
Abstract
Background
Culex univittatus and Culex perexiguus mosquitoes (Diptera: Culicidae) are competent arbovirus vectors, but with unclear morphological differentiation. In Europe, and in the Iberian Peninsula in particular, the presence of either or both species is controversial. However, in order to conduct adequate surveillance for arboviruses in this region, it is crucial to clarify whether Cx. univittatus is present or not, as well as to critically assess existing differentiation tools. This study aimed to clarify this situation, by morphological and molecular phylogenetic comparison of Iberian specimens deemed as Cx. univittatus, with others of South African origin, i.e. from the type-locality region.
Methods
Thus, morphological characteristics useful to distinguish both species, such as midfemur pale line, hindfemur R ratio, seta g R1 ratio, seta f shape, length of ventral arm of phalosome and number of setae on IX tergal abdominal segment, were observed. A phylogenetic analysis based on cox1 mtDNA, of which there were no sequences from Cx. univittatus yet available in the GenBank database, was performed.
Results
This analysis showed that Iberian and South African specimens are morphologically similar, except for the length of the ventral arm of the phalosome, which was higher in the Iberian specimens. Although the Iberian specimens could not be accurately identified using BOLD Systems, phylogenetic analysis still grouped these closer to South African Cx. univittatus, than to Cx. perexiguus from Turkey and Pakistan, despite the observed segregation of both taxa as two individual monophyletic clusters with shared common ancestry.
Conclusions
This survey demonstrates that the West Nile virus vector Cx. univittatus is present in the Iberian Peninsula.
Similar content being viewed by others


Background
Mosquitoes are responsible for the transmission of several pathogens causing diseases with high morbidity and/or mortality [1]. Among them, the genus Culex comprises about 768 taxa, including some of the most ubiquitous, as well as important vectors of human pathogens which, in the present context of global warming and environmental changes, pose particular concern [1, 2]. Within the subgenus Culex lies the Univittatus subgroup, with four closely related taxa that exhibit external morphological similarities in all life stages [2, 3]: Culex (Culex) univittatus Theobald, 1901, Culex (Culex) perexiguus Theobald, 1903, Culex (Culex) neavei Theobald, 1906 and Culex (Culex) fuscocephala Theobald, 1907, the latter being an Oriental species [2]. Culex univittatus is a competent vector of arboviruses with public health importance, such as West Nile, Sindbis and Usutu viruses, in South Africa [4]. Culex perexiguus has also been found infected with West Nile, Sindbis and/or Usutu viruses, in Israel, Egypt and Saudi Arabia (reviewed in [3]). Culex univittatus/perexiguus was found as competent vector for West Nile in Portugal, Italy and Spain [5–7]. Culex neavei from South Africa seems to be a less competent vector of both West Nile and Sindbis viruses [8]. The methods of mosquito species identification in these viral surveys were not always stated, as referred below. However, this group has been subjected to extensive systematic treatment, with some taxa sunk under synonymy, or considered forms or varieties, until finally Cx. perexiguus was reinstated to full species rank [3, 9], as was Cx. neavei [8–10]. Thus, White [9] proposed a differentiation key that, based on the morphological studies by Jupp [10], indicates that analysis of morphological characters such as mid and hind femurs, and male genitalia, would allow the separation of Cx. univittatus, Cx. perexiguus and Cx. neavei. The characters used to distinguish Cx. univittatus and Cx. perexiguus are summarized in Additional file 1.
Culex univittatus, originally described from Salisbury, Zimbabwe (lectotype designated by White [9]), is widely distributed in the temperate highlands of the Afrotropical region, particularly in southern and eastern Africa, in countries such as Angola [11], Ethiopia, Kenya, Zimbabwe, South Africa and Madagascar [9, 10, 12], and Yemen, in the south-western corner of the Arabian Peninsula [3]. However, the occurrence of this species in the lowlands of western Africa, in countries such as Benin, Niger and Burkina Faso [9, 10, 12] has been considered controversial [3]. Culex perexiguus extends throughout the arid areas of West, North and East Africa, across the Sudan savannah belt, Mediterranean basin, Middle East, and south-western Asia, extending eastwards into India [3, 9, 13]. The distribution of Culex neavei is also somewhat controversial, occurring throughout the subtropical and tropical lowlands, either just in southern Africa, Reunion and Madagascar [9], or south of the Sahara [3].
The presence of Cx. univittatus in Europe has been the subject of controversy. It was reported for the first time in Portugal, by Ribeiro et al. [14], and in Spain, by Encinas-Grandes [15]. These reports included a thorough morphological analysis of adults of both sexes as well as larvae, and a sound systematic discussion. However, after examination of specimens from southern Europe (Italy and Greece) and Middle-East (Turkey), based on characteristics of the male genitalia and larvae, Harbach [13] found that these appeared to be Cx. perexiguus, and concluded that the species within the Univittatus subgroup that “occurs in southern Europe should be regarded as Cx. perexiguus rather than Cx. univittatus”. Recent molecular studies based on the analysis of mitochondrial cox1 gene confirmed the presence of Cx. perexiguus in Turkey [16].
Later surveys, usually focused on arboviruses, carried out in the Iberian Peninsula have recorded Cx. univittatus both in Portugal and Spain [17–21], by general external morphological identification, based on the findings of either Ribeiro et al. [14] and Encinas-Grandes [15], but without confirmation by the study of male genitalia. Likewise, Cx. perexiguus has also been recorded in Portugal [22] and in Spain, [23–27], albeit without any mention of particular morphological analysis or how the material was identified, and often exclusively based on the distribution criteria described by Harbach [3, 13]. Nevertheless, other authors [28–30] identified Cx. perexiguus by studying male genitalia, confirming its presence in Spain. The paucity of molecular data concerning these two taxa is also striking. The Barcode of Life Data Systems database (BOLD) [31] does not bear public sequences of either taxon, originating from Europe. Furthermore, the absence of cox1 sequences of Cx. univittatus in the GenBank database is notable, with only 8 sequences of Cx. perexiguus from Turkey and Pakistan.
