
Abstract
Nowadays, the emerging pollutants, such as bisphenol A, are object of increased attention by the citizens and the sectors of industry and academia, as a result of the problems they cause, particularly at the health level. In this study, a set of granulated samples was prepared from a commercial powdered activated carbon, aiming its application in the removal of bisphenol A via the adsorption process. The granulation allowed to obtain granulated adsorbents with higher adsorption capacity (37.0–54.8 mg/g) than the original activated carbon (32.2 mg/g). The samples were structurally and chemically characterised using scanning electron microscopy, X-ray diffraction, nitrogen adsorption at 77 K, Fourier-transformed infrared spectroscopy, point of zero charge and energy-dispersive X-ray spectroscopy techniques. The bisphenol A adsorption tests are carried out in kinetic and equilibrium modes, involving variables such as mass, pH and temperature. The results show that, using different commercial silica binders, it was possible to prepare mechanical, dimensional, chemical and structurally stable granulated adsorbents with a good capacity for bisphenol A adsorption, widening the potential and range of application for these materials.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
As the classical pollutants are being eliminated from daily life, others appear, creating new problems, particularly in the environmental field, with consequences, sometimes not yet entirely predictable, for all living beings, in particular for man. In this new context, we have emerging pollutants, such as bisphenol A, for which is urgent to develop strategies aimed at its mitigation and elimination from systems where it is often found, especially in aqueous systems, with particular prevalence in effluents produced in water treatment plants.
The occurrence of endocrine disrupting chemicals (EDCs) in aqueous environments is of increasing concern due to their effects on humans and the environment, as they mimic the biological activity of natural hormones in the endocrine system and, thus, disrupt the activations of endocrine glands (Soni and Padmaja 2014; Jun et al. 2019). EDCs have the potential to disrupt natural hormone systems and processes, mainly through their action on core hormone receptors, such as estrogen and androgen receptors, where they can have agonistic or antagonistic effects (Fowler et al. 2012). The compound 2,2-Bis(4-hydroxyphenyl) propane or bisphenol A is one of the EDCs known to have estrogenic activity and to be able to interfere with the reproductive systems of wildlife and humans, even at low concentrations.
Bisphenol A is used primarily as a monomer in the production of polycarbonate plastics and epoxy resins. Due to its widespread use in many industrial and commercial products, bisphenol A has been identified in almost all types of environmental matrices that include air, water, sewage sludge, soil, dust, food and beverages, as well as in human samples (Soni and Padmaja 2014). These facts explain that bisphenol A is often found in natural waters at levels of ng/L, making it a trace of this organic contaminant. It is therefore considered a critical contaminant and was selected as a model compound in this study because in natural water it can cause damage to the reproductive systems of humans and animals (Wang and Zhang 2020).
The reality imposes and justifies the increasing research on techniques and materials that can be used with efficacy to remove bisphenol A from water (Wang et al. 2022). Currently, bisphenol A removal methods include biodegradation, catalytic methods, oxidation and adsorption. Due to its efficiency, simplicity of implementation and low cost, adsorption is still among the most advantageous of these (Wang et al. 2022). Thus, it is justified research for new adsorbents and improved presentations of traditional adsorbents, such as geological sediments, granular/modified adsorbents, single/multi-wall carbon nanotubes, biochar, alumina and silica, zeolites, natural-based adsorbents such as chitosan and, finally, activated carbon, the latter the adsorbent par excellence. Activated carbon is a carbon-based adsorbent material, essentially amorphous, although punctuated by microcrystalline structures. These materials have been used as excellent and versatile adsorbents in various gas and liquid phase applications due to their highly developed porosity and extensive surface area. The pore volume in activated carbons is generally defined as being greater than 0.2 cm3/g and the internal surface area greater than 400 m2/g (measured by the Brunauer–Emmett–Teller equation, BET). The physicochemical properties of the material and the subsequent adsorptive behaviour are highly dependent on the nature of the raw material used (the precursor), the activating agent and the conditions of the carbonisation and activation processes (Cansado et al. 2010; Carvajal-Bernal et al. 2015; Torrellas et al. 2015; Gomes et al. 2016; Ricón-Silva et al. 2016; Tchikuala et al. 2017). Lignocellulosic materials constitute the largest source of the precursors used (about 45% by weight of the total raw materials used for the manufacture of activated carbons). The advantages result not only from the low contents of inorganic materials, decisive for obtaining activated carbons with low ash content, but also from the relatively high volatile content required to control carbon porosity. Both characteristics are common to most lignocellulosic materials used for the production of activated carbons (Torrellas et al. 2015). Activated carbon remains the most widely used adsorbent in water treatment, both for human consumption and for wastewater, due to its unique porous textures and surface characteristics that typically facilitate the removal of target pollutants (Naganathan et al. 2021). That is why adsorption of organic contaminants by activated carbon is one of the most effective and widely used methods to purify water (Bautista-Toledo et al. 2005).
However, there is still a potential for improvement as the ideal adsorbents require a good adsorption capacity, which almost always involves a high pore volume, large surface area, adjusted pore size and ease of regeneration. Other aspects, not less important, are related with the granulometry of the adsorbent, which often requires that the adsorbents, most of them in powder form, have to be resized to allow their application in different treatment systems, namely industrial ones, which require the use of columns (Hung et al. 2005). In this case, the use of adsorbent powder is somewhat limited by the problems inherent to the small particle size which will translate into compaction and clogging of the adsorption columns. Due to the advantages of these adsorbents, it is of interest the development of procedures that might maintain or even improve their initial properties. Inherent to this requirement, the search for affordable binders that when used do not significantly distort the nature of the initial adsorbent material. Among these binders, we find silicas, in particular, in the form of aqueous dispersions of particles in the nanometer size range, which typically exhibit a narrow particle size distribution. Their design and process control ensure quality and consistency, which improves the performance of their products as inorganic binders, enhancing refractory binding agents and surface modifiers. Its excellent purity combined with a wide range of options, make the silica family an excellent choice for many applications.
