
Abstract
Mosquitoes (Diptera: Culicidae) pose a significant threat to public health worldwide, especially in tropical and subtropical regions, where they act as primary vectors in transmission of infectious agents. In Peru, 182 culicid species have been identified and several species of the genus Culex are known to transmit arboviruses. However, knowledge of mosquito diversity and distribution remains limited, with many studies focusing on specific regions only. Here, we describe a new morphological variation of Cx. (Culex) coronator Dyar and Knab, 1906, and report the presence of Culex (Carrollia) bonnei Dyar, 1921 in the central region of Peru, Huanuco. Specimens were obtained through larvae collections and identified through morphologic characterization, including dissection of male genitalia, and molecular analyses. In total, 17 mosquitoes were analyzed, and the genitalia of the male specimens allowed the identification of Cx. coronator and Cx. bonnei. Partial sequences of the CoxI gene corresponding to these two species were obtained (N = 10). Phylogenetic analysis revealed that the sequences of Cx. coronator grouped in a monophyletic clade with sequences ascribed to other species corresponding to the subgenus Carrollia, while Cx. bonnei specimens formed a monophyletic clade with homologous sequences from GenBank. This study underscores the importance of continued efforts to study the diversity and distribution of mosquitoes in Peru, including their potential role as vectors of human pathogens, to underpin effective disease control and prevention strategies, highlighting the importance of a complemented morphological and molecular analysis.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Mosquitoes of the Culicidae family represent a threat to public global health, especially in tropical and subtropical regions, where they participate as the main vectors in the transmission of infectious agents that affect humans and animals. Of the circa 3700 species of mosquitoes worldwide, 1069 species are registered in the neotropical region (Wilkerson et al. 2021), of which in Peru, 182 species have been officially reported; of these, 44 belong to the genus Culex Linnaeus, 1758 (Ayala et al. 2020, 2021) and many of these are considered arbovirus vectors, especially the species of the subgenus Melanoconion (Turell et al. 2005; Yanoviak et al. 2005; Turell et al. 2006, 2008; Evangelista et al. 2013; Hang et al. 2016; Treangen et al. 2016; Turell et al. 2021). In Peru, the arboviruses transmitted by Culex spp. mosquitoes include Mayaro virus (MAYV) (Andreolla et al. 2022), Venezuelan equine encephalitis virus (VEEV) (Aguilar et al. 2007; Vilcarromero et al. 2010), Oropouche virus (OROV) (Silva et al. 2019; Martins et al. 2020), Peruvian horse sickness virus (PHSV), Yunnan virus (YUOV) (Méndez et al. 2015), and Guaroa virus (GROV) (Aguilar et al. 2010).
Furthermore, to correctly access the risk of vector borne diseases (VBD), and carry out its surveillance and control, it is important to know the potential vector species that are present. Therefore, it is necessary to both update the registry of the mosquito species in addition and characterize their range of distribution and ecology (Pagac et al. 2021). Morphological analysis of male mosquito genitalia allows accurate identification of most mosquito species (Yadav et al. 2014; Sallum et al. 2020), as they are less susceptible to damage. In addition, the morphological analysis of the genitalia of male mosquito specimens helps to differentiate specimens that are members of species groups whose female specimens are indistinguishable, and even for which, sometimes, genetic analyses of some markers fail to disclose species identity unambiguously (Shaikevich and Vinogradova 2014).
Otherwise, with the development of molecular biology techniques, the study of mosquito DNA has become an important tool for the resolution of many taxonomic discussions (Bejarano 2001). One of the molecular tools used in the identification of mosquitoes is via DNA barcoding (Hebert et al. 2003a), the most widely used being the 658 bp partial sequence of the mitochondrial cytochrome c oxidase I subunit (CoxI) gene (Hebert et al. 2003b).
The department of Huanuco is located in the central-eastern region of Peru, covering an area of 37,266 km2, which represents 2.9% of the national territory. This department has two natural regions: the sierra (mountain) with 22,150 km2 and the mountain jungle, with 15,116 km2, and an altitude which can range between 160 and 3850 m above sea level.
The climate in this department according to the Köppen-Geiger classification is “Af” (tropical climate with forest rain) (Peel et al. 2007). In this department, 36 species of mosquitoes have been reported (Ayala et al. 2020). Of these, six species correspond to the genus Culex (Culex archegus Dyar, 1929; Cx. corniger Theobald, 1903; Cx. declarator Dyar and Knab, 1906; Cx. habilitator Dyar and Knab, 1906; Cx. mollis Dyar and Knab, 1906 and Cx. urichii Coquillett, 1906). However, the species record has had few updates and most of these have resulted from studies which were carried out more than four decades ago (Morales 1971), creating a large gap in our current knowledge of the culicine distribution in the region.
This work reports a new morphological variation of Cx. coronator, and the first record of Cx. bonnei in the mountainous region of the department of Huanuco, Peru.