Culex univittatus and Cx. perexiguus are considered mainly ornitophilic, although Cx. univittatus feeds also on humans, and more frequently than Cx. perexiguus, thus presenting a higher potential for arboviruses transmission between birds and humans or other mammals [3]. Furthermore, they also present different breeding place preferences, with Cx. univittatus immature stages found only in freshwater natural biotopes, while Cx. perexiguus tolerates moderate pollution or salinity and also use artificial containers [3]. In the Iberian Peninsula, Cx. univittatus is deemed as mainly ornithophilic, but also mammo- and particularly anthropophilic [14, 15]. However, Cx. perexiguus in Spain seems to be primarily ornithophilic and less mammophilic [26, 28] or precisely the opposite [27]. Thus, the body of evidence for each of these species vector competence, bionomic features, vectorial capacity and transmission efficiency, is still lacking particularly in this geographic region. Due to these differences and to the presence of arboviruses with medical importance such as West Nile and/or Usutu in Portugal and Spain [5, 7], the clarification of whether only Cx. univittatus or Cx. perexiguus or both species are present in the Iberian Peninsula, is imperative for the operation of surveillance programmes in this region. These programmes must accurately identify the presence and relative abundance of every vector species.
Therefore, the purpose of this study was to simultaneously use cox1 mtDNA as a molecular marker, coupled with a morphological analysis, including that of male genitalia, to identify specimens of Univittatus subgroup in the Iberian Peninsula, in order to ascertain which of its species is/are present in the extreme of western Europe, contributing to the clarification of earlier conflicting results. In parallel, these specimens were compared with specimens from the highlands of South Africa, also known as the Highveld, where Cx. univittatus is the only species of this subgroup known to occur. To the best of our knowledge, the studies that involved the analysis of mosquitoes collected in the Iberian Peninsula have neither used molecular data to corroborate the identification of these two species, nor combined this approach with morphological analysis.
Methods
Mosquito selection
Mosquitoes (n = 80) tentatively identified as Cx. univittatus, were collected in Portugal (n = 47) (districts of Santarém and Setúbal), between 2010 and 2013; in Spain (n = 15) (Extremadura Region) between 2012 and 2013; and in South Africa (n = 18) (Gauteng and Limpopo Provinces) in 2014 (Fig. 1), either by CDC miniature light-traps, mechanical hand aspirators (indoor resting mosquitoes), or tent traps (Additional file 2). Captured specimens were initially stored at -20 °C, brought to the respective laboratories and observed under a stereomicroscope and morphologically identified according to keys of Ribeiro & Ramos [32] for Portugal, of Becker et al. [1] for Spain, and of Jupp [33] for South Africa. While females were stored again at -20 °C until DNA extraction to be used for pathogen screening, males (n = 22) were kept in silica gel (Additional file 2).

Map with the three main areas of study, Portugal, Spain and South Africa
Morphology study and data analysis
Specimens were observed according to the keys of White [9] for the Univittatus subgroup. However, as many of these mosquitoes had already been used for pathogen screening, or were physically damaged, morphological characterization could only be done in a subsample of all the collected specimens. Critical to observe, included the presence or absence of a pale stripe in mid femur and determination of hind femoral index (R) (percentage of its length taken by the dorso-anterior black stripe) [10].
Male terminalia were sectioned from the abdomen and immersed in Marc André solution [34], for 5 days at room temperature. When clarified, genitalia were dissected under a stereomicroscope, in solidifiable formic acid-PVA mounting medium [34], and mounted between slide and cover slip. Gonocoxites were separated and, in each one, two structures were observed in its subapical lobe: (i) seta f, whose tip was denoted as either thin/unswollen (whether rounded or pointed), or wide/swollen (usually rhomboid and where the tip was much wider than its “neck”, c. ≥ 2.5× its “neck”) [10]; (ii) seta g, also known as “the leaflet”, for which R1 index was calculated, as the ratio of the greatest width (s) to the length (l), expressed as a percentage (s/l*100) [10].
In the phallosome, the length of the ventral arm (VA) (also known as outer division or spine), as well as the width of the lateral plate (LP) (also known as aedeagal plate), at the point of attachment of the former, were measured. Based on the fact that Cx. univittatus has a long spine-like VA, reaching beyond the caudal margin of LP [3], a ratio was calculated, consisting of VA/LP. The ninth abdominal segment was also dissected, mounted and the number of setae on its dorsal or tergal side, recorded.
Slide mounts were observed with Nomarski differential interference contrast under an Olympus microscope (BX51), and photographed with an Olympus SC30 digital camera. Normality of data distribution was assessed with Kolmogorov-Smirnov, and Shapiro-Wilk tests, and homogeneity of variances with Levene’s test. Student’s t-test and Mann-Whitney U-test, were used to compare means or medians, respectively, whether the data had normal distribution and homogeneity of variances, or not, respectively. Fisher’s exact test compared discontinuous or ordinal variables, such as the frequency of specimens with seta f, whose tip was denoted as either thin/unswollen or wide/swollen. The statistical package SPSS 20.0 [35] was used. Beeswarm graphs (one-dimensional scatter) of seta g R1 ratio and VA/LP ratio, from both groups of specimens were plotted using the Beeswarm R package (version 0.2.3) to the R statistical software (version 3.2.4) [36].
DNA extraction and amplification of cox1 mtDNA
Genomic DNA was extracted using the CTAB (Cetyltrimethylammonium bromide) method, as described by Ferreira et al. [20]. Phenol/chloroform/isoamyl alcohol was used for DNA purification. DNA was ethanol precipitated and suspended in TE buffer (pH 7.0) and stored at -20 °C until use. Negative controls were performed for each extraction procedure.