This work followed this approach and evaluated the potential use of commercial solutions of colloidal silica, Ludox®AS-40, Ludox®AM and Ludox®HS30, as binders for activated carbon of lignocellulosic origin, in a first phase. Subsequently, it was evaluated the possibility of using the prepared granulated samples in the adsorption process of bisphenol A in aqueous solutions and the stability of the adsorbents after the adsorption process.
This study was conducted in the laboratories of Chemistry and Biochemistry departments on University of Évora (Portugal) and University of Porto (Portugal) from September 2022 to January 2023.
Materials and methods
Materials
In this work a commercial activated carbon (AC), of lignocellulosic origin was used, which was purchased from the company J.M.Vaz Pereira-Portugal (Lot 30460 VDS 2021/04). The silica used as binder was supplied by W.R. Grace & Company-USA, namely in its commercial forms: Ludox®AS-40 (40 wt% suspension in H2O), Ludox®AM (30 wt. % suspension in H2O) and Ludox®HS30 (30 wt% suspension in H2O). Bisphenol A was purchased from Sigma-Aldrich-Portugal (99 + % purity). The gases used in the process of thermal stabilisation and activation of the granulates were purchased from Gasin-Portugal, respectively, nitrogen (X50—99.95%) and carbon dioxide (X50—99.8%). The reagents HCl (ACS reagent, 37%) and NaOH (pellets, ≥ 98%) were purchased from Sigma-Aldrich Inc-Portugal, and NaNO3 (> 99.5%) was purchased from Riedel-VWR-Portugal.
Granulation by liquid binder spreading
The granulation step was carried out using 10% silica solutions prepared by dilution of the original commercial silica solutions Ludox® (AS-40, AM and HS30), in order to facilitate its spreading in spray form, with a minimum of silica to ensure the agglomeration step.
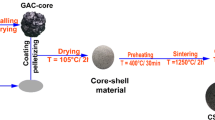
The process of agglutination of the activated carbon in powder form, (AC)p, was carried out manually in a plastic container in which ~ 10 g of (AC)p were spread and on which the 10% solution of the binder was dispersed. The process of manual stirring, subjecting the whole set to a manual continuous centrifugal movement, and the addition of the binder was repeated as many times as necessary until granule formation was observed. The granules were then transferred to an oven where they were subjected to a pre-drying process on two phases. On the first stage, the samples were dried at a temperature of 50 °C for 24 h and then at 100 °C for 72 h.
Thermal stabilisation and activation of granular adsorbents
The granules obtained in the previous step were submitted to a heating process in a carbon dioxide atmosphere up to 500 °C (under a heating rate of 10 °C/min), remaining at this temperature for 15 min, which allowed consolidating their physical and mechanical stability. This procedure also aimed to ensure that the addition of the binder in the binding process did not compromise the access to the porous structure of the AC. Finally, the samples were cooled to room temperature under nitrogen flux.
The prepared granulated samples were stored in plastic containers, without contact with the environment, and identified as (AC + AS40)g, (AC + AM30)g and (AC + HS30)g. The designation of the samples was done according to the generic format (AC + Binder)g, where “AC” represents the original activated carbon in powder form, “Binder” represents the abbreviated designation of the commercial silica ,and “g” represents granule (the final form of the sample).
Adsorbents characterisation
All samples were structurally and chemically characterised using a number of techniques, namely, Scanning electron microscopy (SEM), X-ray diffraction (XRD), Nitrogen adsorption at 77 K, Fourier-transformed infrared spectroscopy (FTIR), Point of zero charge (pHpzc) and energy-dispersive X-ray spectroscopy (EDS).
The surface morphology of the carbon adsorbents and their chemical composition was analysed via SEM analyses by means of a Fei Quanta 400 FEG ESEM/EDAX Pegasus X4M—Thermo Fisher Scientific/USA (Centro de Materiais da Universidade do Porto—CEMUP). XRD measurements were carried out on a PANalytical’s X'Pert PRO MRD—Malvern Panalytical Ltd/UK (CENIMAT-I3N—Universidade NOVA de Lisboa) using CuKα (wavelength 1.540598 Å) as X-rays source, a generator voltage of 45 kV, tube current of 40 mA, with a step size of 0.033° and 50.165 s per step.
The textural properties were analysed by means of N2 adsorption at 77 K using the equipment MicroActive for TriStar II Plus 2.02—Micromeritics®/USA (Faculdade de Ciências da Universidade do Porto). The apparent surface area was determined by the BET method (Brunauer et al. 1938), the total pore volume was determined directly from the isotherm at a relative pressure p/p° = 0.95, and the mean pore size was determined by the BJH method (Hossen et al. 2020). The analysis of the samples functional groups was made by FTIR using a Perkin-Elmer spectrum two FTIR spectrophotometer–Perkin Elmer/USA (Departamento de Química e Bioquímica—Universidade de Évora) by the KBr disc method with a resolution of 4 cm−1 and 15 scans between 4000 and 450 cm−1. The pH at the point of zero charge PZC of all the samples was determined using a modified version of a simple method proposed by Noh and Schwarz (Noh and Schwarz 1989), as described in previous work (Carrott et al. 2008).
Adsorption of bisphenol A
The organic probe molecule bisphenol A adsorption studies included the determination of calibration curves, kinetic and equilibrium tests. To avoid possible interferences, all adsorption tests were carried out under controlled conditions. The parameters analysed included the pH of the adsorption medium, the mass ratio between the adsorbent and the adsorption solution volume, the effect of temperature and the mechanical stability of the adsorbent.
