
Abstract
Culture and characterization of fetal derived cells have received particular attention because of their easy access and possible source of stem cells for the study of development and differentiation. The present study was carried out to establish buffalo fetal fibroblast cell culture, and their longevity, expression patterns of pluripotency markers with prolonged passage and in vitro induced differentiation ability. Buffalo fetal fibroblasts were isolated from sub-dermal region and their primary culture was initiated in re-calcified buffalo plasma drops. On sufficient growth of primary culture, these cells were trypsinized and passaged at 80% confluency with a split ratio of 1:2 for multiplication of cells. Cryopreservation of cells was also performed at intervals of passages (P) 5 from confluent cultures, and the representative cells were allowed to proliferate in continuous cultures. These cells started emerging and anchoring to cell culture flasks within 24 h and survived up to P47 in 185 days with average passage time of 3.9 days. Expression of alkaline phosphatase and pluripotency genes viz., OCT-4, NANOG and SOX-2 were examined up to P45. Further, changes in relative expression of transcriptional factors were determined by quantitative real time PCR and found up-regulation of all the three genes up to P15 followed by up-regulation of SOX-2 up to P45 but down-regulation of Nanog. Upon induced differentiation, these cells differentiated into adipogenic and osteogenic cells as confirmed by oil red O and alizarin red stains, respectively. This study indicates that buffalo fetal fibroblast cells have characteristics of stem cells.
Similar content being viewed by others


Introduction
Fibroblasts are the most ubiquitous cells in complex organisms. They are the main cells of structural framework for animal tissues and play an important role in repair and healing of damaged organs. In mammals, the epidermis of skin is continually refurbished every day. The turnover time of the epidermis is about 60 days in human and 7 days in mice [20]. This rapid remodeling is maintained by stem cells that are capable of self-renewal and supply differentiated cells in a constant manner. Over last two decades, interest on stem cell research is due in large part to the recognition that a broad variety of adult tissues contain stem cells, and these somatic stem cell populations exhibit pluripotent potential. Generation of stem cells from adult tissues like skin has received particular attention because of their accessibility and the possibility that patient could act as a stem cell donor [25]. Moreover, multipotent stem cells that can form neural and adipose cells have been isolated from the fetal skin of mouse [32] and pig [4]. These cells also expressed the neural progenitor marker, nestin, as well as genes that are critical for pluripotency such as Oct-4 and Stat3 [4]. These findings indicate that multiple classes of stem cells with different differentiation potentials are present in the skin, making skin a valuable source of stem cells for the study of development, differentiation, and easier accessibility for in vitro model system to investigate the properties and opportunities of non-embryonic stem cells. Skin-originated stem cells have been differentiated to form cells with characteristics of neuron, astrocytes and adipocytes in porcine [4, 14], mouse [7] and human [21] but no reports are available on generation of stem cells derived from fetal skin fibroblasts from buffalo, one of the important species of farm animals.
Furthermore, buffalo skin provides an easy accessible source of tissue for the isolation of fibroblast cells. Small skin biopsies are sufficient and can be obtained in a minimal invasive way. Although various attempts have been made to establish ES cell-like cell lines from farm animals like sheep [19], pig [35] and cattle [28, 36]. Recently, ES cell-like cells have been reported from buffalo [1, 6, 13, 27, 33], but true stem cell lines in majority of the farm animals are not established. Reason being due to lack of defined species-specific stemness markers [16] or lack of understanding of species specific mechanism that promote cell pluripotency [31] in domestic animals. However, pre-implantation development in mammals shows remarkable differences between species, possibly influencing the mechanism responsible for the formation of a pluripotent cell population. For instance, mouse embryos form an egg cylinder after implantation, whereas human, bovine, and porcine embryos have a planar morphology [3], which could explain why ES cell lines from species such as cattle and pig have not been established [8]. The establishment of ES cell lines is also associated with ethical concerns. To overcome these problems, fibroblast derived stem cells offer a great potential to investigate cell differentiation, cell fate, and the associated cell signaling pathways. In order to obtain more insight in fetal skin fibroblast cells, the present study was carried out to establish buffalo fibroblast cell culture, and their longevity, expression patterns of pluripotency markers with prolonged passage and in vitro induced differentiation ability.