Amplification of cox1 mtDNA from both male and female specimens was performed using LCO1490 and HCO2198 specific primers, described by Folmer et al. [37]. PCR was performed in 20 μl reaction mixture containing GreenGoTaq® Flexi Buffer (Promega), 5 mM of MgCl2 (Promega), 0.2 mM of each dNTP (Promega), 0.3 pM of each primer, 0.04 U/μl of GoTaq® DNA Polymerase (Promega) and 1 ng/μl of template DNA. The thermal cycler was set at 95 °C for 5 min, followed by 40 cycles of denaturation for 30 s at 95 °C, annealing for 30 s at 48 °C, extension for 45 s at 72 °C, and a final extension for 5 min at 72 °C. The amplified products of approximately 650 bp were analysed by electrophoresis in 1.5% agarose gels stained with Ethidium bromide and observed under UV light.
DNA sequencing and sequence analysis
PCR products amplified from each sample were purified with the QIAquick PCR Purification Kit (Qiagen GmbH, Hilden, Germany) and sequenced by GATC Biotech AG or STAB VIDA in forward and reverse senses, using the same primers as for the PCR. Sequences were edited in Chromas Lite 2.1.1 (Technelysium Pty Ltd) and consensus sequences for each forward/reverse pair were created in BioEdit [38], using CLUSTAL-W version 2.0 [39]. The identity at the species level was investigated based on the analysis of the generated cox1 sequences, taking into consideration both the higher similarity in the BOLD Systems identification tool (http://www.boldsystems.org/index.php/IDS_OpenIdEngine), and results of homology searches using the sequences deposited at GenBank (http://www.ncbi.nlm.nih.gov/genbank/). All newly-generated sequences were submitted to DNA Data Bank of Japan (DDBJ) database (http://www.ddbj.nig.ac.jp) under accession numbers LC088986–LC088999, LC100115, LC102118–LC102131, LC102134–LC102136, and LC102138–LC102162.
In order to better characterize our sequence dataset using phylogenetic analysis, cox1 mtDNA sequences from Culex mosquitoes were retrieved from GenBank (accession numbers, origin and other information about those sequences in Additional file 3). All sequences were aligned using the online version of MAFFT (http://mafft.cbrc.jp/alignment/server/index.html), with the G-INS-i interactive refinement method, and taking into account alignment. Confidence score was inferred with Guidance2 Server (http://guidance.tau.ac.il/ver2/). Only regions or sequences with a score higher than 90% were considered to posterior analysis. A region of 637 bp common to all sequences (as well as a smaller 287 bp internal fragment of the latter) were used for further analysis. MEGA 6 software [40] was used to identify variable sites in the alignment.
Phylogenetic analysis
MEGA 6 software [40] was used to infer the best DNA substitution model for phylogenetic analysis. Maximum Likelihood trees were produced based on the Tamura 3-parameter formula [41], with heuristic searches based on initial trees obtained automatically from Neighbor-Joining to a matrix of pairwise distances estimated using the Maximum Composite Likelihood approach. Bootstrap coefficients were calculated for 10,000 replicates. Estimates of evolutionary pairwise divergence between all sequences, between and within the defined groups, were estimated using the Tamura 3-parameter model [41].
Phylogenetic reconstruction (consensus tree) following a Bayesian approach was also conducted using MrBayes v3.0b4 [42], using the GTR + Γ + I model (GTR-General Time Reversal; Γ-Gamma distribution; I-proportion of invariable sites) and default priors. This analysis consisted of 5 × 107 generations starting from a random tree and four Markov chains with default heating values, sampled every 100th generation. Two separate runs were conducted for each analysis, and the first 10% sampled trees discarded as 'burn-in’. Maximum Clade Credibility trees were constructed using BEASTv1.7.5 [43], using the GTR + Γ + I model, and as coalescent priors a constant population size and a strict molecular clock. These analyses were run for 1 × 108 generations starting from a random tree with sampling at every 5,000th generation. The results of two separate runs were combined using LogCombiner (available at http://beast.bio.ed.ac.uk/logcombiner), and the first 10% discarded as 'burn-in'. For each case, convergence was monitored with Tracer v1.6 (available from http://beast.bio.ed.ac.uk/tracer), ensuring that ESS values were above 200. The obtained phylogenetic trees were manipulated for display using FigTree v.1.4.2. (available at http://tree.bio.ed.ac.uk/software/figtree/).
Results
Morphological study
All specimens in this study had been tentatively morphologically identified as Cx. univittatus according the keys of White [9] and Jupp [10]. Curiously, upon observation of 49 specimens, 62.5% (20/32) from Portugal and Spain (PT & SP), and 58.8% (10/17) from South Africa (SA) displayed a clear continuous pale stripe in mid femur, while 21.9% (7/32) PT & SP, and 35.3% (6/17) from SA had an interrupted but clear line, while 15.6% (5/32) from PT & SP, and 5.9% (1/17) from SA had no line at all (Additional file 2). The presence and form of this character was therefore not significantly different between these two samples, Fisher’s exact test = 1.52, P = 0.414, two-sided exact significance.
As to the R hind-femoral index (n = 31), specimens from PT & SP had a mean of 82.7% (95% CI: 77.8–87.6, range 60–98, n = 16), and specimens from SA had a mean of 81.96% (95% CI: 76.7–87.2, range 66–95, n = 15), hence not significantly different (Student’s t = 0.22, df = 29, P = 0.83, 95% CI for the difference: -6.15–7.61).
Dissected genitalia from 22 males, 11 from Portugal and 11 from South Africa (the sample from Spain did not include males), were analysed (Fig. 2; full collection of photographs in Additional file 4). The tip of seta f was thin in 89.5% and swollen in 10.5% of Portuguese specimens versus thin in 75% and swollen in 25% of South African specimens, hence not significantly different, Fisher’s exact test 2-sided P = 0.407, n = 39 (see Additional file 5 for all data).