Calibration curves in acid medium, unaltered medium and in basic medium, were determined and the desired value of pH was obtained by adding a certain volume of HCl solution (0.5 M) or NaOH solution (1 M) to the bisphenol A solution. In the kinetic assays, a fixed amount of sample (0.25 g) was placed in contact with a volume (250 mL) of bisphenol A aqueous solution into a 250-mL stoppered Erlenmeyer flask. Over time (72 h), the amount of non-adsorbed bisphenol A was dosed until an equilibrium situation was reached. In equilibrium isotherms, a fixed amount (0.05 g) of each carbon and 25 mL of aqueous solution of bisphenol A of desired concentration were placed into a 50 mL stoppered Erlenmeyer, along 24 h. The pH was adjusted to be in acid range, around 2, with a hydrochloric acid solution ([HCl] = 0.5 M).
On the kinetics and equilibrium studies, all the flasks were shaken along the selected time using a thermostated shaker bath, OLS Aqua Pro Orbital and Linear model—GRANT/ UK, at 25 °C.
In the study of the influence of temperature on the bisphenol A adsorption process, a similar procedure was adopted, adjusted to the selected temperatures (2.6, 10, 25, 40 and 50 °C).
The dosage of bisphenol A always involved the filtration of the volume of solution to be dosed and its residual amount was measured using the UV/VIS spectrophotometry on a UV–Visible Nicolet Evolution 300 apparatus, from Thermo Fisher Scientific/USA (Departamento de Química e Bioquímica—Universidade de Évora), at a wavelength of 277.5 nm. Bisphenol A dosage was made in conditions of pH that assure that only one of the compound forms is present (based on previous calibration curves where the characteristics wavelengths were identified).
Results and discussion
Granulation by liquid binder spreading
The application of materials in different domains, particularly in the area of treatment and remediation in liquid systems, is often conditioned by their physical size, mechanical stability and the possibility of resizing them. In particular, in the area of aqueous systems treatment, the use of adsorbent materials in powder form brings several inconveniences (e.g. loss in continuous flow systems, compacting in column systems, among others), which can be overcome when these materials acquire a larger dimension (e.g. granules, composites).
This work goes in this direction and presents the possibility of increasing the size of an adsorbent, (AC)p, by a simple granulation process employing the spray technique, and using commercial silica colloidal solutions as binders, Table 1.
By observation of Table 1 it can be seen that all binders of siliceous nature demonstrated the ability to form granules with the carbon adsorbent in powder form, (AC)p, with mass ratio Silica:(AC)p around ~ 1:5. The granules formed showed stability and mechanical and dimensional strength throughout all the stages, namely, pre-drying and drying and, in particular, throughout the physical activation process with carbon dioxide at 500 °C, as shown in Fig. 1.

Illustrative images of the granules formed with the silicate binders prepared from commercial LUDOX® colloidal silicas
Adsorbents characterisation
Morphologic and textural characterisation
The analysis of the lower resolution images, 100 µm, on the left column of Fig. 2, showed no significant variations in the morphology of the surface of the granulated samples ((AC-AM30)g, (AC-AS40)g and (AC-HS30)g), not even when these were compared with the original sample in powder form, (AC)p. However, when looking at the higher resolution 4 µm images in the right column of Fig. 2, the surface shows changes that appear to result from the addition of the silica binders. A detailed analysis shows a partial covering of some sections of the surface in the different granulated samples. On the surface, lighter stained areas are observed, corresponding to the formation of a layer or film, which shows that the addition of the binder is effective from a physical point of view.

SEM images with different resolutions, 100 µm (left column) and 4 µm (right column), of samples (AC)p, (AC-AM30)g, (AC-AS40)g and (AC-HS30)g, respectively, from top to bottom
The X-ray diffractograms, as shown in Fig. 3, show that the original activated carbon (AC)p presents the typical structure of these carbon materials, with the two characteristic bands, the first corresponding to the planes (002) and positioned at 2θ ~ 25°, and the second resulting from the overlap of the planes (100) and (101), located at 2θ ~ 42° (Tchikuala et al. 2017). Those two broad peaks result from the amorphous graphitic carbon structure characteristic of this type of materials. The diffractogram of the sample granulated with Ludox-HS30, representative of the prepared granules, also shows the aforementioned bands, although an increase in the intensity of the first is evident. This increase can be explained by the overlap of this band, characteristic of carbon materials, with the typical broad peak, at 2θ ~ 22°, of the amorphous silica nanoparticles colloidal solution used as a binder. Further, the XRD diffraction pattern indicates the absence of any ordered crystalline structure.

Representative powder X-ray diffraction patterns of the powder (AC)p and granular (AC-HS30)g activated carbons samples
From Fig. 4, it is possible to understand that the isotherms of N2 adsorption at 77 K of the different samples are essentially of Type I, according to the IUPAC classification. All the isotherms show similar shape, a little distant from the usual rectangular shape, but rather specifically of Type I (b), characteristic of materials having pore size distributions over a broader range including wider micropores and possibly narrow mesopores (< ∼ 5 nm) (Thommes et al. 2015). This broader pore size distribution was confirmed by the existence of a clear Type H4 cycle of hysteresis on all the samples, which means that we are in the presence of micro-mesoporous carbon materials.

