. [34].
Out of eleven RAMS, four most polymorphic primers viz., BA1 (CCA (CAG)5), BA2 (GTG)5, BA8 (AAC)5, and BA9 (AT)5 were used for amplification of 40 isolates following the procedure of Padder et al. [34]. In addition, the test isolates were also amplified by Rep-PCR (with a pair of ERIC and BOX-AIR primers) as described by Mahuku, Riascos [31].
rDNA Analysis
Fifteen isolates of Ascochyta identified as A. pinodes (13), A. pisi (1) and A. pinodella (1), and one each from common bean (Phaseolus vulgaris), urdbean (Vigna mungo) and gram (Cicer arietinum) were used for rDNA analysis (Supplementary Table 2). Primers ITS1 and ITS4 [54] were used to amplify approximately 540 bp of ITS1, 5.85 ribosomal gene, and ITS2 region following the conditions of Padder et al. [35]. All PCR assays contained samples of primers without DNA to serve as negative controls. Five μl of the each amplified product was electrophoresed to confirm the successful amplification. The amplified PCR products were resolved by electrophoresis using 1.2 % agarose gel in 0.5X Tris borate EDTA buffer. The gels were stained with ethidium bromide (0.5 μg/ml) for 10 min after electrophoresis followed by distaining under running tap water. The amplicon (3 separate PCR products from each isolate) was lyophilized and sent for custom sequencing (Bioscience India, Pvt. Ltd.). The sequences (forward and reverse) were retrieved from chromatograms using BioEdit version 7.0 [21]. Nucleotide sequences from PCR-amplified fragments (3 amplicons/isolate) were assembled in BioEdit version 7.0 [21] and the consensus sequence was reconfirmed by comparing it with the original sequencing data output. Consensus sequences were aligned using CLUSTAL W [49] option in BioEdit. All the sequences were submitted to gene bank (NCBI) and accession numbers of sequences were obtained (Supplementary Table 2).
Data Analysis
Binary matrices were analyzed by NTYSYS-PC 1.80 and Jaccards coefficient was used to construct dendrogram using SHAN clustered program, selecting the unweighted pair-group arithmetic mean method (UPGMA) in NTSYS-PC V 1.80 (Rolfh 1993). The dendrogram with best fit to similarity matrix based on cophenetic value (COPH) and matrix comparison (MXCOMP) was chosen.
For phylogenetic analysis, ITS regions of Ascochyta species infecting pea and other legumes in Himachal Pradesh were compared with 19 other Ascochyta sequences from different legumes (Supplementary Table 3) retrieved from NCBI (http://www.ncbi.nlm.nih.gov/nuccore). All nucleotide sequences were aligned using CLUSTAL X 1.8 multiple alignment program [49] and refined manually. The GENEDOC package (www.psc.edu/biomed/genedoc/gdpf.html) was used for formatting the sequences to make them compatible with the desired software. Sequence polymorphism was analyzed, genetic divergence values within and between populations were estimated and a neighbor joining (NJ) tree was constructed for all the haplotypes according to Kimura 2-parameter (K2P) model using PHYLIP ver 3.6 (Felsenstein 1993) in MEGA version 4.1 [48]. The NJ tree was rooted with the rDNA sequences of Mycosphaerella graminicola, M. punctiformis, and M. fijiensis (NCBI GenBank Accession Nos AF181694, AY490760 and AF297225, respectively). Nucleotide diversity (π) and haplotype diversity (h) were estimated using DnaSP 5.0 [29].
Population structure was evaluated using the analysis of molecular variance (AMOVA) model in the ARLEQUIN ver. 3.0 software [17]. Fixation indices (G ST) analogous to F ST [24] were estimated. The analysis used a hierarchical structure in which each country (India, USA, Australia, Israel and Georgia) and each species (A. pisi, A. pinodes, A. pinodella, P. koolunga, A. rabiei and Ascochyta sp.) were considered as populations. The statistical significance of the total and pairwise fixation indices was estimated by comparing the observed distribution with a null distribution, generated by 10,000 permutations of the data matrix. Multiple tests of the same null hypothesis were subjected to table-wide sequential Bonferroni correction to avoid elevated Type I error.
Results and Discussion
Speciation of Ascochyta Isolates Based on Symptoms, Cultural and Morphological Characteristics
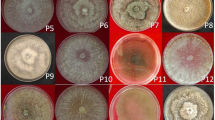
Species identification criteria (Supplementary Table 1) based on symptoms, cultural characters, conidial size and septation, chlamydospores and formation of pseudothecia were used to identify the Ascochyta species associated with blight complex of pea. As per the symptomatology, test isolates were placed in three groups viz., I, II, and III (Table 1). Group I comprised of six isolates viz., As1, As2, As3, As14, As15 and As23 producing circular, light brown to tan colored leaf spots measuring 2–6 mm in diameter with a distinct dark margin delineating the border. Isolates of this group induced production of sparse light brown and erumpent pycnidia as minute pinheads. Group II with 34 isolates caused irregular spots measuring 3–15 mm with broad concentric ring pattern in alternate shades of light yellowish brown and grayish purple. Such spots showed no distinct margin like that of isolates in Group I; however, in a few cases, zonation was observed around the spots. Most of the isolates in this group often showed blighted appearance due to coalescing of 2–3 spots. Pycnidia were scattered as small pinheads. Lawyer [28] clearly differentiated the three species with tan colored spots delineated by distinct margin as A. pisi, concentric lesions/spots as A. pinodes, and the one causing foot rot as A. pinodella. These symptoms resemble the description made by various workers [25, 28, 30, 36, 41]. It was interesting to note that of the four isolates placed in group I exhibited two types of leaf spots, i.e., small tan colored (2–6 mm) to large irregular up to 15 mm spots with concentric rings and resembled group II; in addition, these isolates showed marked variation in symptoms under artificial and natural conditions, and are thus categorized as A. pisi and A. pinodella. Sagar [41] attributed such differences to the environment prevailing during symptom development. Stone [46] and Vaughan [51] considered A. pinodes as an ascigerous stage of A. pisi, while Jones [25] argued that the two fungi are confused in literature.