Methods
Mosquito sampling
On October 7, 2022, 20 mosquito larvae were collected in two breeding places. The first was an artificial container with abundant organic matter (leaves), approximately 92 cm in height and 60 cm in circumference that was used to collect rainwater for the cultivation of coffee (9′30″31.08″S/75′58″51.12″W) (Fig. 1A), in which five larvae were collected. The other was a natural water deposit on the ground formed by a puddle (9′30″46.00″S/75′58″42.95″W) (Fig. 1B), in which 15 larvae were collected, the two collection points belonged to the village of Expedición, district of Chinchao, province and department of Huanuco (Fig. 2). The Expedición village is located within the accessible high jungle ecozone in the central region of Peru where plots of coffee cultivation can be found.

Photograph of mosquito larvae breeding places: A artificial container with abundant organic matter, B natural water deposit on the ground formed by a puddle

Map of Peru, as depicted in the right panel, illustrating the administrative departments where specimens of Culex coronator and Culex bonnei (depicted by black and red silhouettes, respectively) were previously collected. Within the Huanuco department, as shown in the left panel, the approximate locations where Culex coronator and Culex bonnei were collected are denoted by red and black dots, respectively, specifically within the Chinchao district
The collected specimens were placed in 200-ml flasks, with water from these habitats, labelled and transported to the Simulid Laboratory of the Hermilio Valdizan National University, Huanuco. Larvae were fed with fish food and kept at room temperature with 12 h of natural light. Up on emergence of adults, these were preserved in Eppendorf tubes with silica gel. Adult specimens were identified to genus and subgenus level with the help of dichotomic keys (Lane 1953; Bram 1967; Valencia 1973). For species identification, the genitalia of the male specimens were dissected and mounted on a slide with Neo-Mount fixation medium (anhydrous mounting medium for microscopy) and observed under a Leica DM1000LED microscope with a Leica MC190HD digital camera and were analyzed following the same identification keys.
Molecular analysis
Total genomic DNA was extracted from 10 adult whole mosquitoes, following the methodology of Collins et al. (1987). For the partial amplification of the CoxI gene, the specific primers LCO1490 and HCO2198 were used, with the PCR conditions as described by Folmer et al. (1994). The amplified PCR products were purified and sequenced by the Sanger method (STABVida, Lda. 2825-182 Caparica, Portugal). The sequences obtained were edited with the Chromas tool version 2.6.6 (https://technelysium.com.au/wp/, accessed on January 20, 2023).
The analysis of CoxI sequences included the search for their homologues in the public genomic databases (GenBank/ENA/DDBJ) performed with the BLASTn tool (https://blast.ncbi.nlm.nih. gov/Blast.cgi, accessed January 20, 2023) and the taxonomy search engine in the BOLDSystems v4 database (https://www.boldsystems.org/index.php/IDS_OpenIdEngine, accessed January 20 of 2023). CoxI sequence analysis also included the reconstruction of their evolutionary relationships by phylogenetic inference. The latter included the construction of multiple sequence alignments using the G-INS-i iterative refinement method implemented in MAFFT v7 (Katoh et al. 2019). The obtained alignments were treated with Gblocks (http://phylogeny.lirmm.fr/phylo_cgi/one_task.cgi?task_type=gblocks, accessed January 20, 2023) after selecting the most permissive editing options.
Phylogenetic analyses were carried out using two different approaches: the maximum likelihood (ML) optimization criterion and a Bayesian framework. For both approaches, the choice of the best nucleotide substitution model (GTR + Γ: GTR-General Time Reversal; Γ-Gamma distribution) was carried out using the MEGA X software (Kumar et al. 2018). For the ML phylogenetic reconstruction, the IQ-tree software (Nguyen et al. 2015) was used and the topological support of the branches in the obtained phylogenetic trees was assessed using bootstrap analysis and an approximate likelihood ratio test [aLRT], as implemented in IQ-tree. In both cases, 1000 replicates of the original sequence data were used, and bootstrap or aLRT values ≥ 75 (% of total number of replicates) were used as indicative of strong topological support.
For the Bayesian phylogenetic inference analysis, the BEAST v1.10.4 software (Suchard et al. 2018) was used. This analysis consisted of two independent Markov Monte Carlo chains (MCMC) that were run until 1 × 108 states were reached with sampling occurring at each 10,000 MCMC step (10% of which were later discarded as burn-in). Chain convergence was assessed using Tracer v1.7.1 software (http://beast.bio.ed.ac.uk/tracer, accessed 23 January 2023), which was also used to verify an adequate effective sample size (ESS) greater than 200 (after removal of burn-in). The distribution of the tree was summarized using the TreeAnnotator v1.8.3 software as a maximum clade credibility (MCC) tree, using median heights as the heights of the nodes in the tree. All phylogenetic trees were visualized using the FigTree v1.4.2 software (http://tree.bio.ed.ac.uk/software/figtree/, accessed 23 January 2023). Posterior probability values ≥ 0.80 were considered to indicate strong topological support. In both trees, the CoxI sequence of the species Aedes aegypti (Linnaeus, 1762) (MK265729.1) was used as outgroup.