N2/77 K adsorption isotherms determined on powder and granular activated carbons samples. From top to bottom at p/p0 0.9: (AC-HS30)g, (AC)p, (AC-AS40)g and (AC-AM30)g. Open points are the adsorption branch and closed points are the desorption branch of the isotherms
A more detailed analysis of these experimental isotherms and the results obtained by applying the BET and BJH methods, as given in Table 2, and taking as reference the commercial activated carbon of lignocellulosic origin in the form of powder (AC)p, it is found that the granulation step induced changes in the structural parameters. Thus, when the granulation is done with the Ludox-AM, there is a reduction in the apparent surface area from 825 to 631 m2/g. In the case of the other adhesives, the granulation process leads to lower variations and in opposite directions. Thus, in the case of Ludox-AS40, there is a small reduction to 769 m2/g, while in the case of Ludox-HS30 there is a small increase to 861 m2/g. These variations show that the combined effect of the granulation process and subsequent physical activation allows, at least in the last two cases, the preparation of granulated adsorbent materials without compromising the structural parameters. Thus, it is possible to predict that the adsorptive potential of the granulated ACs will not be compromised when compared with the starting adsorbent (AC)p.
In terms of pore size, all activated carbons show an average pore width in the range of 2.6 nm to 2.9 nm, which will not be a limiting factor in the adsorption process of bisphenol A, since this range of values is higher than the molecular size of this compound (~ 1.27 × 0.86 × 0.68 nm) (Balci and Erkurt 2017). However, it should be retained that there was a decrease in the microporous volume in the case of the granulated samples. This can be explained by the blocking of some porosity, particularly of smaller size, by the silica binder during the agglomeration step. Traces of some low-pressure hysteresis on isotherms confirms the presence of some constricted micropore entrances. On the other hand, it can be seen that the physical activation process at 500 °C allowed the clearing, almost in its totality, of the microporous structure, without compromising the mechanical and dimensional stability of the granular adsorbents.
Chemical characterisation
All samples were analysed by FTIR, EDS and pHpzc to obtain more detailed information in order to better understand the implications of the agglomeration process with the silica binders, of the activation stage with CO2 and, also, to predict the adsorption potential of the prepared granular adsorbents.
Figure 5 shows the FTIR spectra of the studied powder and granular activated carbons samples (AC)p and (AC + Binder)g. The spectrum of the powder activated carbon is simple, with no significant peaks and only a few low intensity bands. However, on the spectra it is possible to identify the presence of aromatic structures with the C–H bond stretching around 1500 cm−1, and phenol groups that can be related to the C–O bond stretching with small traces of the bands between 1200 and 1100 cm−1 (Gomes et al. 2016). Also, a small band in the range 3470–3250 cm−1 corresponds to the overlap of the O–H stretching bands of hydrogen-bonded to water molecules (Tchikuala et al. 2017).

FTIR spectra of powder and granular activated carbons samples
The spectra of the granulated samples are similar to each other, regardless of the type of colloidal silica used as binder. However, they show some changes when we compare their spectra with the one of the sample (AC)p. This means that the granulated material reflects, some chemical changes resulting from the incorporation of colloidal silica, as expected. In this set of granulated samples, the semi large band centred around 3470–3250 cm−1 increases in intensity, which can be explained by the activation step with CO2 and by the SiO–H stretching of surface silanol hydrogen-bonded to molecular water (Si–AH…H2O), from silica binder (Oweini and Rassy 2009; Kadja et al. 2017). Some adsorbed water molecules caused the appearance of absorption bands at 1650–1630 cm−1 resulting from deformation vibrations (Oweini and Rassy 2009; Kadja et al. 2017). The narrow and intense band that appears in the range 1250–1000 cm−1 is related to the presence of Si that can be involved in Si–O covalent bonds and Si–O–Si asymmetric stretching vibrations (Oweini and Rassy 2009; Kadja et al. 2017). The symmetric stretching vibrations are reflected at a small band around 790 cm−1.
In parallel with the SEM analysis of the studied carbon materials, the EDS analysis of distinct sections of those samples was also carried out, Fig. 6 and Table 3. Although the chemical analysis obtained was mainly qualitative, it provided information that validates the assumptions of this work, in particular the use of colloidal silica as effective binders and allowed foreseeing the possibility of using the granulates prepared as adsorbent materials for liquid phase applications. Thus, the EDS results show that the chemical composition of the materials is composed essentially by carbon and oxygen, in the order of ~ 89% and ~ 11%, Table 3, which is typical of adsorbent materials of lignocellulosic origin. Also, the addition of the binder of siliceous origin introduces the presence of silica, with values between ~ 10% and ~ 7% of Si, which do not compromise the nature of the starting carbonaceous adsorbent (AC)p. Moreover, these results allow us to verify the existence of a correlation between the amount of Si in the samples and the total apparent surface area ABET determined from the isotherms of N2 at 77 K, Table 2 and 3. The fact that the values of this apparent surface area decrease, from 861 to 631 m2/g, with the increase of the amount of Si detected, from ~ 7 to 10%, can be interpreted by the principle that this higher amount of Si blocked part of the porosity and also reduced the external surface area of the initial porous material. This interpretation is also in agreement with the reduction of the microporous volume, from 0.18 to 0.15 cm3/g, observed when the amount of Si increases. A more detailed analysis of the results for different zones of the same sample, as an example the (AC-HS30)g sample, shows that the clearer zones of the SEM images, area Z3 in Fig. 6e and Table 3, correspond to areas of higher Si concentration, confirming that granule formation and stability was ensured by interfaces or spots of the type –(AC)–Silica–(AC)–.

EDS analysis of powder and granular activated carbons samples (AC)p, (AC-AM30)g, (AC-AS40)g, (AC-HS30)g, respectively, (a–d). SEM images with two distinct areas marked (Z2 and Z3), on (e)
Another parameter usually estimated is the pH at the point zero charge, pHpzc, which is an intrinsic property of the adsorbent. Its value is a determining factor in the adsorption process. From the analysis of Table 3 it can be seen the basic nature of the initial activated carbon sample, (AC)p, with a pHpzc = 8.30, and that the agglomeration step with the siliceous binders leads to a reduction in the pHpzc of the various samples independently of the type of commercial silica used. This pH reduction to values close to the neutral region, 6.7 to 7.7, is a result of the previous dilution of the commercial aqueous colloidal silica to values around 10% (wt. SiO2). This diluted solution is used as a binder on the granulation stage.