All the 40 isolates were divided into three distinct groups on the basis of colony characteristics viz., color, zonation and pattern of pycnidia formation (Table 1). Group I isolates (As2, As3, As4 and As16) formed light colored white colonies possessing few pycnidia without any zonation, whereas Group II accommodating 34 isolates produced brown colonies with concentric ring pattern of alternate dark and brown zonation. Pycnidia were more or less in concentric ring pattern. Two isolates viz., As1 and As30 forming dark gray to black colonies with sparse pycnidia were categorized in group III. Comparison of these characters with other workers revealed that group I, II, and III resembled A. pisi, A. pinodes, and A. pinodella [28, 30, 33]. The variation in colony color has been attributed to the chlamydospore producing capacity of the three species. Onfroy et al. [33] correlated the gray/black color of A. pinodella isolates to profuse chlamydospore production within 15 days as compared to A. pinodes forming fewer chlamydospores even after 25–30 days.
The conidial characters (Supplementary Table 4) categorized various Ascochyta isolates into three groups viz., I, II, and III accommodating 4, 35, and 1 isolates, respectively (Table 1). On the basis of size of conidia, three groups were distinguished measuring 3–5 × 10–16 μm (54.2 × 14 μm), 3–6.5 × 18–16 μm (4.5 × 12.3 μm), and 2–3 × 3.5–8 μm (4.5 × 12.3 μm) for group I, II, and III, respectively. Onfroy et al. [33] advocated that bicellular conidial production to distinguish pea blight pathogens as average cellular conidia formation was 97.9 ± 2.4 for A. pinodes, but only 19.6 ± 6.4 % in A. pinodella. The spore dimensions clearly matched group I isolates to A. pisi, group II to A. pinodes and group III to A. pinodella as described by Lawyer (1984). Onfroy et al. [33] observed longer conidia in A. pinodes isolates and smaller conidia in A. pinodella.
Based on chlamydospore formation, the test isolates were divided into three groups (Table 1). Two isolates As9 and As18 producing no chlamydospores were placed in group I, whereas 35 isolates producing few chlamydospores frequently in about 25-30 day old cultures were placed in group II. However, in group III, 3 isolates induced profuse chlamydospore formation resulting in dark brown to black colored colonies. Lawyer [28] while distinguishing three Ascochyta spp. causing pea blight described chlamydospore formation as a major criterion—with A. pisi, A. pinodes, and A. pinodella—producing no or few chlamydospores in old cultures and profuse chlamydospores turning the culture medium black, respectively. The present findings are in conformity as group I, II, III resembled A. pisi, A. pinodes and A. pinodella, respectively. Similar pattern of chlamydospore formation distinguishing the three species has also been reported [25, 30, 33, 41].
Data on pseudoperithecia development on oat meal agar at 15 ± 1 °C revealed that out of 40 isolates only three viz., As10, As22, As34 formed pseudoperithecia and were placed in group III (Table 1). Pseudoperithecia in As10 and As22 were globose, dark brown to black in color having apical ostiole with an average size of 150 × 112.5 μm. Asci were hyaline, bitunicate, cylindrical to sub-clavate measuring 49.69 × 12.19 μm on average (range 41.25–56.25 × 11.25–15 μm). Pseudoperithecia in As34 measured 168.75 × 150 μm with average size of asci being 56.25 × 10.94 μm (range 45–67.25 × 9.37–11.25 μm). The ascostroma was about 131.25 × 112.5 μm. The ascospores were hyaline, bitunicate, two celled, mostly constricted as septum, round and narrower at tips, with guttulate measuring 16.87 × 7.50 μm. Remaining 37 isolates were placed in group I. Efforts to induce perfect stage in Ascochyta spp. under artificial conditions were unfruitful and the pseudoperithecial dimensions, size and shape of ascus, and size of ascospores resembled A. pinodes [25, 28, 41]. Lawyer [28] stated that principal morphological characteristic distinguishing A. pinodes is formation of pseudoperithecia on PDA/Oat meal agar at 16 ± 1 °C. However, Onfroy et al. [33] contested this as a reliable criterion, as perfect stage depends on environmental and/or cultural factors. Bowen et al. [9] succeeded in induction of pseudoperithecia formation in A. pinodella, a rare phenomenon otherwise.