The analysis of the average intraspecific and interspecific genetic divergence was calculated using genetic distances corrected with the Kimura 2-parameter (K2P) model, as implemented in the MEGA X software.
Results and discussion
A total of 17 adult mosquitoes emerged from collected larvae; of these, 12 were identified as Cx. (Culex) coronator Dyar and Knab, 1906 (five males and seven females) and five as Cx. (Carrollia) bonnei Dyar, 1921 (three males and two females), based on morphological features and confirmed with the analysis of the genitalia of the male specimens (Figs. 3 and 4). For our study, only male specimens of Cx. coronator and all specimens of Cx. bonnei (Table 1). The slide with the mounted genitalia was deposited within the collection of the medical entomology laboratory of the National University Hermilio Valdizan, Huanuco, Peru.

A, B Male mosquito genitalia Culex (Culex) coronator Dyar and Knab, 1906 with 100 × and 200 × magnification, sSe, single separated seta; ACL, apical cluster of setae; SLs, subapical lobe setae

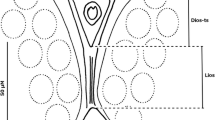
Male mosquito genitalia Culex (Carrollia) bonnei Dyar, 1921 with 100 × magnification; the red lines indicate the structures: distal division, accessory division, proximal division, and seta a and b
Culex coronator was previously considered part of the Coronator complex, along with Cx. camposi Dyar 1925, Cx. ousqua Dyar, 1918, Cx. usquatissimus Dyar, 1922, and Cx. usquatus Dyar, 1922 (Harbach 2011; Wilkerson et al. 2021). However, Laurito et al. (2018), after dissecting the genitalia of lectotype male specimens of Cx. coronator and examining slides of holotype males of Cx. camposi and Cx. ousqua, as well as the male lectotype of Cx. usquatissimus and Cx. usquatus, reported a great variation, and together with the molecular evidence led them to conclude that Cx. coronator is a single polymorphic species with no support for the specific status of those five nominal forms (Laurito et al. 2018), supporting earlier findings by Dyar (1925).
Culex coronator has a wide distribution, extending from Argentina to the United States (Wilkerson et al. 2021), and can be found over a large range of breeding sites: stagnant or slow-moving water in ground pools and seeps, ditches, culverts, artificial containers, ground depressions, tire ruts, and even dredge sites, most commonly in open, sunlit aquatic habitats (Schluep et al. 2023). In this study, Cx. coronator larvae were collected in a ground puddle, with no other mosquito larvae present. The dissected genitalia of the specimen’s male reported in this study showed different features than those described by Laurito et al. (2018). The subapical lobe of the gonocoxite was observed without division, in addition to three robust setae on the distal margin, with 11 finer setae distributed evenly, with similar length and a small curvature at the distal end (SLs, Fig. 3B). Likewise, there is a small protuberance distal to the subapical lobe with two relatively robust setae (sSe, Fig. 3B). On the other hand, long setae are observed at the apex of the gonocoxite, with a length greater than half of the gonostylus (Fig. 3A, B). These features are very similar to those described by Bram (1967), in specimens which they considered Cx. camposi (Fig. 12 – c, pg. 52), although Laurito et al. (2018) found errors in this identification for not taking into consideration the holotype of Cx. camposi. In view of the conclusions by Laurito et al. (2018), we have but to consider that the specimens here described, found in Peru, are a form of Cx. coronator, compatible with the morphological variations of this species already reported by other authors (Demari et al. 2014; Laurito et al. 2018).
Culex coronator was implicated as a potential vector of the West Nile Virus (WNV), Saint Louis Encephalitis Virus (SLEV), Venezuelan Equine Encephalitis Virus (VEEV), Murutucu Virus (MURV), and Itaqui Virus (ITQV), in Peru, Brazil, and the United States (Consoli and Oliveira 1994; Alto et al. 2014; Demari et al. 2015; Turell et al. 2021). In Peru, Cx. coronator was reported in the eastern region (Loreto, Cusco, and Ucayali), although some of these reports were attributed as Cx. usquatissimus, Cx. usquatus, and Cx. camposi (Morales 1971; Ayala et al. 2020 and Bram 1967). However, to date, no CoxI gene sequences have been reported for this species in Peru.
Partial sequences of the CoxI gene of all five male specimens Cx. coronator (LC750482-LC750486) were obtained. By using the BLASTn tool, these sequences showed 99.84–100% identity with homologous sequences from Cx. usquatissimus, Cx. usquatus, and Cx. coronator. When using the Boldsystems taxonomy tool, an identity of 99.36 to 100% with the homologues from Cx. usquatissimus, Cx. usquatus, Cx. coronator, and Cx. maxi Dyar, 1928 was observed. Regarding the phylogenetic reconstruction analysis, the generated trees showed that our Cx. coronator was grouped with GenBank sequences attributed as Cx. coronator, Cx. usquatus, and Cx. camposi (Figs. 5 and 6); in addition, the divergence analysis between these sequences is rather low (0.79 ± 0.21 to 0.72 ± 0.18) (Table S1-1) coinciding with what has already been reported (Demari et al. 2015, 2017; Laurito et al. 2013, 2018; Vesgueiro et al. 2011).