Materials and Methods
Primary Culture and Cell Growth
Buffalo gravid uteri at 50–100 days gestation were obtained from abattoir, washed 2–3 times with isotonic saline fortified with 400 IU/ml penicillin and 500 μg/ml streptomycin and transported to the laboratory within 6 h. The fetus was located by uterine incision and taken out. The tissue was collected from fetus (n = 9), and cells were cultured in three replicates for each fetus processed. The fetal sub-dermal biopsies were taken from upper part of foreleg and minced by surgical blade into smaller pieces and washed 4–6 times with DPBS. The tissue pieces were transferred on re-calcified buffalo plasma droplets (20 μl) in 25-cm2 cell culture flasks. After placing the tissue pieces on the drops, it was allowed to coagulate for ~30 min at 37°C for the attachment of tissue to the surface of the culture flask. The adhered tissue pieces were cultured in culture medium containing DMEM with 10% FBS, 2 mM l-glutamine, 1% (v/v) nonessential amino acids, 1% (v/v) vitamins, 1% antibiotics (penicillin, streptomycin and amphotericin) in a CO2 incubator (5% CO2 in humidified 95% air at 38°C). On confluency of primary cultures, these cells were trypsinized (trypsin–EDTA 0.25%) and passaged with a split ratio of 1:2 for multiplication of cells. In this study the average passage time was taken as population doubling time.
Cryopreservation and Thawing
Cells from confluent cultures were cryopreserved at the interval of 5 passage and the representative cells were allowed to proliferate in continuous culture. Cryopreservation of cells was performed as per protocol reported previously [37]. Briefly, the confluent cultures were treated with 0.25% trypsin–EDTA and washed by centrifugation (200×g at 4°C, 5 min) with cell culture medium to remove trypsin–EDTA. The cell pellet thus obtained was resuspended in pre-cooled (4°C) cryopreservation medium (culture medium with 10% DMSO and 20% FBS) in 1 ml cryovials. These cryovials were placed at −40°C for ~24 h before plunging in liquid nitrogen (−196°C). After 7 days of cryopreservation, the cells were thawed in a water bath (37°C) for ~15 s. The cell contents were suspended in culture medium and centrifuged twice at 200×g for 10 min. The cell pellet thus obtained was resuspended in culture medium and plated in 25 cm2 culture flask. A fraction of cells was used to evaluate the cell survival rate with trypan blue dye exclusion method using Neubar haemocytometer chamber under phase contrast microscope (Nikon Eclipse Ti, Japan).
Expression of Pluripotency Markers
Alkaline phosphatase activity and expression of pluripotency genes OCT-4, NANOG and SOX-2 were studied at P2, 5, 10, 15, 20, 25, 30, 35, 40 and 45.
Alkaline Phosphatase (AP)
AP activity was studied using AP staining kit (Sigma Chemical Co.) following manufacturer’s instructions. Briefly, monolayer of fibroblast cells was washed twice with PBS, fixed in citrate–acetone–formaldehyde fixative for 1 min, washed thrice with de-ionized water and incubated at room temperature for 15 min in the presence of alkaline dye under dark condition. The cells were rinsed again with de-ionized water, counter stained with Neutral Red and observed under phase contrast microscope. Cells accepting red stain were considered AP positive.