Based on symptom type, colony color, and morphological traits it was not possible to distinguish Ascochyta species. So identification of test isolates was done using combination of various parameters (Supplementary Table 2). Depending upon matching characters for each species out of 40 isolates, 36 were designated as A. pinodes, 1 each of As1 and As2 as A. pinodella and A. pisi. However, in case of two isolates As3 and As14 (Supplementary Table 2) no inference could be drawn as same cultural characteristics matched both A. pisi and A. pinodes. The dependability of major differential criteria used for identification of various Ascochyta spp. is debatable [10, 18, 39, 47]. Different criteria used in present study resolved the identity in most of the cases and as a consequence, 36 isolates were designated as A. pinodes indicating its wide prevalence in Himachal Pradesh. Preliminary studies on symptomatology and pathogen morphology conducted in Himachal Pradesh have shown A. pisi as most prevalent in Kullu and Kangra valley of Himachal Pradesh [2]. Chaudhary [11] also identified A. pisi as the predominant species causing Ascochyta blight and pod spot in three out of four districts of Himachal Pradesh, though possibility of other species was not ruled out. Change in agroclimatic conditions over the years due to global warming could have shifted toward the dominance of A. pinodes over A. pisi. This was further supported by the absence of Ascochyta blight in high altitude areas of Mandi, Chamba, and Kinnaur by Chaudhary [11]. Conversely, areas exhibiting maximum disease incidence previously were found to be disease free in the present study. Similar shift in pathogen populations has also been reported from Canada. Wallen et al. [52] found A. pinodes causing pea blight epiphytotic although, formerly A. pisi was the predominant species.
Two pathogen isolates could not be differentiated as most of their characters matched both A. pisi and A. pinodes. These isolates are considered intermediates between two species or misidentified due to lack of dependable differential criteria. Such a case of misidentification is upheld by Faris-Mokaiesh et al. [18] who identified two isolates as A. pinodes in the beginning but the PCR–RFLP profiles of IGS region later clustered them in A. pisi. They emphasized the need to integrate such molecular tools in the disease diagnosis due to their stability. The probability of these being intermediate species is enhanced by Onfroy et al. [33] who showed two isolates intermediate between A. pinodes and A. pinodella. The RAPD profiles also separated these two isolates forming a separate clade. However, ITS sequencing of these isolates put them in A. pinodes group strengthening the view point of Faris-Mokaiesh et al. [18] to include sequencing of ITS and IGS region as one of the criteria used.
Molecular Characterization
Speciation Based on rDNA Sequence Pattern and Their Phylogenetic Analysis
rDNA segment of 15 isolates of different Ascochyta species from pea (Chamba, Kinnaur, and Mandi populations), and one isolate each from common bean, urdbean, and chickpea (Supplementary Table 2) were used to establish the identity of the test isolates determined on morpho-cultural characteristics. rDNA amplification of test isolates with universal primer pairs ITS1 and ITS4 amplified amplicons of 540–550 bp which upon sequencing revealed that sequences of test isolates consisted of 510–545 bp. The obtained sequences were first subjected to BLAST search against Ascochyta spp. sequences available online in NCBI GeneBank using blastn program and their identity was confirmed as Ascochyta. The rDNA analysis resolved several ambiguities encountered while determining the taxonomic position of some isolates using morpho-cultural traits. Comparative analysis of sequences of the test isolates infecting pea and reference isolates from gene bank revealed that identity of 13 of the 15 isolates was in confirmation with that of the morphological traits (Supplementary Table 2). Eleven isolates viz., As4, As5, As12, As8, As15, As24, As26, As34, As39, As25 and As16 were more close to A. pinodes and 2 isolates As1 and As41 were identical to A. Pinodella (Fig. 1).

Phylogenetic relationship of Ascochyta species using the ITS gene nucleotide sequence alignment. Bootstrap values supporting the branches are shown at nodes; Branch lengths are proportional to divergence. ASP Ascochyta spp.; AR Ascochyta rabiei; AP Ascochyta pisi; PMP Ascochyta pinodella (Phoma medicaginis var. pinodella); MP Ascochyta pinodes (Mycosphaerella pinodes); PSP Phomo spp.; AUS Australia; PK Phoma koolunga; IND India; ISR Israel; USA United States of America; GRG Georgia
One isolate As2 misidentified as A. pisi based on morphological traits showing more sequence homology with A.pinodes was placed in species A. pinodes and submitted to the NCBI gene bank. Similarly, isolates As3 and As14 rDNA sequences ascribed the isolates to A. pinodes group after blast analysis and were submitted as A. pinodes in the NCBI gene bank. Chickpea isolate As44 showing maximum homology to A. rabiei sequences was identified as A. rabiei. Two other sequences of As42 and As43 isolated from common bean and urd bean were submitted as Ascochyta spp.
The use of sequence information from rDNA repeat units is well established in fungal taxonomy for characterizing isolates and resolving taxonomic ambiguities and species definition as this gene cluster occurs within the chromosomes having multiple copies in a single nucleus [19]. These rDNA arrays are homogenized through evolution and the functional nature of these gene blocks minimize mutation within sequences rendering its utility in taxonomy [4]. Hence, nucleotide sequence comparison of ITS region of 18 test Ascochyta isolates with 22 other sequences available online in NCBI gene bank established the identity of present isolates as Ascochyta spp. The higher sequence homology of As2 (FJ159103) with A. pinodes established its identity as A. pinodes though it was identified as A. pisi on morphological basis. Similarly, As3 (FJ188410) was also designated as A. pinodes due to its higher sequence homology with A. pinodes. Both As2 (FJ 159103) and As3 (FJ188410) were close to A. pinodes due to lower genetic distance (0.000–0.012) with A. pinodes as compared to A. pisi (0.024 with EU383949 and 0.021 with DQ383954). Moreover, these two isolates were grouped in clade I along with A. pinodes and A. pinodella. Faris-Mokaiesh et al. [18] also confirmed the identity of two isolates MP-007 Fra and MP-014 Fra as A. pisi, erroneously identified as A. pinodes, in PCR–RFLP analysis of IGS region of rDNA. The true identity of pathogen isolates has also been confirmed with molecular methods in Beauvaria brongniartii [32]. Similarly, As44 (FJ032643) isolated from chickpea showed minimum genetic distance of 0.000–0.002 with A. rabiei sequences (EU595358, EU573020, DQ383949, and DQ822480) available at NCBI gene bank.