Adsorption of bisphenol A
Influence of the mass, pH and kinetics parameters
The initial bisphenol A adsorption experiments included the analysis of parameters such as the effect of the adsorbent mass, pH of the adsorption medium, determination of calibration curves and the evaluation of the kinetic process of the adsorptive-adsorbent systems studied.
The UV–Visible profile spectra of bisphenol A on aqueous solution at different pH were determined in order to identify the characteristic wavelengths, Fig. 7. The bisphenol A present a significant UV absorption at ~ 225, ~ 276 and ~ 282 nm in acid and unchanged (slightly acid) medium pH, and at ~ 243 and ~ 294 nm in basic medium. Based on these results, all the bisphenol A dosage measures were made in acid controlled medium pH (~ 2), to avoid interferences, namely, from the chemical nature of the carbon adsorbent samples.

a Bisphenol A chemical structure and select physicochemical properties (MS-molecular size, MW-molecular weight, WS-water solubility, b bisphenol A UV–visible absorption spectra on aqueous solution under different pH and calibration curve data on acid medium (pH ~ 2, [C]Bisphenol A = 50 mg/L)
The influence of the adsorbent mass was also one of the parameters analysed in a representative granular sample, (AC-AS40)g. From the analysis of Fig. 8, it was found that the amount adsorbed tends to stabilise with the increase of the adsorbent mass. This stabilisation is clear for a mass between 0.1 and 0.2 g which means that the favourable limit value of the ratio (mass of adsorbent/volume of adsorbate) for the system and under these experimental conditions (volume of adsorbate = 50 mL) was reached. Based on these results, it was defined that the equilibrium adsorption tests would be performed with the ratio 0.05 g of adsorbent sample and 25 mL of bisphenol A solution.

Influence of adsorbent sample mass and pH on the normalised adsorption capacity of a representative sample, (AC-AS40)p
Simultaneously, the influence of pH on bisphenol A adsorption was evaluated considering three different situations, acid medium (pH ~ 2), unaltered medium (pH ~ 6) and basic medium (pH ~ 11), for an adsorbent mass of 0.05 g. The results show that adsorption is favoured when the system is in the acidic pH range. These results agree with the previously determined pHpzc for the various samples, Table 3, with values between 6.7 and 7.7, which already allowed predicting that adsorption would be favoured for values below a pH ~ 6.
Another decisive experimental parameter in this kind of studies is the equilibrium time, namely, the time required for the adsorbed amount to be maximised. In this work, the kinetic behaviour of the carbon adsorbents was analysed for representative samples, specifically, the original activated carbon (AC)p and the granulated sample (AC-AS40)g. The distinct behaviour of the adsorbents is noticeable in Fig. 9. While in the case of (AC)p sample the equilibrium condition is reached very quickly, between 10 and 12 h, in the case of the granulated adsorbent this condition tends to be reached for a contact time equal or slightly higher than 24 h. This difference and the specific shape of the kinetic curves, Fig. 9, can be explained by the greater accessibility to the porous structure and the external surface when comparing samples in powder and granulated form, respectively, (AC)p and (AC-AS40)g. In the case of (AC)p, the bisphenol A adsorptive will access more easily and quickly to the active centres and pores of the adsorbent, leading to a faster increase of the adsorbed amount and in larger quantity. The analysis of the kinetics profile in the case of sample (AC-AS40)g shows the existence of a small step in the initial phase, around 8 h, which can be explained by the difficulty of bisphenol A in accessing the adsorption sites, which ends up being overcome over time (> 12 h).

Representative adsorption kinetics determined on powder and granular activated carbons samples, (AC)p and (AC-AS40)g, (mSample = 0.25 g, [C]Bisphenol A = 50 mg/L, pH = 6)
Equilibrium adsorption isotherms
The experimental isotherms presented in Fig. 10 show a different behaviour between the starting sample, (AC)p, and the samples prepared by granulation with silica binders, (AC-Binder)g. It is perceptible that the granulation step had consequences, first at the adsorption process level, profile of isotherm, and at the level of the maximum amount adsorbed, (Qads)max. In the case of sample (AC)p, the bisphenol A adsorption is significant at low concentrations ([Ceq] < 10 mg/L), gradually increasing in the intermediate concentration range ([Ceq] ~ 10 to 40 mg/L), followed by a tendency for the stabilisation of the adsorbed amount ([Ceq] > 40 mg/L). This behaviour is typical of the great majority of bisphenol A adsorption processes on porous carbon adsorbents. The granulated samples present a similar behaviour with respect to the step-shaped of the isotherm, from which it is possible to see that the adsorption presents four phases: (i) a first, ([Ceq] < 5 mg/L), in which the adsorption is relatively fast, (ii) a second ([Ceq] ~ 5 to 10 mg/L) in which there is a tendency to form a small plateau, (iii) a third, ([Ceq] ~ 7. 5 to 10 mg/L), in which the amount adsorbed increases considerably, (iv) a fourth, ([Ceq] > 40 mg/L), when the amount adsorbed increases slowly, with a predictable tendency to stabilise. In the case of sample (AC-AS40)g this behaviour is displaced towards the higher equilibrium concentration range, also reflecting in a higher adsorbed amount. The tendency to plateau, both initial and final, is not so clear in this sample, continuing a slow and gradual increase in the amount adsorbed.