Phylogenetic analysis by Bayesian inference under the GTR + G model; the analysis involved 64 nucleotide sequences. Support values correspond to a posteriori probability. The size bar indicates 0.03 replacements per site. The sequences obtained in this work have been designated with the “EM” code, in red and blue, while the black circle indicates that they are sequences corresponding to male mosquitoes, associated with genitalia assembly. Reference sequences downloaded from the public databases and are shown by their respective access number, as well as country of origin. Vertical lines mark the Culex and Carrollia subgenera

Phylogenetic analysis with maximum likelihood tree under the GTR + G model; the analysis involved 64 partial CoxI nucleotide sequences from Culex; consensus tree probability was − 3073.525. Support values for the branches were estimated with aLRT/Bootstrap with 1000 repetitions for each method. The size bar indicates 0.04 replacements per site. The sequences obtained in this work have been designated with the “EM” code, in red and blue, while the black circle indicates that they are sequences corresponding to male mosquitoes, associated with genitalia assembly. Reference sequences downloaded from the public databases and are shown by their respective access number, as well as country of origin. Vertical lines mark the Culex and Carrollia subgenera
Culex bonnei, of the subgenus Carrollia Lutz, 1904, is so far considered a mosquito species without medical importance. The subgenus Carrollia is divided into two groups: the Bihaicolus Group composed of five species (Cx. bihaicolus Dyar & Núñez Tovar 1928; Cx. guerreroi Cova García, Sutil, & Pulido 1971; Cx. infoliatus Bonne-Wepster & Bonne 1920; Cx. metempsytus Dyar 1921 and Cx. rausseoi Cova García, Sutil O, & Pulido F. 1972) and the Iridescens Group composed of thirteen species, which in turn is divided into two subgroups: Iridescens Subgroup with eleven species (Cx. antunesi Lane & Whitman 1943; Cx. babahoyensis Levi-Castillo 1953; Cx. bonnei Dyar 1921; Cx. cerqueirai Valencia 1973; Cx. insigniforceps Clastrier & Claustre 1978; Cx. iridescens (Lutz 1905); Cx. kompi Valencia 1973; Cx. secundus Bonne-Wepster & Bonne 1920; Cx. soperi Antunes & Lane 1937; Cx. wannonii Cova García & Sutil O. 1976 and Cx. wilsoni Lane & Whitman 1943) and Urichii Subgroup with two species (Cx. anduzei Cerqueira & Lane 1944 and Cx. urichii (Coquillett 1906)) (Valencia 1973; Wilkerson et al. 2021). In our study, we followed on the characteristics of the mosquito male genitalia, in which the subapical lobe was observed, in agreement with that described by Valencia (1973), with three divisions: the distal division formed by a small bump, the accessory division in the form of a robust and long column that ends with four long and flattened setae, and the elongated and slightly curved proximal division with two to three simple distal setae and seta a and b with dilated and curved apex (Fig. 4). In Peru, five species of Culex of the Carrollia subgenus were reported, including Cx. bonnei, Cx. urichii, Cx. infoliatus, Cx. iridescens, and Cx. bihaicolus (Ayala et al. 2020, 2021). So far, Cx. bonnei has only been only reported in northern countries of South America (Wilkerson et al. 2021) reaching the 3rd parallel south in the region northeastern from Peru (Iquitos) (Lopes et al. 1985; Pecor et al. 2000). In this study, we reported for the first time Cx. bonnei in the central region of Peru (Huanuco) to the 9th parallel south. This species has been reported in many types of breeding: Broken or cut bamboo, tree holes, fallen palm spathes, fallen cacao pod, fallen fruit, artificial containers metal and plastic (Valencia 1973; Lopes et al. 1985; Patrick et al. 2002). In this study, the larvae were collected in an artificial container of plastic; furthermore, we did not find it associated with other species.