Reverse Transcription PCR (RT-PCR)
Expression of pluripotency genes was analyzed by RT-PCR as reported earlier [37]. Briefly, total RNA was isolated from cells at different passages using Gen Elute Mammalian Total RNA miniprep Kit (Sigma, RTN70). RNA concentration was measured at 260 nm absorbance using spectrophotometer (Picodrop, UK). DNase-I treated RNA served as a template for reverse transcription and amplification. RT-PCR was performed using one step RT-PCR Kit (Life Technologies, India Pvt. Ltd.) using random and oligo dT primers. The RT reaction was initiated with 3–5 μg of total RNA in RNase-free water, random primers (100 μM) and oligo dT (50 μM), heated at 65°C for 5 min., and then immediately kept on ice. The heat-treated RNA was added to RT reaction mixture containing Super Script® III Reverse Transcriptase (200 U/μl), 5× RT buffer, di-thiothretol (0.1 M), and dNTPs (10 mM). The conditions for reverse transcription included heating at 65°C for 5 min, incubation on ice for 1 min, 25°C for 5 min, and again at 50°C for 50 min, followed by inactivation of reaction at 80°C for 15 min. The first strand complementary DNA (cDNA) obtained was further amplified using gene-specific primers. PCR mix (25 μl) was prepared by using cDNA, 10× PCR buffer, 25 mM MgCl2, 10 mM dNTP mix, 10 μM forward and reverse primers each, and Taq DNA polymerase (5 U/μl). The conditions for amplification included 36 cycles each consisting of denaturation at 94°C for 30 s, annealing at touch down of 56–54°C for 30 s, elongation at 72°C for 30 s, and final extension at 72°C for 5 min. The PCR primers and the reaction conditions used are as summarized in Table 1. A set of reaction without template cDNA was used as −ve control for PCR and in vitro produced blastocysts as positive control. β-Actin and GAPDH were amplified at each stage as house keeping marker genes. The amplified DNA fragments were resolved on 2% agarose gel containing ethidium bromide (0.5 μg/ml final concentration) and visualized under gel documentation system (Alpha Imager, Alpha Innotech, USA). The gene-specific bands were excised and purified using AuPrep Gel Extraction Kit (Life Technologies India Pvt. Ltd.) for further analysis.
Quantitative PCR
Real-time quantitative PCR was conducted with Q-PCR 600548 Kit (Stratagene, La Jolla, CA, United States) using SYBR green fluorescence dye as reported earlier [37]. Briefly, the reaction mixture was set up using nuclease-free PCR grade water to adjust the final volume to 20 μl including experimental DNA with 2× SYBR Green Master mix, 10 μM Primer (Forward + Reverse) followed by PCR steps comprising of initial denaturation at 95°C for 10 min, subsequent 40 cycles each consisting of denaturation at 95°C for 30 s, annealing step at 55–60°C for 1 min and elongation at 72°C for 1 min. The primers used for RT-PCR were the same as used for reverse transcription (Table 1). The thermal cycler was set to detect and report fluorescence both during the annealing step and the extension step of each cycle. cDNA created using RT-PCR from P2 was used as calibrator for comparison of quantitative expression of OCT-4, NANOG and SOX-2 genes.
Karyotype
Actively proliferating cells were incubated with colchicine (0.1 μg/ml) for 4 h at 37°C. The cells were washed twice with DPBS, trypsinized, suspended in a chilled hypotonic solution (68 mM KCl) and incubated for 20 min at 37°C and then fixed for 10 min in chilled fixative (methanol and glacial acetic acid, 3:1). The pellet was finally suspended in 5 ml of chilled fixative for another 10 min. Metaphase spreads were prepared by dropping the cell suspension onto ice cold glass slides. The air-dried cell spreads were stained with Giemsa stain and observed under oil immersion (1,000×) for chromosomal sketch.
In Vitro Induced Differentiation
For differentiation, cultured fetal fibroblast cells were dissociated by trypsinization, centrifuged and then cultured in lineage specific differentiation media. The cells were plated onto tissue culture grade 6-well plates. For osteogenic differentiation, medium containing DMEM supplemented with 10% FBS, 10−7 M dexamethasone, 50 μM ascorbic acid and 10 mM β-glycerol phosphate was used. The differentiation of cells was assessed morphologically and stained with alizarin red which indicate the calcium mineralization in cells. To induce adipogenic differentiation, the cells were cultured in DMEM containing 10% FBS, 2 mM l-glutamine, 1% (v/v) nonessential amino acids, 1% (v/v) vitamins, 1% antibiotics and supplemented with 10 mM nicotinamide. The presence of intracellular lipid globules indicative of adipogenic differentiation was assessed by staining cells with oil-Red O solution on day 14 and 21. The medium was replaced twice a week.