Experimental adsorption isotherms of bisphenol A on powder and granular activated carbons samples
Regarding the maximum adsorbed amounts, Table 4, an increase of them is observed when comparing these samples with the original activated carbon in powder form (Qads ~ 32.2 mg/g). The maximum value of 54.8 mg/g is obtained with the sample (AC + AS40)g. These results indicate that the agglomeration of the powder adsorbent leads to the formation of constrictions that hinder the initial adsorption step of bisphenol A, which is justified by a physical obstruction of part of the porous structure of the adsorbent when the binder is added. Specifically, part of the microporous structure was partially blocked. However, when the initial bisphenol A concentration increases, there are strength and concentration gradients between the solution and the internal part of the solid adsorbent, that lead to the reorganisation of the adsorbent molecules, which ends up forcing the entrance in the part of the porous structure that presents constrictions. Additionally, the presence of silica in the adsorbent and the formation of new functional groups also seem to contribute to an increase in the amount of bisphenol A adsorbed.
Among the most widely used models to analyse the experimental isotherms of adsorption from the liquid phase there are the Langmuir and Freundlich models, with the first being applied in the region of the monolayer, while the second includes the higher equilibrium concentrations, with a multilayer formation (Cloirec et al. 1997; Haghseresht and Lu 1998).
The Langmuir equation can be written as:
where nmL is the monolayer capacity and KL the Langmuir constant. Plotting (Ceq/Qads) against Ceq, the parameters nmL and KL are calculated. Equation (1), which is generally limited to a restricted range of low concentrations, presents some limitations especially when the adsorption is not limited to a monolayer and the adsorbent surface is heterogeneous.
The Freundlich equation, Eq. (2), is based on a system adsorptive-adsorbent that gives isotherms of a non-linear nature:
where nads is the adsorbed amount at a solute equilibrium concentration Ceq and the constant KF and exponent nF are adjustable parameters system dependents. A plot of ln(nads) versus ln(Ceq) gives the values of KF and nF. The applicability of this equation to systems involving heterogeneous surfaces and organic compounds is very acceptable over a wide range of concentrations (Adibfar et al. 2014).
From Table 4, it is possible to notice that there is an agreement between the experimental values of the adsorbed quantities (Qads) and the estimated values, particularly in absolute terms in the case of the Langmuir model (nmL). However, for all equations we obtain a decreasing sequence of adsorbed amount that obeys the following relation: (AC-AS40)g > > (AC-HS30)g > > (AC-AM30)g > (AC)p. The Freundlich distribution coefficient, KF, related with the amount adsorbed, shows lower values, in absolute terms, than those obtained experimentally, which must be understood on the basis that KF only corresponds to the amount adsorbed in the case that Ceq is equal to 1 mg/L. When we focus on the values of KL and nF parameters, the consistency with the experimental isotherms is maintained, in particular in the case of KL, where it is clear the existence of two types of samples. The first, (AC)p with a lower value of KL (~ 11) and another of the granular samples (AC-Binder)g with higher values (~ 15 to 18), reflecting the greater extent of the interaction between adsorbate and the adsorbent surface. The similar values of exponent nF, as an empirical constant parameter related to the heterogeneity of the adsorbent surface, obtained in our systems show that the incorporation of silica did not cause a significant variation of the surface of (AC)p as could have been expected. The modifications resulted essentially from a combined effect of changes in pore structure and surface chemistry, thus preserving the essence of the original carbon material.
Temperature influence
Normally, temperature plays a fundamental role in the adsorption process affecting, in a direct way, the solubility of adsorptive in water and, indirectly, the degree of its dissociation. On Fig. 11, it is possible to see the profile of the amount adsorbed of bisphenol A with increasing temperature, on the range 2.6 to 50 ºC, for different initial concentrations, 50, 100 and 125 mg/L. Our adsorptive-adsorbent systems follow the usual pattern of an increase in the amount adsorbed with the rise of the initial concentration. This can be explained by an enhancement of the adsorption drive between the adsorbent and the solute (Lee et al. 2016; Idrees et al. 2018). This means that the process is spontaneous and endothermic. However, this increase seems to follow the form of a step, being more significant between 10 and 40 °C. Some of the observed oscillations result from the fact that the adsorption process in these systems, (AC-Silica)g—Bisphenol A, is of a combined nature, i.e. the retention of bisphenol A in these adsorbents occurs by chemical adsorption processes, despite also having a strong role for physical adsorption.

Effect of temperature on the adsorption of bisphenol A onto representative granular activated carbon samples, a (AC-AS40)g and b (AC-AM30)g, for different initial concentrations (m(Sample)g = 0.05 g, pH = 2)
Adsorbent stability
The visual observation of the granular samples after the adsorption process shows that they maintain dimensional and mechanical stability. This direct observation is also reinforced by XRD and FTIR analyses of the samples before and after their application in the aqueous phase, which reveals identical diffractograms and spectra, Figs. 12 and 13. As an example for (AC + HS30)g sample, it is clear from the diffractograms profile, Fig. 12, that the amorphous nature of this type of porous carbon adsorbents is retained, since only a slightly decrease on the first characteristic band, 2θ ~ 25°, is noted.

Representative powder X-ray diffraction patterns of the granular (AC-HS30)g activated carbons sample, before and after its application on the adsorption of bisphenol A ([Cinitial]Bisphenol A = 160 mg/L, pH = 2, T = 25 °C)

FTIR spectra of powder (AC)p and granular (AC-HS30)g activated carbons samples, before and after its application on the adsorption of bisphenol A ([Cinitial]Bisphenol A = 160 mg/L, pH = 2, T = 25 °C)
From a chemical point of view, the FTIR analysis of sample (AC-HS30)g shows a spectrum where the profile is maintained, when we compare the previous data with those obtained after the application of the sample on the adsorption of bisphenol A. Although the chemical composition is maintained, a reduction in the intensity of the spectrum is also clear. This can mean that some amount of the less strongly bound functional chemical groups characteristic of this carbon material were released into the water during the adsorption process.