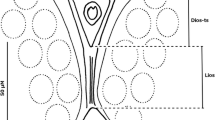
Dissected and mounted genitalia of male Cx. univittatus from South Africa (a-c; specimen SAfr-GAU-117-w3) and Cx. univittatus from Portugal (d-f; specimen Port-2630.69/3435). Gonocoxites with g and f setae (a, d; magnification 100×), phalosome (b, e; magnification 200×) with ventral arm (VA) and lateral arm (LA), and ninth segment tergum (c, f; magnification 200×) with setae (S). Scale-bars: a, d, 100 μm; b, c, e, f, 50 μm
Leaflet or seta g R1 ratio [10] varied between 32 and 54%, mean 45% (± 0.07 SD), for Portuguese specimens, and 34–57%, mean 45% (± 0.05 SD), for South African specimens (Fig. 3a), hence not significantly different between these two populations; Student’s t = 0.15, df = 33, P = 0.8 (see Additional file 5 for all data).

Beeswarm scatter plots (a, b), and Scatter diagram (c). Gonocoxite’s leaflet or seta g R1 ratio [10] (a) and phallosome’s ratio of the length of the ventral arm (VA) over the width of the lateral plate (LP), at the point of attachment of the former, VA/LP, from Portuguese and South African mosquitoes (b). All ratios for individual structures are plotted. The horizontal lines show the 25, 50 and 75% quartiles. Scatter diagram of g R1 ratio versus VA/LP ratio of mean values for each specimen (c)
For the phallosome, the ratio of the length of the ventral arm (VA) over the width of the lateral plate (LP) at the point of attachment of the former, VA/LP, varied between 1.235–1.746, mean 1.451 (± 0.17 standard deviation, SD) for Portuguese specimens, and from 1.081–1.529, mean 1.321 (± 0.13 SD) for the South African specimens (Fig. 3b). This difference was shown to be statistically significant by Student’s t-test [t = 4.18, df = 42, P < 0.001, 95% CI of the difference: 0.09–0.27 (data with normal distribution and homogeneity of variances)] (see Additional file 5 for all data). A scatter diagram of paired values of seta g R1 index and VA/LP ratio is plotted in Fig. 3c, showing an overlap of these compound ratios for the two population samples.
The ninth tergal lobe had 8–11 (median 9) setae for Portuguese specimens, and 7–15 (median 10) setae for South African specimens (Fig. 2). This difference, however, was not statistically significant, using the Mann-Whitney U-test (U = 262, P = 0.286) (see Additional file 5 for all data).
cox1 mtDNA amplification and sequence analysis
From all 80 specimens, it was only possible to have good cox1 mtDNA amplification for 56 samples (Additional file 2). From these specimens, consensus sequences were obtained and examined in BLASTn and BOLD System. Their analysis with BLASTn revealed homologies ranging between 90 and 96% with cox1 sequences of Cx. perexiguus (accession numbers: KJ012105.1, KF406802.1, KJ012109.1), worthy of being noted the absence of cox1 sequences of Cx. univittatus in the GenBank database (the only sequence found for Cx. univittatus in GenBank was a 375 bp mtDNA sequence from 12 NADH dehydrogenase subunit 4 gene, accession number: EF030093.1). Therefore, BLASTn could not allow an accurate identification of our samples, as even with the MegaBlast option implemented, sequence homology values of 96% do not allow for clear-cut species identification.
When submitted to the BOLD System identification tool, all the sequences from South Africa presented more than 98.46% similarity with Cx. univittatus sequences, though for the great majority of them (all but 3), sequence similarity values with Cx. univittatus was equal or higher than 99.23%. Thus, all samples from South Africa were considered as Cx. univittatus by the BOLD system platform (Additional file 2). Sequences from the Iberian Peninsula, in BOLD System platform revealed similarities with Cx. univittatus that varied from 97.22 to 98.01%. After these higher similarities, in 21 samples, the next matches corresponded to sequences of Cx. univittatus, and then, to Cx. perexiguus (in this order) (Additional file 6). However, in 7 samples from Portugal, as well as in all samples from Spain, this order was inverted, with higher similarity corresponding to Cx. univittatus, immediately followed by similarities (< 97%) with Cx. perexiguus (Additional file 2), and thereafter to Cx. univittatus again. Therefore, it was not possible to clearly identify the Iberian specimens as Cx. univittatus or Cx. perexiguus. However, in phylogenetic trees obtained through BOLD Systems identification, our sequences appear clustering with Cx. univittatus sequences, and in a different clade of Cx. perexiguus sequences, including 4 sequences from Spain (Additional file 6).
Phylogenetic analysis
Additionally, a 637 bp region of the alignment obtained was analysed, leading to the finding of 76 polymorphic sites in sequences from the Univittatus subgroup (Additional file 7). Those polymorphisms revealed the existence of substantial differences between sequences from the Iberian Peninsula (Portugal and Spain), with respect to other species from this subgroup, including Cx. univittatus from South Africa and Cx. perexiguus from Turkey and Pakistan. From all 56 DNA sequences, only 44 were considered for phylogenetic analysis, since all sequences with less than 648 bp were excluded. All the aligned positions with a Guidance score lower than 90% were also excluded (Additional file 2).
By phylogenetic analysis (Fig. 4) it was perceptible that sequences from Portugal and Spain are closer to each other, forming a joint clade that groups the Iberian specimens together (bootstrap value, BS = 97), which is a sister clade of another one which clustered the South African sequences, considered as bona fide Cx. univittatus (BS = 99). This monophyletic clade that reveals common ancestry for the Iberian and South African sequences is only subsequently joined with that defining Cx. perexiguus (BS = 93). Therefore, the analysis of cox1 coding sequences here performed strongly suggests that Portuguese and Spanish specimens are closer to the South African Cx. univittatus, than to Cx. perexiguus from Turkey and Pakistan (Fig. 4). Furthermore, phylogenetic analyses carried out under a Bayesian framework, as implemented in MrBayes (consensus tree with default priors) or Beast (strict molecular clock and a constant population size) confirmed the topology of the ML tree. Both trees present identical topologies, for which only the consensus tree is shown as Additional file 8.