Five partial CoxI gene sequences were obtained corresponding to the specimens identified as Cx. bonnei (3 males and 2 females) (LC750487–LC750491), and these showed 90.53–99.84% identity with homologues from Cx. bonnei in BLASTn tool, and an identity of 99.35 to 99.84% with CoxI sequences from this same species when the Boldsystems tool was used, thus, differently from the previous specimens, confirming its taxonomic identity. In the phylogenetic analysis, these sequences were clearly clustered with the Cx. bonnei from Ecuador used as reference, forming a monophyletic group strongly supported, in a clade grouping the subgenus Carrollia, sister to the subgenus Culex clade (Figs. 5 and 6). Regarding the divergence analysis, the results showed an intraspecific variation of 0.13 ± 0.09, a lower result to that obtained by Demari et al. (2011) who reported a value of 0.6, while interspecific divergence value that varied between 4.83 ± 0.92 and 10.23 ± 1.44 with Cx. bihaicola, Cx. infoliatus, Cx. urichi, and Cx secundus sequences (Table S1-1). On the other hand, it is observed that the grouping of the sequences does not correspond to the proposed informal groups. In other studies, only one or two species of this subgenera were included in their phylogenetic analysis, which limited the observation of this grouping (Demari et al. 2011; Linton et al. 2013; Viveros et al. 2022).
Studies regarding the analysis of mosquitoes from Peru are limited, outdated, and concentrated only in some regions of the country, which generates a gap in the knowledge of the distribution, ecology, and diversity of the species over the Peruvian national territory (Ayala et al. 2020). In the department of Huanuco, 15 genera including 36 species of Culicidae have been registered, and of these, six are Culex species. Our study reports a new form of Cx. coronator, in addition to reporting for the first time Cx. bonnei in this region in Peru, besides contributing with partial CoxI gene sequences of these species originating from Peru to GenBank, highlighting the importance of using both morphological and molecular tools in mosquito identification (Montalvo et al. 2022, Mixão et al. 2016). However, we suspect an underestimation of mosquito biodiversity, since no further studies on culicids have been carried out in this region since 1971, even though they are important vectors of human and/or animal pathogens and whose presence is affected by the activities of humans (Johnson et al. 2008; Gorris et al. 2021).
Data availability
The slides with the mounted dissected genitalia of the mosquitoes in this study are deposited in the Faculty of Veterinary Medicine and Zootechnics of the Hermilio Valdizan National University.
References
Aguilar PV, Robich RM, Turell MJ, O’Guinn ML, Klein TA, Huaman A, Guevara C, Rios Z, Tesh RB, Watts DM (2007) Endemic eastern equine encephalitis in the Amazon region of Peru. Am J Trop Med Hyg 76(2):293–298
Aguilar PV, Morrison AC, Rocha C, Watts DM, Beingolea L, Suarez V, Vargas J, Cruz C, Guevara C, Montgomery JM, Tesh RB, Kochel TJ (2010) Guaroa virus infection among humans in Bolivia and Peru. Am J Trop Med Hyg 83(3):714–721. https://doi.org/10.4269/ajtmh.2010.10-0116
Alto BW, Connelly CR, O’Meara GF, Hickman D, Karr N (2014) Reproductive biology and susceptibility of Florida Culex coronator to infection with West Nile virus. Vector-Borne Zoonotic Dis 14(8):606–614
Andreolla AP, Borges AA, Bordignon J, Duarte dos Santos CN (2022) Mayaro virus: the state-of-the-art for antiviral drug development. Viruses 14(8):1787
Ayala Y, Carrasco-Badajoz C, Ramírez R, Iannacone J (2020) Diversidad y distribución de mosquitos (Diptera: Culicidae) en el Perú y su relación con las enfermedades metaxénicas. Rev Fac Med 70(3):e92324–e92324. https://doi.org/10.15446/revfacmed.v70n3.92324
Ayala Y, Carrasco-Badajoz C, Huicho-Yanasupo N, Zamalloa-Vilca C, Arque-Chunga W, Ortega-Morales AI, Ramírez R, Fernadez-Salas I (2021) First national record for Culex iridescens in Peru. J Am Mosq Control Assoc 37(2):90–92. https://doi.org/10.2987/20-6976.1
Bejarano EE (2001) Nuevas herramientas para la clasificación taxonómica de los insectos vectores de leishmaniosis: Utilidad de los genes mitocondriales. Biomedica 21(2):182–191