Alizarin Red Staining
Medium was aspirated from culture wells and cells were fixed in 4% paraformaldehyde for 1 h, washed twice with water, added Alizarin red solution to cover the cells and then incubated at room temperature for 30 min after incubation, stain was removed and cells were washed 4 times thoroughly with water and finally ~1 ml water was left to prevent dryness of cells. The cells were visualized under phase contrast microscope.
Oil-Red O Staining
Cells were washed with PBS, fixed in 4% paraformaldehyde for 30 min, washed again with PBS, then incubated with filtered 0.16% oil red O diluted in isopropanol (w/v) for 10 min. The oil red O stain was aspirated and the dishes were washed with water for 2–3 min, and subsequently visualized and photographed under phase contrast microscope and images were taken immediately following staining.
Results
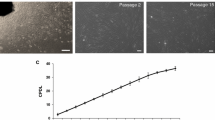
In the present study, fibroblast cells derived from buffalo fetal skin started emerging and anchoring to cell culture flasks within 24 h (Fig. 1a) after placing the minced tissue on re-calcified plasma drops. Majority of cells adhered to the surface of culture flasks. The attached cells expanded with spindle-shaped morphology resulting in primary cultures (Fig. 1b) with first confluency time of 4 days. In initial cultures the population doubling time up to P10 was 2.7 days and then it increased gradually in later passages up to 4.5 days. These fibroblast cells had been cultured continuously for P47 that took 185 days with average passage time of 3.9 days. After P45 cell growth was slowed down and finally stopped growing in culture after P47. These cells changed their morphology with increased size, retarded proliferation rate and finally stopped dividing. Upon comparison of viability of fibroblast cells before cryopreservation and post-thaw showed 98.9 and 83.8% respectively as assessed by trypan blue dye exclusion method (Fig. 1c). The culture behavior and morphology of freeze–thawed cells were similar as that of fresh cells before cryopreservation.

Derivation and culture of fibroblast cells from fetus skin. a Fibroblast cells emerging from fetal skin tissue at 24 h of culture initiation (×100). b Confluent culture (80–90%) of fibroblast cells in primary culture (×100). c Viability of fibroblast cells assessed by trypan blue dye (100 ×)
The expression of AP (Fig. 2a) taken as marker of pluripotency was found positive in P2 to P45 when checked at an interval of P5. Beside this, a normal chromosomal profile (Fig. 2b) was observed up to P45 indicating genomic integrity of the cells during prolonged culture. RT-PCR analysis of the total RNA isolated from cultured fibroblast cells showed that these cells expressed the transcription factors of pluripotency, OCT-4, NANOG and SOX-2 (Fig. 2c). The expression was observed positive in the cells during various passages i.e. at P2, P5, P10, P15, P20, P25, P30, P35, P40 and P45 and these results were also confirmed by sequencing of the PCR products of all three genes at passage no 15. The amplimer sequences were aligned with published sequences from other species and analyzed using the Basic Local Alignment Search Tool (BLAST; National Center for Biotechnology Information, US National Library of Medicine, Bethesda, MD, USA; http://www.ncbi.nlm.nih.gov). The sequence of Oct-4 (Bubalus bubalis) had 93% identity with Oct-4 (Bos taurus) mRNA and 90% with Pig DNA sequence from available clone CH242-102G9 on chromosome 7. Sox-2 blast showed 98% homology with Bos taurus and 96% identity with pig DNA sequence from clone CH242-330B10 