Phylogenetic analysis by Maximum Likelihood. The tree with the highest log likelihood (-2167.4061) is shown. The size bar indicates 0.02 substitutions per site. The analysis involved 84 nucleotide sequences, with a total of 637 positions in the final dataset. The tree has been rooted using Ae. (Och) caspius as the outgroup. Abbreviations: SAfr, South Africa; Port, Portugal; Spai, Spain
Our phylogenetic analysis also provides a clear differentiation between all our sequences and those from other Culex taxa, namely Cx. fuscocephala, another member of the Univittatus subgroup. On the other hand, no sequences of Cx. neavei were included as none were available in GenBank. A second phylogenetic analysis, including sequences of lesser quality, and from a smaller part of the alignment (287 bp), but including more male specimens, in relation to Fig. 4, with combined morphological genitalia analysis, corroborated these findings, with only a slight decrease in bootstrap values and maintaining the same tree topology (Additional file 9).
The analysis of pairwise distance values also indicates that cox1 mtDNA sequences from Portugal and Spain are very similar to one another, some comparisons revealing sequence identity (genetic distance of 0), which is in accordance with results from determination of polymorphic sites presented in the previous section. Estimates of average evolutionary divergence over sequence pairs within groups (Additional file 10) allowed the confirmation of low divergences within samples from Portugal and Spain (0.001; standard error, SE 0.0), supporting our decision to perform the analysis grouping sequences from both countries. Estimates of evolutionary divergence over sequence pairs between groups showed that sequences from Portugal and Spain have a distance of 0.022 (SE 0.005) with sequences of Cx. univittatus from South Africa and of 0.042 (SE 0.008) with Cx. perexiguus from Turkey and Pakistan (Table 1).
Discussion
The Univittatus subgroup of Culex spp. mosquitoes includes Cx. univittatus and Cx. perexiguus, which are vectors of arboviruses such as West Nile, Sindbis or Usutu. These viruses are responsible for various febrile or neurological syndromes, either in humans or other animals, in several countries of the northern and southern hemisphere especially in Europe and Africa [4–8]. However, controversy has existed regarding which species of this subgroup is/are present in Europe. In the Iberian Peninsula, the presence of Cx. univittatus has been documented [14, 15], while specimens from Italy, Greece and Turkey were stated to be Cx. perexiguus [13, 16]. In the context of arboviral circulation [5–7], the correct identification of infected/infectious species is mandatory since all cascade of bionomical and epidemiological studies, as well as control strategies that might be applied, depend on this knowledge.
In this study, comparative morphological and molecular analyses were carried out, based on the study of specimens putatively identified as Cx. univittatus from Portugal and Spain, as well as others previously identified as Cx. univittatus, from a region where this is the only species of the Univittatus subgroup known to be present (the Highveld region of South Africa) [10].
The morphological analysis revealed no significant differences in the proportion of mosquitoes from either population that possessed a continuous pale line on the mid femur. Although this character is one of those used to separate Cx. univittatus from both Cx. perexiguus and Cx. neavei [9], contrary to what might be expected, Cx. univittatus from the Highveld South Africa often have an interrupted discontinuous pale line, possibly indicating that this feature should not be considered a determinant or reliable character for the identification of this species. The hind femoral R index which allows the differentiation between Cx. univittatus and Cx. neavei [10], was not different between these mosquito populations of South Africa and Iberia Peninsula, either. Obviously, the observation of both these characters is strongly conditioned by the conservation status of the captured specimens, which in CDC traps is often poor, hence should be evaluated with caution.
In the males, the study of the genitalia, allowed us the confirmation that the shape of the tip of seta f, the seta g R1 ratio, and the number of setae in the ninth tergal lobe, are also similar between the Portuguese and the South African specimens. In the phalosome, although the ventral arm seemed to be longer in Portuguese specimens than in South African ones, the range of variation was overlapping, and in any case would point further in the direction of Cx. univittatus, rather than to Cx. perexiguus which has a short ventral arm [3, 9]. Altogether, these morphological characters, particularly those from the genitalia, seem to indicate that the Iberian specimens should be identified as Cx. univittatus, confirming previous studies [14, 15].
Identification of our samples through the BOLD Systems platform clearly assigned the South African specimens as Cx. univittatus. Likewise, the sequences of Turkish Cx. perexiguus from GenBank [16] were clearly identified as Cx. perexiguus. For the Iberian specimens, no sequence was clearly identified as either species, although their higher similarity was always closer to Cx. univittatus. Furthermore, trees generated through the BOLD System identification tool showed a differentiation of the sequences of Portuguese and Spanish origin with respect to four sequences of Cx. perexiguus from Spain. Although the latter were not available for further sequence inspection, the separation between Cx. univittatus and Cx. perexiguus was further reinforced by phylogenetic analysis. However, in the Barcode of Life Data Systems database [31], 184 sequences of Cx. perexiguus were referred with origins such as Jordan, Tanzania, Turkey, Kenya, India and Pakistan, with no mention to the existence of those Cx. perexiguus sequences from Spain that appeared in the trees (http://www.boldsystems.org/index.php/Taxbrowser_Taxonpage?taxon=culex+perexiguus&searchTax= accessed on 28-6-2016).
Likewise, in BOLD Systems platform there are only 21 sequences of Cx. univittatus from Tanzania, 1 from South Africa and 1 from Kenya. Therefore it is not surprising that Iberian samples did not reach maximum similarity or clear identification (http://boldsystems.org/index.php/Taxbrowser_Taxonpage?taxon=culex+univittatus&searchTax=, accessed on the 28-06-2016). These results show one of the limitations of barcoding identification that is the dependence on available similar sequences for evaluation. Other authors have also reported some difficulties in distinguishing some mosquito close related species by barcoding [44, 45]. Thus, the phylogenetic analysis based on the larger region of cox1 mtDNA performed in this survey was essential to clarify the identification of Iberian specimens.