Conclusion
Granulated activated carbons prepared by spray-dispersion of colloidal solutions of low concentration silica binders (10%) present structural and chemical characteristics that allow their application in the adsorption of bisphenol A from aqueous solutions.
The tests showed that all the binders used, Ludox® (AS-40, AM and HS30), allowed the preparation of granular carbon adsorbents with a final percentage of silica between 6.84 and 10.00%, not significantly changing the structural and chemical nature of the original powdered activated carbon, (AC)p. In structural terms, when the granular activated carbons were compared with the original powdered activated carbon, small variations were observed, namely, a reduction in the microporous volume, a slight increase in the average pore size and a tendency to reduce the total apparent area for the samples (AC-AM30)g and (AC-AS40)g to 631 and 769 m2/g, respectively (while in the case of the sample (AC-HS30)g it was observed an increase to 861 m2/g). Equally at the chemical characterisation level, the FTIR analyses clearly identify a set of bands characteristic of the presence of silica in the granulated samples.
The adsorption tests of bisphenol A from the liquid phase show that the process is favoured when the pH of the adsorption medium is acid, when a madsorbent/Vsolution adsorptive ratio of ~ 0.05 g/25 mL and when the temperature increases. Similarly, it was concluded that an adsorptive-adsorbent contact time of 24 h is sufficient to reach the equilibrium. Some structural constraints, particularly at the level of microporosity, cause an additional difficulty in the process of diffusion of the adsorbent, bisphenol A, and consequently of its access to the active sites of adsorption.
The analysis of the adsorption tests at various temperatures showed that in our systems the adsorption of bisphenol A is a spontaneous and endothermic process.
XRD and FTIR analyses confirm the previous direct observation that powder and granular adsorbents maintain its structural and chemical stability after their application in liquid phase.
The achievements previously mentioned allow the possibility of testing these adsorbents in granular form an in real-life scenarios, namely, on fixed bed column systems. Furthermore, it is possible to predict that the outlined procedure enables the extension of the number and range of applications for these adsorbent materials to different systems, with potential replication for other adsorbents.
Data availability
The data used on this study are available from the corresponding author on reasonable request.
References
Adibfar M, Kaghazchi T, Asasian N, Soleimani M (2014) Conversion of poly (ethylene terephthalate) waste into activated carbon: chemical activation and characterization. Chem Eng Technol 37:979–986. https://doi.org/10.1002/ceat.201200719
Balci B, Erkurt FE (2017) Adsorption of bisphenol-A by eucalyptus bark/magnetite composite: modeling the effect of some independent parameters by multiple linear regression. Adsorpt Sci Technol 35(3–4):339–356. https://doi.org/10.1177/0263617416676819
Brunauer S, Emmett PH, Teller E (1938) Adsorption of gases in multimolecular layers. J Am Chem Soc 60:309–319. https://doi.org/10.1021/ja01269a023
Cansado IPP, Mourão PAM, Ribeiro Carrott ML, Carrott PJM (2010) Activated carbons prepared from natural and synthetic raw materials with potential applications in gas separations. Adv Mater Res 107(1):1–7. https://doi.org/10.4028/www.scientific.net/AMR.107.1
Carrott PJM, Ribeiro Carrott MML, Vale TSC, Marques L, Valente Nabais JM, Mourão PAM, Suhas PAM (2008) Characterisation of surface ionisation and adsorption of phenol and 4-nitrophenol on non-porous carbon blacks. Adsorpt Sci Technol 26(10):827–841. https://doi.org/10.1260/026361708788708252
Carvajal-Bernal AM, Gómez F, Giraldo L, Moreno-Piraján JC (2015) Adsorption of phenol and 2,4-dinitrophenol on activated carbons with surface modifications. Microporous Mesoporous Mater 209:150–156. https://doi.org/10.1016/j.micromeso.2015.01.052
Fowler PA, Bellingham M, Sinclair KD, Evans NP, Pocar P, Fischer B, Schaedlich K, Schmidt JS, Amezaga MR, Bhattacharya S, Rhind SM, O’Shaughnessy PJ (2012) Impact of endocrine-disrupting compounds (EDCs) on female reproductive health. Mol Cell Endocrinol 355:231–239. https://doi.org/10.1016/j.mce.2011.10.021
Gomes JAFL, Azaruja BA, Mourão PAM (2016) From MDF and PB wastes to adsorbents for the removal of pollutants. Appl Surf Sci 380:119–126. https://doi.org/10.1016/j.apsusc.2016.02.038
Haghseresht F., Lu, GQ (1998) Adsorption characteristics of phenolic compounds onto coal-reject-derived adsorbents. Energy Fuels 12:1100–1107
Hossen MR, Talbot MW, Kennard R, Bousfield DW, Mason MD (2020) A comparative study of methods for porosity determination of cellulose based porous materials. Cellulose 27:6849–6860. https://doi.org/10.1007/s10570-020-03257-9
Hung YT, Lo HH, Wang LK, Taricska JR, Li KH (2005) Granular activated carbon adsorption. In: Wang LK, Hung YT, Shammas NK (eds) Physicochemical treatment processes. Handbook of environmental engineering, vol 3. Humana Press, Totowa
Idrees M, Batool S, Kalsoom T, Yasmeen S, Kalsoom A, Raina S, Zhuang Q, Kong J (2018) Animal manure-derived biochars produced via fast pyrolysis for the removal of divalent copper from aqueous media. J Environ Manag 213:109–118