on chromosome 13. Alignment of Nanog amplimer had 95% homology with B. bubalis homeobox transcription factor and 91% with B. taurus homeobox transcription factor Nanog mRNA. Expression of β-actin and GAPDH were used as house keeping gene for RT-PCR and normalizing genes for quantitative real time PCR. Moreover, changes in relative expression of transcriptional factors were determined by quantitative real time PCR and found up-regulation of OCT-4 (5.82), NANOG (1.64) and SOX-2 (5.84) fold shown in Fig. 3a–c respectively up to P15 followed by up-regulation of SOX-2 up to P45 but down-regulation of OCT-4 and NANOG. Furthermore, we also examined the ability of fibroblast cells for in vitro induced differentiation into adipocyte and osteocyte cells. When fibroblast cells were cultured in osteogenic differentiation medium, the cells started changing morphology into osteoblasts (Fig. 4a) after 7 days of incubation and it was observed till 21st day of culture. The cells were stained with Alizarin red on day 14 and 21 of culture in osteogenic conditions; showed positive expression of alizarin confirmed the depiction of calcium deposits in differentiated cells (Fig. 4b). When cells were cultured under adipogenic conditions, they differentiated into adipocytes (Fig. 4c) and exhibited high intensity of oil red O stain in the cytoplasm of cells on 14 day onwards of culture, signifying the presence of lipid vacuoles (Fig. 4d).

Characterization of buffalo fetal fibroblast cells at different passages. a Positive AP staining of buffalo fetal fibroblast cells at passage 5 (×100), b RT-PCR analysis of gene expression in fetal fibroblast cells, where Lane 1 100 bp Ladder, 2 OCT-4, 3 OCT-4 +ve control, 4 NANOG 5 NANOG +ve control, 6 SOX-2, 7 SOX-2 +ve control, 8 β-actin, 9 β-actin +ve control, 10 GAPDH, 11 GAPDH +ve control, 12 Negative control, 13 100 bp Ladder

Real time-PCR analysis of genes expression at P15 compared with P2 in fetal skin derived fibroblast cells. a Relative expression level of OCT-4, b NANOG, and c SOX-2

In vitro induced differentiation of fetal skin derived fibroblast cells. a Osteocytes (×100), b osteogenic differentiated cells depicting alizarin red (×200), c adipocytes (×200), and d adipogenic differentiated cell exhibiting oil red O stain (×100)
Discussion
In the present study primary cultures were initiated from buffalo fetal skin tissues after fixing on tiny drop of re-calcified buffalo plasma as an initial adherent support. The tissues placed directly on the culture flasks did not result in primary cultures. The emergence and anchoring of cells in culture flasks was observed within 24 h. Similar procedure was used to obtain primary culture of fibroblasts from sub dermal tissues and essentiality of autologous plasma micro-drops for fixing and providing initial stimulation of fetal fibroblast cells have been reported by Kues et al. [12, 11]. In the present study, DMEM containing 10% serum supported growth of cultured fibroblast cells up to P47 with average passage time of 3.9 days mimic the result obtained by Kues et al. [12] for culture of fetal fibroblasts from murine and porcine. Also porcine fibroblasts cultured in high serum supplementation activated OCT-4 gene and lost contact inhibition, resulting in the formation of three-dimensional colonies [12]. In contrast, our study showed long term cell cultures up to 185 days and their proliferation rate was more up to 10 passages, then decreased later possibly due to accumulations of more non-dividing cells. Our findings also confirm results of earlier study conducted by Gupta et al. [5].