Bram RA (1967) Classification of Culex subgenus Culex in the new world (Diptera: Culicidae). Proceedings of the United States National Museum
Collins FH, Mendez MA, Rasmussen MO, Mehaffey PC, Besansky NJ, Finnerty V (1987) A ribosomal RNA gene probe differentiates member species of the Anopheles gambiae complex. Am J Trop Med Hyg 37(1):37–41
Consoli RA, de Oliveira RL (1994) Principais mosquitos de importância sanitária no Brasil. Editora Fiocruz
Demari B, Vesgueiro FT, Sallum MAM, Marrelli MT (2011) Taxonomic and phylogenetic relationships between species of the genus Culex (Diptera: Culicidae) from Brazil inferred from the cytochrome c oxidase I mitochondrial gene. J Med Entomol 48(2):272–279. https://doi.org/10.1603/ME09293
Demari B, Suesdek L, Sallum MAM, Marrelli MT (2014) Wing geometry of Culex coronator (Diptera: Culicidae) from South and Southeast Brazil. Parasit Vectors 7:174. https://doi.org/10.1186/1756-3305-7-174
Demari B, Foster PG, de Oliveira TMP, Bergo ES, Sanabani SS, Pessôa R, Sallum MAM (2015) Mitochondrial genomes and comparative analyses of Culex camposi, Culex coronator, Culex usquatus and Culex usquatissimus (Diptera: Culicidae), members of the coronator group. BMC Genomics 16:831. https://doi.org/10.1186/s12864-015-1951-0
Demari B, Multini LC, Suesdek L, Oliveira TMP, Sallum MAM, Marrelli MT (2017) Wing morphometry and genetic variability between Culex coronator and Culex usquatus (Diptera: Culicidae), two sibling species of the coronator group. J Med Entomol 54(4):901–908. https://doi.org/10.1093/jme/tjx033
Dyar HG (1925) Some mosquitoes from Ecuador (Diptera, Culicidae). Insecutor Inscitiae Menstruus 13(1–3)
Evangelista J, Cruz C, Guevara C, Astete H, Carey C, Kochel TJ, Morrison AC, Williams M, Halsey ES, Forshey BM (2013) Characterization of a novel flavivirus isolated from Culex (Melanoconion) ocossa mosquitoes from Iquitos, Peru. J Gen Virol 94(Pt 6):1266–1272. https://doi.org/10.1099/vir.0.050575-0
Folmer O, Black M, Wr H, Lutz R, Vrijenhoek R (1994) DNA primers for amplification of mitochondrial Cytochrome C oxidase subunit I from diverse metazoan invertebrates. Mol Mar Biol Biotech 3:294–299
Gorris ME, Bartlow AW, Temple SD, Romero-Alvarez D, Shutt DP, Fair JM, Kaufeld KA, Del Valle SY, Manore CA (2021) Updated distribution maps of predominant Culex mosquitoes across the Americas. Parasit Vectors 14(1). https://doi.org/10.1186/s13071-021-05051-3
Hang J, Yang Y, Kuschner RA, Evangelista J, Astete H, Halsey ES, Kochel TJ, Forshey BM (2016) Genome sequence of Bellavista virus, a novel orthobunyavirus isolated from a pool of mosquitoes captured near Iquitos, Peru. Genome Announc 4(6):e01262-e1316
Harbach RE (2011) Classification within the cosmopolitan genus Culex (Diptera: Culicidae): the foundation for molecular systematics and phylogenetic research. Acta Trop 120(1):1–14. https://doi.org/10.1016/j.actatropica.2011.06.005
Hebert PDN, Cywinska A, Ball SL, deWaard JR (2003a) Biological identifications through DNA barcodes. Proc R Soc B: Biol Sci 270(1512):313–321. https://doi.org/10.1098/rspb.2002.2218
Hebert PDN, Ratnasingham S, deWaard JR (2003b) Barcoding animal life: cytochrome c oxidase subunit 1 divergences among closely related species. Proc Biol Sci 270(Suppl 1):S96-99. https://doi.org/10.1098/rsbl.2003.0025
Johnson MF, Gómez A, Pinedo-Vasquez M (2008) Land use and mosquito diversity in the Peruvian Amazon. J Med Entomol 45(6):1023–1030. https://doi.org/10.1093/jmedent/45.6.1023
Katoh K, Rozewicki J, Yamada KD (2019) MAFFT online service: multiple sequence alignment, interactive sequence choice and visualization. Brief Bioinform 20(4):1160–1166. https://doi.org/10.1093/bib/bbx108
Kumar S, Stecher G, Li M, Knyaz C, Tamura K (2018) MEGA X: molecular evolutionary genetics analysis across computing platforms. Mol Biol Evol 35(6):1547–1549. https://doi.org/10.1093/molbev/msy096
Lane J (1953) Neotropical Culicidae. Volumes I & II
Laurito M, de Oliveira TM, Almirón WR, Sallum MAM (2013) COI barcode versus morphological identification of Culex (Culex) (Diptera: Culicidae) species: a case study using samples from Argentina and Brazil. Mem Inst Oswaldo Cruz 108:110–122. https://doi.org/10.1590/0074-0276130457
Laurito M, Briscoe AG, Almirón WR, Harbach RE (2018) Systematics of the Culex coronator complex (Diptera: Culicidae): morphological and molecular assessment. Zool J Linn Soc 182(4):735–757