Jun RM, Hwang HS, Heo J, Han J, Jang M, Sohn J, Park CM, Yoon Y (2019) Removal of selected endocrine-disrupting compounds using Al-based metal organic framework: performance and mechanism of competitive adsorption. J Ind Eng Chem 79:345–352. https://doi.org/10.1016/j.jiec.2019.07.009
Kadja GTM, Fabiani VA, Aziz MH, Fajar ATN, Prasetyo A, Suendo V, Ng EP, Mukti RR (2017) The effect of structural properties of natural silica precursors in the mesoporogen-free synthesis of hierarchical ZSM-5 below 100 °C. Adv Powder Technol 28:443–452. https://doi.org/10.1016/j.apt.2016.10.017
Le Cloirec P, Brasquet C, Subrenat E (1997) Adsorption onto fibrous activated carbon: applications to water treatment. Energy Fuels 11(2):331–336. https://doi.org/10.1021/ef9601430
Lee CG, Park JA, Choi JW, Ko SO, Lee SH (2016) Removal and recovery of Cr(VI) from industrial plating wastewater using fibrous anion exchanger. Water Air Soil Pollut 227:287
Naganathan KK, MohdFaizal AN, AhmadZaini MA, Ali A (2021) Adsorptive removal of bisphenol a from aqueous solution using activated carbon from coffee residue. Mater Today Proc 47:1307–1312. https://doi.org/10.1016/j.matpr.2021.02.802
Noh JS, Schwarz JA (1989) Estimation of the point of zero charge of simple oxides by mass titration. J Colloid Interface Sci 130(1):157–164. https://doi.org/10.1016/0021-9797(89)90086-6
Oweini RA, Rassy HE (2009) Synthesis and characterization by FTIR spectroscopy of silica aerogels prepared using several Si(OR)4 and R′′Si(OR′)3 precursors. J Mol Struct 919:140–145. https://doi.org/10.1016/j.molstruc.2008.08.025
Rincón-Silva NG, Moreno-Piraján JC, Giraldo L (2016) Equilibrium, kinetics and thermodynamics study of phenols adsorption onto activated carbon obtained from lignocellulosic material (Eucalyptus Globulus labill seed). Adsorption 22(1):33–48. https://doi.org/10.1007/s10450-015-9724-2
Soni H, Padmaja P (2014) Palm shell based activated carbon for removal of bisphenol A: an equilibrium, kinetic and thermodynamic study. J Porous Mater 21:275–284. https://doi.org/10.1007/s10934-013-9772-5
Tchikuala E, Mourão P, Nabais J (2017) Valorisation of natural fibres from African baobab wastes by the production of activated carbons for adsorption of Diuron. Proc Eng 200:399–407. https://doi.org/10.1016/j.proeng.2017.07.056
Thommes M, Kaneko K, Neimark AV, Olivier JP, Rodriguez-Reinoso F, Rouquerol J, Sing KS (2015) Physisorption of gases, with special reference to the evaluation of surface area and pore size distribution (IUPAC Technical Report). Pure Appl Chem 87(9–10):1051–1069. https://doi.org/10.1515/pac-2014-1117
Toledo IB, Ferro-García MA, Rivera-Utrilla J, Moreno-Castilla C, Fernández FJV (2005) Bisphenol A removal from water by activated carbon. Effects of carbon characteristics and solution chemistry. Environ Sci Technol 39:6246–6250. https://doi.org/10.1021/es0481169
Torrellas SA, Lovera RG, Escalona N, Sepúlveda C, Sotelo JL, García J (2015) Chemical-activated carbons from peach stones for the adsorption of emerging contaminants in aqueous solutions. Chem Eng J 279:788–798. https://doi.org/10.1016/j.cej.2015.05.104
Wang J, Zhang M (2020) Adsorption characteristics and mechanism of bisphenol A by magnetic biochar. Int J Environ Res Public Health 17:1075. https://doi.org/10.3390/ijerph17031075
Wang Y, Pang Y, Yang Y, Shen X (2022) Facile synthesis of recyclable magnetic covalent organic frameworks for adsorption of bisphenol A from aqueous solution. J Porous Mater 29:1411–1421. https://doi.org/10.1007/s10934-022-01266-6
Acknowledgements
The authors thanks to the following projects for financial support received: LIFE Programme of the European Union for funding the LIFE BIOAs project (LIFE19ENV/IT/000512). National Funds through FCT/MCTES—Portuguese Foundation for Science and Technology within the scope of the project UIDB/50006/2020|UIDP/50006/2020. National Funds through FCT/MCTES from MED & CHANGE Research Centres within the scope of the project UIDB/05183/2020. To the W.R. Grace & Company (Columbia, MD, USA) for supplying the silicates binders
Funding
Open access funding provided by FCT|FCCN (b-on).
Author information
Authors and Affiliations
Contributions
PM contributed to conceptualisation, methodology, writing, review, editing, data analysis and funding acquisition; CC contributed to laboratory test and data analysis; IC contributed to review and editing; JC contributed to acquisition of XRD analysis, review and editing; LR contributed to acquisition of N2, SEM–EDS analysis, review and editing; and FP contributed to review, editing and funding acquisition.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Consent for publication
We undertake and agree that the manuscript submitted to your journal has not been published elsewhere and has not been simultaneously submitted to other journals.
Additional information
Editorial responsibility: Jing Chen.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Mourão, P., Cassavela, C., Cansado, I. et al. Adsorption of bisphenol A by granular activated carbon prepared with different silicates as binders. Int. J. Environ. Sci. Technol. 21, 3719–3734 (2024). https://doi.org/10.1007/s13762-023-05212-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13762-023-05212-0