The present findings also provided evidence for the presence of stem cell characteristics by expressions of AP, OCT-4, NANOG, SOX-2 and differentiation ability. This is interesting because the expression of these markers is a characteristic of ES cells [2, 22] and pluripotency of ES cells is governed via a regulatory complex of these essential transcription factors [14]. A trinity of nuclear regulators Oct-4, Nanog and Sox-2 govern pluripotency in vivo and in vitro [17]. These genes are dominant in maintaining pluripotent state of cells while suppressing the functional expression and activity of lineage specific factors [17, 26]. The expression of AP is considered as primary indicator of ES like cells by various researchers in almost all species. In this study, fibroblast cells were found to express AP, as also reported by Kues et al. [12] in high serum fibroblast cultures. The consistent expression of these genes by buffalo fetal fibroblast throughout the culture period of 185 days provides strong support that these cells have characteristics of undifferentiated pluripotent scenery. Kues et al. [12] reported the presence of somatic stem cell population in explant cultures derived from mouse and pig fetuses which had extended proliferative capacity along with expression of stem cell specific markers including Oct-4. These transcription factors have also been expressed in fetal fibroblast and buffalo amniotic fluid derived cells [9, 36]. Furthermore, quantitative real time PCR analysis for differential expression patterns revealed up-regulation of OCT-4, NANOG and SOX-2 up to P15, whereas, expression of NANOG was down-regulated in the cells at P45 indicating that the maintenance of OCT-4 expression at a critical concentration is necessary to sustain ES cell self-renewal and the increased expression triggers differentiation into endoderm or mesoderm, while its suppression causes ES cells to become trophectoderm [18]. Whereas, expression level of Nanog is highly variable in ES cells in contrast with apparent homogeneity of Oct-4, Sox2 and down-regulation of Nanog indicate exit of culture from self renewal as evident for many ES cells [24]. Similar down-regulation of NANOG was seen in this study at P45 which exit the growth of fetal fibroblasts from culture. There are no data available for comparison on expression level of these stem cell marker genes in buffalo. However, the variation in level of expression of these pluripotency genes in buffalo amniotic fluid cells at different passages has been reported earlier [36]. Recently, generation of induced pluripotent stem (iPS) cells from somatic cells with defined transcription factors in mouse [30] and human [29] provide a promising source of patient specific cells for cell replacement therapies as well as in vitro models for a variety of genetic diseases [34]. Now it has been demonstrated by various workers, that cells having ability to express Oct-4 and Sox2, would allow much easier and effective induction of pluripotency by the introduction of just one transcription factor, Kfl-4 or c-Myc [10, 23]. So, the expression of OCT-4, NANOG and SOX-2 in buffalo fetus derived skin fibroblast cells could be used as a potential source for generation of iPS cells in this species. The fibroblast cells studied were subsequently shown to differentiate into adipocyte and osteocyte cells. In adult porcine skin derived stem cell-like cells, similar differentiation ability to ectoderm and mesoderm was also observed [14]. Further study conducted on differentiation potential of human dermal skin-derived fibroblasts cultured under appropriate inducible conditions showed the differentiation of both adipogenic and osteogenic lineages [15]. In our observation, differentiation of cells in adipogenic lineage started after 1 week, whereas, osteogenic lineage appeared after 2 weeks in inductive conditions. But in other study, time reported varied 3–4 weeks for differentiation from human fibroblastic mesenchymal stem cell-like cells into adipogenic and osteogenic lineages [15].
This study has further raised an important query regarding the use of fetal fibroblasts as feeder layers for establishing ES cell-like cells in domestic animals. Since these cells are expressing the pluripotency marker genes in domestic species, it needs to give a fresh look whether the pluripotency gene expression is from the ES cells or fibroblast cells used as feeder layer. The quantitative gene expression shall further help in determining more appropriate stage of fetal fibroblast cells as donor cells and to understand its mechanism of reprogramming in nuclear transfer experiments.