Multiple alignments showed that complete coding regions viz., ITS1, 5.85 ribosomal gene and ITS2 had comparable size with reference isolates from NCBI gene bank with few exceptions. Some of these differences were specific, e.g., changes in ITSI region of As43 (FJ156067) and 5.8S ribosomal RNA gene of As42 (FJ032642), As43 (FJ156067) and As44 (FJ-32643). The size variation within the isolates could be due to genetic changes as reported by DePriest [15] in lichenized-fungi or due to different number of simple sequence repeats [27].
A total of 540–550 bp of the rDNA fragment was successfully sequenced in 15 individuals of Ascochyta. In total, four unique haplotypes with 15 variable sites (3.94 %) were identified in 15 samples of A. pinodes, and 14 unique haplotypes with 36 variable sites (8.8 %) were identified in 40 samples of various Ascochyta species infecting different legumes. Among the polymorphic sites, most were transitions and only a few were transversions. The mean number of nucleotide composition was A = 27.80 %, T = 28.50 %, C = 29.50 %, and G = 14.20 %. Kinnaur and Mandi isolates showed a single haplotype with 0.00 haplotype and nucleotide diversity, whereas in Chamba population, two haplotypes were observed with 0.50 and 0.00567 haplotype and nucleotide diversity, respectively (Table 2). Among the 40 sequences including 18 sequences from Himachal Pradesh, 14 unique haplotypes were detected (Table 2). All the isolates of A. pinodes belonged to a single haplotype irrespective of countries; whereas, 4 and 2 haplotypes were observed in A. pisi and A. pinodella, respectively. A. pinodes, A rabiei, and P. koolunga showed 0.00 haplotype and nucleotide diversity. In case of A. pisi, Ascochyta species and A. pinodella 0.900, 1.00, and 0.40 haplotype diversity and 0.50346, 0.02617, and 0.00455 nucleotide diversity was observed, respectively. AMOVA of clustering at species (Table 3) level showed that the genetic differentiation among species was highly significant (Φ ST = 0.834, P < 0.001; 50175 permutations), with 83.38 and 16.62 % of the genetic variability being among species and within species (populations), respectively. Similarly, AMOVA from different countries was highly significant (Φ ST = 0.364, P < 0.016; 32453 permutations) with 36.99 and 63.01 % variability being among countries and within countries, respectively (Table 3). High haplotype and low nucleotide diversity was observed at species level primarily because haplotypes were separable by a maximum of 15 nucleotide differences in three populations from Himachal Pradesh and 36 nucleotide differences from the populations of exotic Ascochyta species. Such a variation indicates that a recent expansion event(s) eclipsed any previous phylogenetic structure in Ascochyta species infecting pea in the region. The present study supports such a phenomenon in Himachal Pradesh as A. pisi was dominant over the other species causing the disease up to 1992 [41]. The pathogen has moved hitherto to disease-free areas. The low nucleotide diversity of A. pinodes also suggests homogeneity in pathogenic populations in Himachal Pradesh. Analysis of molecular variance (AMOVA) obtained from clustering at species and country level showed maximum variation. Existence of such a variation at species level may be due to different Ascochyta species as distinct populations.
In order to study inter-relationship between different Ascochyta spp., all the sequences of 18 isolates were compared with other 19 reference sequences of Ascochyta available online in NCBI gene bank by molecular evolutionary genetic analysis (MEGA) software version 4.0 and an optimal tree was generated by NJ method, and different taxa were clustered together in a bootstrap test with 1,000 replicates (Fig. 1). The evolutionary distance computed using maximum composite likelihood method (MCLM) were in units of base substitution per site. The optimal tree had branch length of 3.30834065 base substitutions per site and the bootstrap value greater than 50 % for each branch.
It is clear from the dendrogram that all the Ascochyta isolates were divided into five clades viz., I–V. In clade I, all the A. pinodes and A. pinodella isolates (both custom sequenced or available at NCBI gene bank) clustered together indicating that the two species cannot be differentiated on the basis of ITS region. This clade was the largest group accommodating 22 out of the 40 taxa used in the present study. However, it was interesting to note that Ascochyta sp. infecting bean (FJ032642) clustered with clade indicating its close relationship with A. pinodes and A. pinodella.
Further, one isolate As44 identified as A. rabiei clustered together in clade III with A. rabiei sequences available in the gene bank. Thus A. rabiei made a distinct clade. One A.pisi sequence clustered together in clade III which also included Ascochyta sp. from Vicia villosa (DQ383956) and Pisum elatius (DQ383955) isolated from common bean indicating their close association with A. pisi isolates. All the four isolates of Phoma koolunga formed a single clade IV. The isolate As43 infecting Vigna mungo was separated and formed the clade V.