Linton Y-M, Pecor JE, Porter CH, Mitchell LB, Garzón-Moreno A, Foley DH, Pecor DB, Wilkerson RC (2013) Mosquitoes of eastern Amazonian Ecuador: biodiversity, bionomics and barcodes. Mem Inst Oswaldo Cruz 108:100–109
Lopes J, Ramon Arias J, Charlhwood JD (1985) Estudo ecológico de Culicidae (Díptera) silvestres criando em pequenos recipientes de água em mata e em capoeira no município de Manaus-AM. Ciênc Cult (Säo Paulo), pp 1299–1311
Martins J, del Valle-Mendoza J, Silva-Caso W, Sandoval I, del Valle LJ, Palomares-Reyes C, Carrillo-Ng H, Peña-Tuesta I, Aguilar-Luis MA (2020) Oropouche infection a neglected arbovirus in patients with acute febrile illness from the Peruvian coast. BMC Res Notes 13(1):67. https://doi.org/10.1186/s13104-020-4937-1
Méndez MR, Attoui H, Florin D, Calisher CH, Florian-Carrillo JC, Montero S (2015) Association of vectors and environmental conditions during the emergence of Peruvian horse sickness arbivirus and Yunnan arbivirus in northern Peru. J Vector Ecol 40(2):355–363. https://doi.org/10.1111/jvec.12174
Mixão V, Bravo Barriga D, Parreira R, Novo MT, Sousa CA, Frontera E, Venter M, Braack L, Almeida APG (2016) Comparative morphological and molecular analysis confirms the presence of the West Nile virus mosquito vector Culex univittatus in the Iberian Peninsula. Parasit Vectors 9:601. https://doi.org/10.1186/s13071-016-1877-7
Montalvo E, Abílio AP, Guarido MM, Valadas V, Novo MT, Kampango A, Sousa CA, Fafetine J, Venter M, Thompson PN, Braack L, Cornel AJ, Parreira R, Almeida APG (2022) Morphological and molecular characterization using genitalia and CoxI barcode sequence analysis of Afrotropical mosquitoes with arbovirus vector potential. Diversity 14(11):940. https://doi.org/10.3390/d14110940
Morales F (1971) A list of the mosquitoes of Peru (Diptera, Culicidae). Mosq Syst 3:138–145
Nguyen L-T, Schmidt HA, von Haeseler A, Minh BQ (2015) IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol Biol Evol 32(1):268–274. https://doi.org/10.1093/molbev/msu300
Pagac BB, Spring AR, Stawicki JR, Dinh TL, Lura T, Kavanaugh MD, Pecor DB, Justi SA, Linton Y-M (2021) Incursion and establishment of the Old World arbovirus vector Aedes (Fredwardsius) vittatus (Bigot, 1861) in the Americas. Acta Trop 213:105739. https://doi.org/10.1016/j.actatropica.2020.105739
Patrick ML, Ferreira RL, Gonzalez RJ, Wood CM, Wilson RW, Bradley TJ, Val AL (2002) Ion regulatory patterns of mosquito larvae collected from breeding sites in the Amazon rain forest. Physiol Biochem Zool 75(3):215–222. https://doi.org/10.1086/342004
Pecor JE, Jones J, Turell MJ, Fernandez R, Carbajal F, O’Guinn M, Sardalis M, Watts D, Zyzak M, Calampa C (2000) Annotated checklist of the mosquito species encountered during arboviral studies in Iquitos, Peru (Diptera: Culicidae). J Am Mosq Control Assoc-Mosq News 16(3):210–218
Peel MC, Finlayson BL, McMahon TA (2007) Updated world map of the Köppen-Geiger climate classification. Hydrol Earth Syst Sci 11(5):1633–1644. https://doi.org/10.5194/hess-11-1633-2007
Sallum MAM, Obando RG, Carrejo N, Wilkerson RC (2020) Identification key to the Anopheles mosquitoes of South America (Diptera: Culicidae). III. Male genitalia. Parasit Vectors 13(1):542. https://doi.org/10.1186/s13071-020-04300-1
Schluep SM, Burkett-Cadena ND, Mathias DK, Buckner EA (2023) Culex coronator (Dyar & Knab) (Insecta: Diptera: Culicidae). EENY-794/IN1392, 3/2023. EDIS, 2023(2). https://doi.org/10.32473/edis-in1392-2023
Shaikevich EV, Vinogradova EB (2014) The discovery of a hybrid population of mosquitoes of the Culex pipiens L. complex (Diptera, Culicidae) on the Kos Island (Greece) by means of molecular markers. Entomol Rev 94(1):35–39. https://doi.org/10.1134/S0013873814010047
Silva W, Aguilar-Luis MA, Palomares-Reyes C, Mazulis F, Weilg C, del Valle LJ, Espejo-Evaristo J, Soto-Febres F, Martins-Luna J, del Valle-Mendoza J (2019) First outbreak of Oropouche Fever reported in a non-endemic western region of the Peruvian Amazon: molecular diagnosis and clinical characteristics. Int J Infect Dis 83:139–144. https://doi.org/10.1016/j.ijid.2019.04.011
Suchard MA, Lemey P, Baele G, Ayres DL, Drummond AJ, Rambaut A (2018) Bayesian phylogenetic and phylodynamic data integration using BEAST 1.10. Virus Evol 4(1). https://doi.org/10.1093/ve/vey016