References
Anand T, Kumar D, Singh MK, Shah RA, Chauhan MS, Manik RS, Singla SK, Palta P (2011) Buffalo (Bubalus bubalis) embryonic stem cell-like cells and pre-implantation embryos exhibit comparable expression of pluripotency-related antigens. Reprod Domest Anim 46:50–58
Baal N, Reisinger K, Jahr H, Bhole RM, Linag O, Münstedt K, Rao CV, Preissner KT, Zygmunt MT (2004) Expression of transcription factor Oct-4 and other embryonic genes in CD133 positive cells from human umbilical cord blood. Thromb Haemost 92:767–775
Behringer RR, Wakamiya M, Tsang TE, Tam PP (2000) A flattened mouse embryo: leveling the playing field. Genesis 28:23–30
Dyce PW, Hai Z, Jesse C, Julang L (2004) Stem cells with multilineage potential derived from porcine skin. Biochem Biophys Res Commun 316:651–658
Gupta N, Taneja R, Pandey A, Mukesh M, Singh H, Gupta SC (2007) Replicative senescence, telomere shortening and cell proliferation rate in Gaddi goat’s skin fibroblast cell line. Cell Biol Int 31:1257–1264
Hunag B, Li T, Wang XL, Xie TS, Lu YQ, da Silva FM, Shi DS (2010) Generation and characterization of embryonic stem like cell lines derived from in vitro fertilization buffalo (Bubalus bubalis) embryos. Reprod Domest Anim 45:122–128
Karl JL, Ian AM, Pleasantine M, Smith KM, Mahnaz A, Fanie BH, Jeff B, Adrienne J, Nao RK, Jean GT, David RK, Patricia AL, Victor R, Chi-Chung H, Freda DM (2004) A dermal niche for multipotent adult skin-derived precursor cells. Nat Cell Biol 6:767–775
Keefer CL, Pant D, Blomberg L, Talbot NC (2007) Challenges and prospects for the establishment of embryonic stem cells lines of domestic ungulates. Anim Reprod Sci 98:147–168
Kim J, Lee Y, Kim H, Hwang KJ, Kwon HC, Kim SK, Cho DJ, Kang SG, You J (2007) Human amniotic fluid-derived cells have characteristics of multipotent stem cells. Cell Prolif 40:75–90
Kim JB, Zaehres H, Wu G, Gentile L, Ko K, Sebastiano V, Araúzo-Bravo MJ, Ruau D, Han DW, Zenke M, Schöler HR (2008) Pluripotent stem cells induced from adult neural stem cells by reprogramming with two factors. Nature 454:646–650
Kues WA, Anger M, Carnwath JW, Paul D, Motlik J, Niemann H (2000) Cell cycle synchronization of porcine fetal fibroblasts. Effects of serum deprivation and reversible cell cycle inhibitors. Biol Reprod 62:412–419
Kues WA, Peterson B, Mysegades W, Carnwath JW, Niemann H (2005) Isolation of murine and porcine fetal stem cells from somatic tissue. Biol Reprod 72:1020–1028
Kumar D, Anand T, Singh KP, Singh MK, Shah RA, Chauhan MS, Singla SK, Palta P, Manik RS (2011) Derivation of buffalo embryonic stem-like cells from in vitro-produced blastocysts on homologous and heterologous feeder cells. J Assist Reprod Genet 28:679–688
Lermen D, Erwin G, Paul WD, Hagen VB, Paul M (2010) Neuro-muscular differentiation of adult porcine skin derived stem cell-like cells. PLoS ONE 5:e8968
Lorenz K, Marit S, Eva S, Thomas R, Juergen S, Schulz-Siegmund M, Augustinus B (2008) Multilineage differentiation potential of human dermal skin-derived fibroblasts. Exp Dermatol 17:925–932
Munoz M, Rodriguez A, De Frutos C, Caamano JN, Diez C, Facal N, Gomez E (2008) Conventional pluripotency markers are unspecific for bovine embryonic-derived cell-lines. Theriogenology 69:1159–1164
Niwa H (2007) How is pluripotency determined and maintained. Development 134:635–646
Niwa H, Miyazaki J, Smith AG (2000) Quantitative expression of Oct-3/4 defines differentiation, dedifferentiation or self-renewal of ES cells. Nat Genet 24:372–376