The phylogenetic analysis led to the grouping of 37 isolates into five different clades when a consensus tree using bootstrap procedure was drawn. The ITS ribotyping clustered all the A. pinodes and A. pinodella into a single clade I. This indicated that A. pinodes and A. pinodella could not be differentiated on the basis of ITS region. There was considerable homogeneity between the isolates because pairwise genetic distance among themselves deviated only up to 0.05 or 0.012 as in As5 (FJ188409). Faris-Mokaiesh et al. [18] reported considerable homogeneity between the two species and could not differentiate them on the basis of PCR–RFLP of ITS and IGS regions of rDNA gene cluster. Fatehi et al. [20] depending on mean genetic distance between A. pinodella and A. pinodes group sequences and ß-tubulin PCR–RFLP’s suggested to reduce two epithets of the two species to synonyms (con-specific), as was observed in the present study. However, there are ambiguities which prevent them from being called as con-specific. First, the differences in symptom expression, cultural and morphological traits point them to be separate species. Fatehi et al. [20] questioned the precision of ITS sequence homology because there is 1–2 % variation within species and around 5 % variation between species, but no absolute value is considered as evidence of species identity. This means that ITS region does not separate these two species and there is a need to sequence other regions of their genome. In fact, [7, 36] were unable to differentiate these two species on the basis of ITS ribotyping, but could easily differentiate into separate clades on the basis of high mobility group (HMG) domain of MAT gene and glyceraldehyde-3-phosphate dehydrogenase (G3PD) phylogeny.
The third species in the Ascochyta blight complex of peas, A. pisi formed a separate clade II which also included As42 (FJ032642), an Ascochyta spp. isolated from common bean. ITS sequences were clearly heterogenous enough to separate A. pisi from other two species. Faris-Mokaiesh et al. [18] on the basis of PCR–RFLP of IGS whereas Barve et al. [7] and Peever et al. [36] on the basis of ITS sequences also differentiated A. pisi from A. pinodes and A. pinodella. Moreover, A. pisi showed lower mean/net genetic distance with A. rabiei group than A. pinodes/A. pinodella group indicating its closeness to A. rabiei than two species infecting the same host. Moreover, the branch separating A. pisi (clade II) and A. rabiei (clade III) from A. pinodes/A. pinodella (clade I) clades was long with more than 50 % maximum likelihood bootstrap values establishing its high divergence from clade I species. This implies that A. pisi has followed a different evolutionary route than A. pinodes/A. pinodella having evolved from a different ancestor. On the other hand, close affinity between A. pinodella and A. pinodes indicates common ancestory. Fatehi et al. [20] on basis of mitochondrial genomes alluded them to be evolved from a single, less specific ancestor. Based on HMG phylogeny, Barve et al. [7] found that A. pisi was closer to A. rabiei clade than other two species infecting pea. On the basis of G3PD sequencing, Peever et al. [36] found A. rabiei and A. pisi to be in the same clade.
Based on phylogenetic analysis, one isolate As44 (FJ032643) infecting chickpea was clustered in clade III having four other A. rabiei isolates from NCBI gene bank with pairwise genetic distance of 0.000–0.002, separating it from other three species infecting pea. Similarly, As43 isolated from Vigna mungo formed separate clade IV. This means that ITS ribotyping was able to separate the Ascochyta spp. on the basis of host specificity.
The genetic distance within individuals of a species was 0.007, 0.005, and 0.001 for A. pisi, A. pinodella, and A. pinodella, respectively. The lower genetic distance within pathogenic populations of a species indicates lower reproductive barrier between them. The lowest genetic distance within individuals of A. pinodes indicates its ability for sexual reproduction in nature. In fact, A. pinodes being homothallic [40] has no barrier to sexual exchange between individuals. A higher genetic distance within A. pinodella on the other hand, indicates to the contrary. In A. pinodella, formation of sexual fruiting body (pseudoperithecia) is not usually observed in nature. There is only a single report of teleomorphic stage of A. pinodella [9]. On the basis of RFLP data and comparison of crosses made with single ascospore versus pooled ascospores inoculum, they demonstrated that this fungus is heterothallic. The high genetic distance (0.007) within individuals of A. pisi indicates that this barrier is stronger than other two. A. pisi has no reported teleomorph yet and Peever et al. [36] on the basis of unpublished data on PCR amplification of MAT 1–2 HMG-box region of A. pisi isolates indicated that nature of heterothallism is similar to other heterothallic Ascochyta spp. This has several implications in Himachal Pradesh. The higher ability of A. pinodes populations to undergo sexual reproduction will generate higher diversity within individuals. This means A. pinodes has better adaptability to survive under adverse conditions. This can be ascribed to the predominance of A. pinodes on pea currently over the earlier dominance of A. pisi [11].
One isolate (FJ032642) from common bean clustered together with clade accommodating A. pinodes and A. pinodella. Their pairwise and mean/net genetic distance with other Ascochyta spp. being low indicated their similarity. This implies that either the isolate is A. pinodes or A. pinodella, or there is a chance change in the nucleotide sequence of the ITS region. The former probability is ruled out because of different traits in the isolate viz., symptoms, colony color, and associated vegetative characteristics as compared to other Ascochyta isolates showing genetic segregation of their ITS sequences. However, there is a least likelihood of change in rDNA sequence because of its widespread use in fungal taxonomy. Fatehi and Bridge [19] argued that despite this gene cluster having been derived from single ancestry, the different copies within a replicated nucleus are liable to mutate during crossing over which could be true in the present case. Such changes in gene cluster might include the presence of introns as shown in lichenised fungi [15] or may take the form of sequence differences which preserve the size of particular region as demonstrated in arbuscular mycorrhizal fungi [43] and occurrence of more than one ITS region in filamentous fungi [22].