In order to perform a rigorous phylogenetic analysis (637 bp cox1 fragment), some of the specimens were excluded from the dataset due to a low score in the alignment, or because of their reduced size. This latter reason led to the absence from this so-called main-tree of some sequences obtained from male specimens with morphological analysis associated. For that reason, a second tree corresponding to the analysis of a smaller fragment size (287 bp) was generated. While the number of species differs in both datasets (e.g. Cx. fuscocephala is absent from one of them) the resulting sequence-clustering pattern (tree topology) remained generally congruent with that of the main-tree. Altogether, the phylogenetic analyses here presented allowed the distinction between all the known species within the Univittatus subgoup (Cx. univittatus, Cx. perexiguus and Cx. fuscocephala) except for Cx. neavei, for which cox1 sequences could not be found in the public databases. Fortunately, the BOLD Systems database does include Cx. neavei sequences (8 from Kenya and Nigeria, albeit not public), therefore their absence from the BOLD Systems automatic sequence identification result lists clearly shows that the specimens analysed in this report are not closely related to this species. We therefore consider their absence in our phylogenetic analysis not paramount to the study. The phylogenetic analysis hereby reported confirms that cox1 sequences amplified from mosquitoes from Portugal and Spain cluster together and are closer to Cx. univittatus from South Africa than to Cx. perexiguus from Turkey and Pakistan, in accordance with the BOLD system analysis.
Although the obtained Bayesian trees were based on a smaller sequence dataset than that previously used for the ML phylogenetic reconstruction, both of them showed a clear separation of the Cx. perexiguus and Cx. univittatus clusters, and a clear-cut segregation of the South African Cx. univittatus away from the Iberian Cx. univittatus/perexiguus sequences.
The differentiation observed between the Iberian and the South African cluster is in accordance with the genitalia VA/LP ratio values, which although overlapping, were already statistically different. Such differences are naturally expected in such geographically distant populations, and it would be interesting to study the crossbreeding success of these populations. Nevertheless, both the morphological and molecular analysis were coherent in supporting the identification of these Iberian specimens as Cx. univittatus.
Conclusions
This study represents, to the best of our knowledge, the first molecular and phylogenetic analysis of Cx. univittatus/perexiguus. Although not exhaustive, either geographically or taxonomically, our results clearly confirm the presence of Cx. univittatus in the Iberian Peninsula, as previous morphological studies have shown [14, 15]. Thus, it can be concluded that Cx. perexiguus is not the only species of the Univittatus subgroup existing in Europe. It was also possible to see that these Cx. univittatus specimens are genetically different from those from South Africa, and it would be interesting to study the evolutionary track of the Univittatus subgroup, including its other members and other origins, to clarify their evolutionary relationships. Considering the vector role for arboviruses of both these taxa and the lack of updated characterization of their respective vectorial capacity and transmission efficiency, further bionomic characterization subsequent to correct species identification is needed for a sounder knowledge of arbovirus receptivity in this geographic region.
Abbreviations
- cox1:
-
Cytochrome c oxidade subunit 1 mtDNA
- mtDNA:
-
Mitochondrial DNA
- USU:
-
Usutu Virus
- WNV:
-
West Nile Virus
References
Becker N, Petric D, Zgomba M, Boase C, Minoo M, Dahl C, et al. Mosquitoes and their control. New York: Kluwer Academic/Plenum; 2010.
Harbach RE. Classification within the cosmopolitan genus Culex (Diptera: Culicidae): The foundation for molecular systematics and phylogenetic research. Acta Trop. 2011;120(1–2):1–14.
Harbach RE. The mosquitoes of the subgenus Culex in southwestern Asia and Egypt (Diptera: Culicidae). Contrib Am Ent Inst (Ann Arbor). 1988;24(1):1–240.
McIntosh BM, Jupp PG, dos Santos I, Meenehan GM. Epidemics of West Nile and Sindbis viruses in South Africa with Culex (Culex) univittatus Theobald as vector. S African J Sci. 1976;72(10):295–300.
Esteves A, Almeida AP, Galão RP, Parreira R, Piedade J, Rodrigues JC, et al. West Nile virus in Southern Portugal, 2004. Vector Borne Zoonotic Dis. 2005;5(4):410–3.
Tamba M, Bonilauri P, Bellini R, Calzolari M, Albieri A, Sambri V, et al. Detection of Usutu Virus within a West Nile Virus surveillance program in Northern Italy. Vector Borne Zoonotic Dis. 2011;11(5):551–7.
Vázquez A, Ruiz S, Herrero L, Moreno J, Molero F, Magallanes A, et al. West Nile and Usutu viruses in mosquitoes in Spain, 2008–2009. Am J Trop Med Hyg. 2011;85(1):178–81.
Jupp PG, Harbach RE. Crossmating and morphological studies of Culex neavei and Culex perexiguus (Diptera: Culicidae) to elucidate their taxonomic status. Mosq Syst. 1990;22(1):1–10.
White GB. Notes on a catalogue of Culicidae of the Ethiopian Region. Mosq Syst. 1975;7:303–43.
Jupp PG. The taxonomic status of Culex (Culex) univittatus Theobald (Diptera: Culicidae) in South Africa. J Entomol Soc South Af. 1971;34:339–57.
Ribeiro H, Ramos HDaC. Research on the mosquitoes of Angola (Diptera, Culicidae). X. The genus Culex L., 1758. Check-list with new records, keys to females and larvae, distribution and taxonomic and bioecological notes. Est. Ens. Doc., n 134, Lisboa; Junta de Investigações Científicas do Ultramar, 1980.
Jupp PG. A morphological study of Culex (Culex) univittatus Theobald and Culex (Culex) neavei Theobald from various African countries. Mosq Syst. 1972;4:103–13.
Harbach RE. The identity of Culex perexiguus Theobald versus Cx. univittatus Theobald in southern Europe. Eur Mosq Bull. 1999;4:7.
Ribeiro H, Ramos HC, Pires CA, Capela RA. Research on the mosquitoes of Portugal (Diptera, Culicidae) I. Four new culicine records. An Inst Hig Med Trop (Lisb). 1977;5(1–4):203–14.