Notarianni E, Galli C, Laurie S, Moor RM, Evans MJ (1991) Derivation of pluripotent, embryonic cell lines from the pig and sheep. J Reprod Fertil 43(Suppl):255–260
Potten CS, Loeffler M (1987) Changes with time in the proportion of isolated paired and clustered labelled cells in sheets of murine epidermis. Virchows Arch B Cell Pathol Zell Pathol 53:279–285
Rieske P, Barbara KS, Ausim A (2005) Human fibroblast-derived cell lines have characteristics of embryonic stem cells and cells of neuro-ectodermal origin. Differentiation 73:474–483
Rosner MH, Vigano MA, Ozato K, Timmons PM, Poirier F, Rigby PW, Staudt LM (1990) A POU-domain transcription factor in early stem cells and germ cells of the mammalian embryo. Nature 21:686–692
Shi Y, Desponts C, Do JT, Hahm HS, Schöler HR, Ding S (2008) Induction of Pluripotent stem cells from mouse embryonic fibroblasts by Oct4 and Klf4 with small-molecule compounds. Cell Stem Cell 3:568–574
Silva J, Smith A (2008) Capturing pluripotency. Cell 132:532–536
Slack J (2001) Skinny dipping for stem cells. Nat Cell Biol 3:E205–E206
Smith A (2005) The battlefield of pluripotency. Cell 123:757–760
Sritanaudomchai H, Pavasuthipaisit K, Kitiyanant Y, Kupradinun P, Mitalipov S, Kusamran T (2007) Characterization and multilineage differentiation of embryonic stem cells derived from a buffalo parthenogenetic embryo. Mol Reprod Dev 74:1295–1302
Strelchenko N (1996) Bovine pluripotent stem cells. Theriogenology 45:131–140
Takahashi K, Tanabe K, Ohnuki M, Narita M, Ichisaka T, Tomoda K, Yamanaka S (2007) Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 131:861–872
Takahashi K, Yamanaka S (2006) Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 126:663–676
Talbot NC, Blomberg LA (2008) The pursuit of ES cell lines of domestic ungulates. Stem Cell Rev 4:235–254
Toma JG, Akhavan M, Fernandes KJ, Barnabe-Heider F, Sadikot A, Kaplan DR, Miller FD (2001) Isolation of multipotent adult stem cells from the dermis of mammalian skin. Nat Cell Biol 3:778–784
Verma V, Gautam SK, Singh B, Manik RS, Palta P, Singla SK, Goswami SL, Chauhan MS (2007) Isolation and characterization of embryonic stem cell-like cells in vitro produced buffalo (Bubalus bubalis) embryos. Mol Reprod Dev 74:520–529
Wernig M, Meissner A, Foreman R, Brambrink T, Ku M, Hochedlinger K, Bernstein BE, Jaenisch R (2007) In vitro reprogramming of fibroblasts into a pluripotent ES-cell-like state. Nature 448:318–324
Wheeler MB (1994) Development and validation of swine embryonic stem cells: a review. Reprod Fertil Dev 6:563–568
Yadav PS, Kues WA, Herrman D, Carnwath JW, Niemann H (2005) Bovine ICM derived cells express the OCT4 ortholog. Mol Reprod Dev 72:182–190
Yadav PS, Mann A, Singh V, Yashveer S, Sharma RK, Singh I (2010) Expression of pluripotency genes in buffalo (Bubalus bubalis) amniotic fluid cells. Reprod Domest Anim. doi:10.1111/j.1439-0531.2010.01733.x
Acknowledgments
The authors wish to fully acknowledge Department of Biotechnology, Government of India, for financial support. Authors also acknowledge Wilfried A Kues and Birbal Singh for critically reading the manuscript.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Yadav, P.S., Mann, A., Singh, J. et al. Buffalo (Bubalus bubalis) Fetal Skin Derived Fibroblast Cells Exhibit Characteristics of Stem Cells. Agric Res 1, 175–182 (2012). https://doi.org/10.1007/s40003-012-0013-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40003-012-0013-y