However, after repeated amplification of nearly 524 bp sized fragment as well as other isolates excludes the possibility that new intron of the amplicon size might have changed. Fatehi and Bridge [19] also found such ITS amplicons with different sequence homology but same size in Ascochyta isolates with one major and two minor forms of ITS. The occurrence of more than one amplification products from rRNA gene cluster has also been reported [22, 43]. Harlton et al. [22] suggested that these different ITS regions may be associated with different nuclei where thallus is bi-nucleate or multinucleate. The Ascochyta cultures used were bi-nucleate and thus different ITS cluster has been amplified, associated with a particular nucleus. The inability to detect other types of ITS clusters might be due to competitive nature of PCR reaction or a case of contamination. The possibility of contamination seems to be remote because cultures were axenic and derived from single conidium.
Genetic Diversity Analysis Using Molecular Markers
RAPD Analysis
Eight 10-mer primers viz., OPA-02, OPA-09, OPA-13, OPD-11, OPQ-13, OPQ-18, S-1466, and S-144 producing consistent polymorphic banding were selected out of 140 for RAPD analysis of 40 isolates of the test fungus. The number of scorable and polymorphic bands ranged from 5 to 8. Among the 40 isolates, all the primers showed 100 % except S-144 showing 71.43 % polymorphism. The overall polymorphism was 96.8 %. The scorable bands subjected to cluster analysis using UPGMA option of NTYSYS-pc package version 1.80 generated a dendrogram categorizing different isolates into six clusters (Fig. 2). Cluster I contained two isolates (As1 and As2) collected from district Chamba. Cluster I showed a similarity of 64.9, 61.6, 59.4, 57.2, and 56.0 % to Cluster II, Cluster III, Cluster IV, Cluster V, and Cluster VI, respectively. Cluster II consisted of 27 isolates with maximum isolates belonging to Mandi district (Jinjhiali). However, Cluster II was more close to cluster I. Cluster II showed a similarity of 61.6, 59.4, 57.2, and 56.0 % with cluster III, IV, V, and VI, respectively. All isolates in cluster III and V belonged to districts of their origin, i.e., Mandi and Kinnaur. Cluster II could be differentiated further into two sub-clusters IIa and IIb accommodating 16 and 11 isolates. Most of the isolates were more or less clustered on the basis of their geographic origin.

RAPD dendrogram of 40 Ascochyta isolates infecting pea generated by UPGMA based on Jaccard’s coefficient. Bootstrap values obtained using WinBoot of more than 25.0 % are shown at nodes
RAMS and REP-PCR categorized 40 isolates into seven clusters (Fig. 3). Cluster I contained only two isolates viz., As1 and As2 from Chamba district whereas cluster II, VI, and VII contained only one isolate each, i.e., As3, As39, and As40, respectively. Six isolates viz., As4, As7, As25, As20, As26, and As31 were categorized in cluster III in which all isolates except As4 (Chamba) and As7 (Kinnaur) were from Mandi district. Cluster IV was the largest comprising 25 isolates from all the districts except Chamba. Cluster V comprised four isolates viz., As12, As19, As3, and As2. Isolates As1 and As2 categorized in A. pisi and A. pinodella, respectively, clustered together in both the dendrograms obtained from RAPD, RAMS, and Rep-PCR analysis. RAPD came out to be a better marker in isolate identification pertaining to their geographical origin. The combined analysis showed less diversity within A. pinodes with RAMS and Rep-PCR, whereas RAPD showed maximum diversity.

REP-PCR and RAMS dendrogram generated from UPGMA based on Jaccards coefficient. Bootstrap values obtained using WinBoot of more than 50.0 % are shown at nodes
Due to the inadequacy of overlapping morphological parameters, molecular markers were used to elucidate the variation in Ascochyta complex of pea. The RAPD analysis of 40 isolates of Ascochyta spp. grouped them into six clusters (I–VI). Almost perfect correlation between RAPD clusters and locations could be established. Such grouping of isolates vis-a-vis their geographical origin has several implications in Indian context where information lacks about genetic pathogen variability. Several workers have employed molecular markers to study this pathogen but were unable to find a correlation in pathogen populations in clustering and their origin [18, 33, 53]. The geographical grouping of the isolates could be attributed to reproductive barriers between the regions. Pathogenic populations have to adapt to the environmental conditions of a region to sustain by accumulating genes (here novel alleles). If the populations of one region are unable to intermate with the populations of the other regions, movement of genes/alleles across populations is restricted leading to geographic isolation of alleles. This possibility seems plausible in Himachal Pradesh due to hilly terrain thus creating a natural geographical barrier to the pathogen spread. The reproductive isolation is also maintained due to little exchange of seed between the farmers of different regions as they use the seed of previous year crop. If sexual exchange between individuals occurs within an isolated region, sharing of novel alleles will be there between the individuals of a population. Asexual reproduction among such individuals leads to dominance of novel alleles. Since Ascochyta pinodes is homothallic, several such sexual exchanges between individuals of a region can occur. Also, asexual reproduction producing millions of pycnidiospores could be responsible for dominance of such novel alleles. Such a reproductive isolation ultimately leads to divergence between the isolated regions and uniformity within a population. These observations are in conformity with Zang et al. [55] who also found direct correlation between clusters and countries of origin in the AFLP analysis of 56 A. pinodes isolates of worldwide origin. They grouped isolates of different origin (Australia, New Zealand, and Canada) into separate clades. Further, within isolates from a country separate sub-groups were formed representing different regions.
In conclusion, it is inferred from the present study that A. pinodes dominates the Ascochyta populations in Himachal Pradesh, where pea is one of the most remunerative off-season cash crop. Different morpho-cultural characteristics categorized various isolates into three major groups with few isolates floating from one group to another on the basis of individual characteristics. However, the combined data resolved the ambiguity and placed majority of isolates in A. pinodes. Molecular characterization with RAPD, RAMS, and Rep-PCR grouped isolates according to their geographical origin. Sequencing of ITS region of test isolates and reference gene bank Ascochyta spp. revealed identity and ambiguity.