Encinas-Grandes A. Taxonomía y biología de los mosquitos del área salmantina (Diptera, Culicidae). CSIC – Ed. Salamanca, Spain: Universidad de Salamanca; 1982. p. 437.
Gunay F, Alten B, Simsek F, Aldemir A, Linton YM. Barcoding Turkish Culex mosquitoes to facilitate arbovirus vector incrimination studies reveals hidden diversity and new potential vectors. Acta Trop. 2015;143:112–20.
Almeida AP, Galão RP, Sousa CA, Novo MT, Parreira R, Pinto J, et al. Potential mosquito vectors of arboviruses in Portugal: species, distribution, abundance and West Nile infection. Trans R Soc Trop Med Hyg. 2008;102(8):823–32.
Almeida AP, Freitas FB, Novo MT, Sousa CA, Rodrigues JC, Alves R, et al. Mosquito surveys and West Nile Virus screening in two different areas of southern Portugal, 2004–2007. Vector Borne Zoonotic Dis. 2010;10(7):673–80.
Pérez–Bote JL. Mosquitoes (Diptera, Culicidae) de las vegas del río Guadiana (Extremadura, España). Boln Asoc Esp Ent. 2012;36:61–73.
Ferreira CA, Mixão VDP, Novo MT, Calado MM, Gonçalves LA, Belo S, et al. First molecular identification of mosquito vectors of Dirofilaria immitis in continental Portugal. Parasit Vectors. 2015;8:139.
Bravo-Barriga D, Parreira R, Almeida AP, Calado M, Blanco-Ciudad J, Serrano-Aguilera FJ, et al. Culex pipiens as a potential vector for transmission of Dirofilaria immitis and other unclassified Filarioidea in Southwest Spain. Vet Parasitol. 2016;223:173–80.
Osório HC, Amaro F, Zé-Zé L, Pardal S, Mendes L, Ventim R, et al. Mosquito species distribution in mainland Portugal 2005–2008. J Am Mosquito Contr. 2010;28:187–93.
Alcaide M, Rico C, Ruiz S, Soriguer R, Muñoz J, Figuerola J. Disentangling vector-borne transmission networks: a universal DNA barcoding method to identify vertebrate hosts from arthropod bloodmeals. PLoS ONE. 2009;4(9):e7092.
Vazquez A, Sanchez-Seco MP, Ruiz S, Molero F, Hernandez L, Moreno J, et al. Putative new lineage of West Nile virus, Spain. Emerg Infect Dis. 2010;16(3):549–52.
García–Bocanegra I, Jaén-Téllez JA, Napp S, Arenas-Montes A, Fernández-Morente M, Fernández-Molera V, et al. Monitoring of the West Nile virus epidemic in Spain between 2010 and 2011. Transbound Emerg Dis. 2012;59(5):448–55.
Ferraguti M, Martínez-de la Puente J, Muñoz J, Roiz D, Ruiz S, Soriguer R, et al. Avian Plasmodium in Culex and Ochlerotatus Mosquitoes from Southern Spain: effects of season and host–feeding source on parasite dynamics. PLoS ONE. 2013;8(6):e66237.
Martínez-de la Puente J, Ruiz S, Soriguer R, Figuerola J. Effect of blood meal digestion and DNA extraction protocol on the success of blood meal source determination in the malaria vector Anopheles atroparvus. Malar J. 2013;12:109.
Muñoz J, Ruiz S, Soriguer R, Alcaide M, Viana DS, Roiz D, et al. Feeding patterns of potential West Nile virus vectors in south–west Spain. PLoS ONE. 2012;7(6):e39549.
Roiz D, Roussel M, Muñoz J, Ruiz S, Soriguer R, Figuerola J. Efficacy of mosquito traps for collecting potential West Nile mosquito vectrs in a natural Mediterranean wetland. Am J Trop Med Hyg. 2012;86(4):642–8.
Roiz D, Ruiz S, Soriguer R, Figuerola J. Landscape effects on the presence, abundance and diversity of mosquitoes in Mediterranean wetlands. PLoS ONE. 2015;10(6):e0128112.
Ratnasingham S, Hebert PDN. BOLD: The Barcode of Life Data System (http://www.barcodinglife.org). Mol Ecol Notes. 2007;7(3):355–64.
Ribeiro H, Ramos HC. Identification keys of the mosquitoes of Continental Portugal, Açores and Madeira. Eur Mosq Bull. 1999;3:1–11.
Jupp PG. Mosquitoes of Southern Africa. Culicine and Toxorhynchitinae. Hartebeespoort, Republic of South Africa: Ekogilde Publishers; 1996. p. 156.
Ribeiro H. A solidifiable formic acid–PVA solution for transporting, preserving and mounting mosquito larvae and pupae. Stain Technol. 1967;42(3):158–60.
IBM. SPSS for Windows ® Inc., Statistical Package for Social Sciences®, Chicago, Illinois, USA. 1999.
Team RDC R. A language and environment for statistical computing. Vienna, Austria: R Foundation for Statistical Computing; 2016.
Folmer O, Black M, Hoeh W, Lutz R, Vrijenhoek R. DNA primers for amplification of mitochondrial cytochrome c oxidase subunit I from diverse metazoan invertebrates. Mol Mar Biol Biotechnol. 1994;3(5):294–9.
Hall T. Bioedit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symp Ser. 1999;4:95–8.
Thompson JD, Higgins DG, Gibson TJ. ClustalW: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994;22(22):4673–80.
Tamura K, Stecher G, Peterson D, Filipski A, Kumar S. MEGA6: molecular evolutionary genetics analysis version 6.0. Mol Biol Evol. 2013;30(12):2725–9.
Tamura K. Estimation of the number of nucleotide substitutions when there are strong transition–transversion and G + C-content biases. Mol Biol Evol. 1992;9(4):678–87.