Treangen TJ, Schoeler G, Phillippy AM, Bergman NH, Turell MJ (2016) Identification and genomic analysis of a novel Group C orthobunyavirus isolated from a mosquito captured near Iquitos, Peru. PLos Negl Trop Dis 10(4):e0004440. https://doi.org/10.1371/journal.pntd.0004440
Turell MJ, O’guinn ML, Jones JW, Sardelis MR, Dohm DJ, Watts DM, Fernandez R, Da Rosa AT, Guzman H, Tesh R (2005) Isolation of viruses from mosquitoes (Diptera: Culicidae) collected in the Amazon Basin region of Peru. J Med Entomol 42(5):891–898
Turell MJ, Dohm DJ, Fernandez R, Calampa C, O’guinn ML (2006) Vector competence of Peruvian mosquitoes (Diptera: Culicidae) for a subtype IIIC virus in the Venezuelan equine encephalomyelitis complex isolated from mosquitoes captured in Peru. J Am Mosq Control Assoc 22(1):70–75
Turell MJ, O’guinn ML, Dohm D, Zyzak M, Watts D, Fernandez R, Calampa C, Klein TA, Jones JW (2008) Susceptibility of Peruvian mosquitoes to eastern equine encephalitis virus. J Med Entomol 45(4):720–725
Turell MJ, Dohm DJ, Fernandez R, Klein TA (2021) Vector competence of Peruvian mosquitoes for two orthobunyaviruses isolated from mosquitoes captured in Peru. J Med Entomol 58(3):1384–1388
Valencia JD (1973) Mosquito studies (Diptera, Culicidae) XXXI. A revision of the subgenus Carrollia of Culex. Contrib Am Entomol Inst 9(4):1–134
Vesgueiro FT, Demari-Silva B, dos Santos Malafronte R, Sallum MAM, Marrelli MT (2011) Intragenomic variation in the second internal transcribed spacer of the ribosomal DNA of species of the genera Culex and Lutzia (Diptera: Culicidae). Mem Inst Oswaldo Cruz 106:01–08. https://doi.org/10.1590/S0074-02762011000100001
Vilcarromero S, Aguilar PV, Halsey ES, Laguna-Torres A, Razuri H, Perez J, Valderrama Y, Gotuzzo E, Suárez L, Céspedes M, Kochel TJ (2010) Venezuelan equine encephalitis and 2 human deaths, Peru. Emerg Infect Dis 16(3):553–556. https://doi.org/10.3201/eid1603.090970
Viveros V, Hernández-Triana LM, Ibáñez-Bernal S, Ortega-Morales AI, Nikolova NI, Pairot P, Fooks AR, Casas-Martínez M (2022) Integrated approaches for the identification of mosquitoes (Diptera: Culicidae) from the Volcanoes of Central America physiographic subprovince of the state of Chiapas, Mexico. Vector-Borne Zoonotic Dis 22(2):120–137. https://doi.org/10.1089/vbz.2021.0034
Wilkerson RC, Linton Y-M, Strickman D (2021) Mosquitoes of the world, vol 1–2. Johns Hopkins University Press
Yadav K, Sarkar P, Veer V (2014) Utility of male mosquito hypopygium in species identification. Int J Mosq Res 1:50–54
Yanoviak SP, Aguilar PV, Lounibos LP, Weaver SC (2005) Transmission of a Venezuelan equine encephalitis complex Alphavirus by Culex (Melanoconion) gnomatos (Diptera: Culicidae) in northeastern Peru. J Med Entomol 42(3):404–408
Acknowledgements
We are grateful to the authorities of the Hermilio Valdizan National University, for allowing us to rear mosquito larvae in their facilities and especially to Dra. Gizeth K. Daza Condezo, for allowing us to use the microscope with a camera in her laboratory to obtain the photomicrographs, and the Fundação para a Ciência e a Tecnologia for funds to GHTM—UID/04413/2020 and LA-REAL – LA/P/0117/2020.
Author information
Authors and Affiliations
Contributions
A.P.G.d.A., R.P., and E.M.S.—study conceptualization; A.P.G.d.A., R.P., and E.M.S.—study design and methodology; E.M.S., O.P.M.O., and G.A.O.M.—field surveys and sample collections; E.M.S., O.P.M.O., G.A.O.M., M.A.C.T., T.M., G.S., R.P., and A.P.G.d.A.—sample processing and analysis; E.M.S., R.P., and A.P.G.d.A.—phylogenetics and statistical analysis; E.M.S., A.P.G.d.A., R.P., and G.S.—manuscript drafting. All authors have read and agreed to the published version of the manuscript.
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Additional information
Edited by Rodrigo Gurgel Gonçalves
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Montalvo-Sabino, E., Marquez-Ocaña, O.P., Otiniano-Moreno, G.A. et al. Description of New Morphological Variation of Culex (Culex) coronator Dyar and Knab, 1906 and First Report of Culex (Carrollia) bonnei Dyar, 1921 Found in the Central Region of Peru. Neotrop Entomol (2024). https://doi.org/10.1007/s13744-024-01160-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s13744-024-01160-7