References
Adachi Y, Watanabe H, Tanabe K, Doke N, Nishimura S, Tsuge T (1993) Nuclear ribosomal DNA as a probe for genetic variability in the Japanese pear pathotype of Alternaria alternata. Appl Environ Microbiol 59:3197–3205
Anonymous (1986) Annual progress report, Department of Mycology and Plant Pathology, CSK HPKV, Palampur
Anonymous (2006) Area and production of vegetables in Himachal Pradesh. Directorate of Agriculture (HP), Shimla
Appel DJ, Gordon TR (1995) Intraspecific variation within population of Fusarium oxysporum based on RFLP analysis of the intergeneric spacer of the rDNA. Exp Mycol 19:120–128
Aradhya MK, Chan HM, Parfitt DE (2001) Genetic variability in the pistachio late blight fungus, Alternaria alternata. Mycol Res 105:300–306
Bagley MJ, Anderson SL, May B (2001) Choice of methodology for assessing genetic impacts of environmental stressors: polymorphism and reproducibility of RAPD and AFLP fingerprints. Ecotoxicology 10:239–244
Barve MP, Arie T, Salimath SS, Muehlbauer FJ, Peever TL (2003) Cloning and characterization of the mating type (MAT) locus from Ascochyta rabiei (teleomorph: Didymella rabiei) and a MAT phylogeny of legume-associated Ascochyta spp. Fungal Genet Biol 39(2):151–167
Beasse C, Ney B, Tivoli B (2000) A simple model of pea growth affected by Mycosphaerella pinodes. Plant Pathol 49:187–200
Bowen JK, Lewis BG, Mathews P (1997) Discovery of the telemorph of Phoma medicaginis var. pinodella in culture. Mycol Res 101:80–84
Buchanan PK (1987) A repraisal of Ascochytula and Ascochytella (Coelomycetes). Mycological Papers 156:1–83
Chaudhary MR (1988) Studies on Ascochyta blight and pod spot of pea (Pisum sativum L.) caused by Ascochyta pisi. CSKHPKV, Palampur
Chilvers MI, Rogers JD, Dugan FM, Stewart JE, Chen W, Peever TL (2009) Didymella pisi sp. nov., the teleomorph of Ascochyta pisi. Mycol Res 113(Pt 3):391–400. doi:10.1016/j.mycres.2008.11.017
Davidson JA, Hartley D, Priest M, Herdina MK, McKay A, Scott ES (2009) A new species of Phoma causes Ascochyta blight symptoms on field peas (Pisum sativum) in South Australia. Mycologia 101(1):120–128
Davidson JA, Krysinska-Kaczmarek M, McKay A, Scott ES (2012) Comparison of cultural growth and in planta quantification of Didymella pinodes, Phoma koolunga and Phoma medicaginis var. pinodella, causal agents of ascochyta blight on field pea (Pisum sativum). Mycologia 104(1):93–101
DePriest PT (1993) Small subunit rDNA variation in a population of lichen fungi due to optional group I introns. Gene 134:67–74
Ellsworth DL, Rittenhouse KD, Honeycutt RL (1993) Artifactual variation in randomly amplified polymorphic DNA banding patterns. Biotechniques 2:214–217
Excoffier L, Laval G, Schneider S (2005) Arlequin ver3.0: an integrated software package for population genetics data analysis. Evolutionary Bioinformatics Online 1:47–50
Faris-Mokaiesh S, Boccara M, Denis JB, Derrien A, Spire D (1996) Differentiation of the “Ascochyta complex” fungi of pea by biochemical and molecular markers. Curr Genet 29(2):182–190
Fatehi J, Bridge PD (1998) Detection of multiple rRNA-ITS regions in isolates of Ascochyta. Mycol Res 102:762–766
Fatehi J, Bridge PD, Punithalingam E (2003) Molecular relatedness within the “Ascochyta pinodes-complex”. Mycopathologia 156(4):317–327
Hall TA (1999) BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucl Acids Symp Ser 41:95–98
Harlton CE, Levesque CA, Punja ZK (1995) Genetic diversity in Scelerotium (Athelia) rolfsii and related species. Phytopathology 85:1269–1281
Herzberg M, Fischer R, Titze A (2002) Conflicting results obtained by RAPD-PCR and large-subunit rDNA sequences in determining and comparing yeast strains isolated from flowers: a comparison of two methods. Int J Syst Evol Microbiol 52:1423–1433
Hudson RR, Slatkin M, Maddison WP (2002) Estimation of levels of gene flow from DNA sequence data. Genetics 132:583–589
Jones LK (1927) Studies on the nature and control of blight, leaf and pod spot and foot rot of peas caused by species of Ascochyta. NY State Agri Res Bull 547:45
Kaiser WJ, Viruega JR, Peever TL, Trapero A (2008) First report of Ascochyta blight outbreak of pea caused by Ascochyta pisi in Spain. Plant Dis 92:1365
Kuo BA, Gonzalez IL, Gillespie DA, Sylvester JE (1996) Human ribosomal RNA variants from a single individual and their expression in different tissues. Nucleic Acid Res 24:4817–4824
Lawyer AS (1984) Disease caused by Ascochyta spp. In: Hagedorn DJ (ed) Compendium of pea diseases. The American Phytopathological Society, Minnosota, pp 11–15
Librado P, Rozas J (2009) DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics 25:1451–1452
Linford MB, Sprague R (1927) Species of Ascochyta parasitic on the pea. Phytopathology 17:381–398