Ronquist F, Huelsenbeck JP. MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics. 2003;12:1572–74.
Drummond AJ, Suchard MA, Xie D, Rambaut A. Bayesian phylogenetics with BEAUti and the BEAST 1.7. Mol Biol Evol. 2012;29(8):1969–73.
Bourke BP, Oliveira TP, Suesdek L, Bergo ES, Sallum M. A multi-locus approach to barcoding in the Anopheles strodei subgroup (Diptera: Culicidae). Parasit Vectors. 2013;6:111.
Wang G, Li C, Guo X, Xing D, Dong Y, Wang Z, et al. Identifying the main mosquito species in China based on DNA barcoding. PLoS ONE. 2012;7(10):e47051.
Acknowledgments
The authors thank Prof. Silvana Belo, for supporting part of the study, Prof. João Pinto for hosting DBB in VBD-GHTM, and Doctor Ana Abecassis for phylogenetic advice.
Funding
This study was financed by: (i) Fundação para a Ciência e Tecnologia (FCT, Portugal), through the project grant PTDC/SAU-SAP/113523/2009, and GHTM - UID/Multi/04413/2013, supporting mosquito collection in Portugal, and respective analysis of Portuguese and South African specimens; (ii) research project (IB10044) of the “Consejería de Economía, Comercio e Innovación” of the Extremadura regional Government (Spain), supported mosquito collection in Spain, and respective analysis; (iii) Grant or Cooperative Agreement with the National Institute for Communicable Diseases, Activity 23 (2012–2013): Surveillance for zoonotic vector borne neurological diseases in humans and animals in South Africa, Number [1U19GH000571], funded by the Centers for Disease Control and Prevention, which supported mosquito collections in South Africa. Its contents are solely the responsibility of the authors and do not necessarily represent the official views of the Centers for Disease Control and Prevention or the Department of Health and Human Services. DBB holds a scholarship from the Ministry of Education, Culture and Sports of Spain (FPU grant AP2010-5854). Part of this work was done during the stay of DBB in the Global Health and Tropical Medicine (GHTM) in Lisbon, funded by Banco Santander, SA through program “Becas Iberoamérica. Jóvenes Profesores e Investigadores. Santander Universidades 2014”. APGA was the recipient of a Visiting Professor Program grant from the Department of Medical Virology, Faculty of Health Sciences, University of Pretoria, South Africa, and grant SFRH/BSAB/1364/2013, FCT, Portugal.
Availability of data and materials
DNA sequences used for cox1 mtDNA analysis generated and analysed during the current study are available in the GenBank database, DDBJ, or the European Nucleotide Archive database, under the accession numbers LC088986–LC088999, LC100115, LC102118–LC102131, LC102134–LC102136, and LC102138–LC102162. All other data generated for morphological analysis are available in the Additional files.
Authors’ contributions
APGA conceived this study, within the greater framework of surveillance programs designed by MV, LB and EF. APGA, VM, DBB, MTN and CS collected and/or identified the mosquitoes. APGA performed the morphological study, its photographic and statistical analysis. VM participated in the morphological analysis. VM and DBB carried out the molecular studies. VM and RP performed the phylogenetic analysis. VM and DBB analysed the data and drafted the manuscript. APGA supervised the analysis of the data and redaction of the manuscript. All authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Consent for publication
Not applicable.
Ethics approval and consent to participate
Not applicable.
Author information
Authors and Affiliations
Corresponding author
Additional files
Additional file 1:
Characters used to distinguish Cx. univittatus and Cx. perexiguus. (PDF 34 kb)
Additional file 2:
Samples analysed with respective morphological and molecular data. (XLSX 226 kb)
Additional file 3:
cox1 mtDNA sequences retrieved from the GenBank database for sequence and phylogenetic analysis. (PDF 89 kb)
Additional file 4:
Photos of male genitalia of Cx. univittatus from South Africa and Portuguese specimens. (PDF 3361 kb)
Additional file 5:
Data for the structures of male genitalia results of statistical analyses. (XLSX 252 kb)
Additional file 6:
Example of some results obtained in BOLD Systems identification tool. Percentage of similarity results and phylogenetic trees are shown. (PDF 10649 kb)
Additional file 7:
Variable sites found in a 637 bp region of cox1 mtDNA alignment of Univittatus subgroup. Abbreviations: SAfr, Culex univittatus from South Africa; Port, specimens from Portugal; Spai, specimens from Spain. (PDF 86 kb)
Additional file 8:
Bayesian phylogenetic analysis (consensus tree) based on cox1 mosquito sequences. At specific branch nodes posterior probabilities ≥ 0.90 are indicated. The scale-bar indicates the number of nucleotide substitutions per site. The tree was rooted with a cox1 sequence from Ae. (Och.) caspius. (TIF 2231 kb)
Additional file 9:
Molecular phylogenetic analysis of a small fragment of the cox1 alignment, with a higher number of male sequences, by Maximum Likelihood. The tree with the highest log likelihood (-883.6486) is shown. The percentage of trees in which the associated taxa clustered together is shown next to the branches. The scale-bar indicates 0.02 substitutions per site. The analysis involved 90 nucleotide sequences. There were a total of 287 positions in the final dataset. (TIFF 240 kb)
Additional file 10:
Estimates of average evolutionary divergence in cox1 over sequence pairs within groups. The numbers of base substitutions per site from averaging over all sequence pairs within each group are shown. Standard error estimates are shown in the last column. The analysis involved 84 nucleotide sequences. Codon positions included were 1st + 2nd + 3rd + Noncoding. All positions containing gaps and missing data were eliminated. There were a total of 637 positions in the final dataset. (PDF 59 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Mixão, V., Bravo Barriga, D., Parreira, R. et al. Comparative morphological and molecular analysis confirms the presence of the West Nile virus mosquito vector, Culex univittatus, in the Iberian Peninsula. Parasites Vectors 9, 601 (2016). https://doi.org/10.1186/s13071-016-1877-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13071-016-1877-7