Mahuku GS, Riascos JJ (2004) Virulence and molecular diversity within Colletotrichum lindemuthianum isolates from Andean and Mesoamerican bean varieties and regions. Eur J Plant Pathol 110:253–263
Neuveglise C, Brygoo Y, Vercambre B, Riba G (1994) Comparative analysis of molecular and biological characteristics of strains of Beauvaria brongniartii. Mycol Res 98:322–328
Onfroy C, Tivoli B, Corbière R, Bouznad Z (1999) Cultural, molecular and pathogenic variability of Mycosphaerella pinodes and Phoma medicaginis var. pinodella isolates from dried pea (Pisum sativum) in France. Plant Pathol 48(2):218–229. doi:10.1046/j.1365-3059.1999.00323.x
Padder BA, Sharma PN, Sharma OP, Kapoor V (2007) Genetic diversity and gene flow estimates among five populations of Colletotrichum lindemuthianum across Himachal Pradesh. Physiol Mol Plant Pathol 70:8–12. doi:10.1016/j.pmpp.2007.05.003
Padder BA, Sharma PN, Sharma OP, Kapoor V (2008) Ribosomal DNA analysis of Colletotrichum lindemuthianum virulences from Himachal Pradesh (India). Appl Biol Res 10:6–10
Peever TL, Barve MP, Stone LJ (2007) Evolutionary relationships among Ascochyta species infecting wild and cultivated hosts in the legume tribes Cicereae and Vicieae. Mycologia 99(1):59–77
Perez T, Albornoz J, Dominguez A (1998) An evaluation of RAPD fragment reproducibility and nature. Mol Ecol 7:1347–1357
Pryor BM, Michailides TJ (2002) Morphological, pathogenic and molecular characterization of Alternaria isolates associated with Alternaria late blight of pistachio. Phytopathology 92:406–416
Punithalingam E (1979) Graminicolous Ascochyta species. Mycological papers, vol 142. Commonwealth Mycological Institute, Kew
Punithalingam E, Holliday P (1972) Mycosphaerella pinodes. Descriptions of pathogenic fungi and bacteria, vol 342. Commonwealth Mycological Institute, Kew
Sagar V (1992) Studies on host resistance and management of Ascochyta blight in peas. CSK HPKV, Palampur
Salam M, MacLeod W, Salam K, Maling T, Barbetti M (2011) Impact of climate change in relation to Ascochyta blight on field pea in Western Australia. Australas Plant Pathol 40(4):397–406. doi:10.1007/s13313-011-0050-1
Sanders IR, Groppe AM, Boller K, Wiemken TA (1995) Identification of ribosomal DNA polymorphisms among and within spores of the Glomales: application to studies on the genetic diversity of arbuscular mycorrhizal fungal communities. New Phytol 130:419–427
Sharma PN, Kaur M, Sharma OP, Sharma P, Pathania A (2005) Morphological, pathological and molecular variability of Colletotrichum capsici, the cause of fruit rot of chillies in the sub-tropical region of North-Western India. J Phytopathol 153:232–237
Slade RW, Moritz C, Heideman A, Hale PT (1993) Rapid assessment of single-copy nuclear DNA variation in diverse species. Mol Ecol 2:359–373
Stone RE (1913) The life history of Ascochyta on some leguminous plants. PhD, Cornell University, Ithaca
Sutton BC (1980) The coelomycetes. CAB International Mycological Institute, Kew
Tamura K, Dudley J, Nei M, Kumar S (2007) MEGA4: molecular evolutionary genetics analysis (MEGA) software version 4.0. Mol Biol Evol 24:1596–1599
Thompson JD, Gibson JJ, Plewniak F, Jeanmougin F, Higgins DG (1997) The Clustal X windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acid Res 24:4876–4882
Upholt WB (1977) Estimation of DNA sequence divergence from comparison of restriction endonuclease digests. Nucleic Acid Res 4:1257–1265
Vaughan RE (1913) Mycosphaerella pinodes, the ascigenous stage of Ascochyta pisi. Phytopathology 3:71
Wallen VR, Cuddy TF, Graringer PN (1967) Epidemiology and control of Ascochyta pinodes on field peas in Canada. Can J Plant Sci 47:395–403
Wang H, Hwang SF, Chang KF, Turnbull GD, Howard RJ (2000) Characterization of Ascochyta isolates and susceptibility of pea cultivars to the ascochyta disease complex in Alberta. Plant Pathol 49(5):540–545. doi:10.1046/j.1365-3059.2000.00489.x
White TJ, Bruns T, Lee S, Taylor J (1990) Amplification and direct sequencing of fungal ribosomal RNA genes for phylogenetics. In: PCR protocols: a guide to methods and applications. Academic Press, San Diego
Zang JX, Fernando WGD, Xue AG (2003) Virulence and genetic variability among isolates of Mycosphaerella pinodes. Plant Dis 87:1376–1383
Acknowledgments
The authors are highly thankful to Indian Council of Agricultural Research, New Delhi for financial assistance in the form ad hoc research project.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article
Cite this article
Padder, B.A., Kapoor, V., Kaushal, R.P. et al. Identification and Genetic Diversity Analysis of Ascochyta Species Associated with Blight Complex of Pea in a Northwestern Hill State of India. Agric Res 1, 325–337 (2012). https://doi.org/10.1007/s40003-012-0033-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40003-012-0033-7