
Abstract
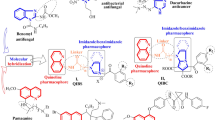
Ciprofloxacin-Piperazine C-7 linked quinoline derivatives 6a–c and 8a–c were synthesized and investigated for their antibacterial, antifungal, and anti-proliferative activities. Ciprofloxacin-quinoline-4-yl-1,3,4 oxadiazoles 6a and 6b showed promising anticancer activity against SR- leukemia and UO-31 renal cancer cell lines. The hybrids 8a–c and compound 6b exhibited noticeable antifungal activities against C. Albicans; 8a experienced the most potent antifungal activity compared to Itraconazole with MICs of 21.88 µg/mL and 11.22 µg/mL; respectively. Most of derivatives displayed better antibacterial activity than the parent ciprofloxacin against all the tested strains. Compound 6b was the most potent against the highly resistant Gram-negative K. pneumoniae with MIC 16.96 of µg/mL relative to the parent ciprofloxacin (MIC = 29.51 µg/mL). Docking studies of the tested hydrides in the active site of Topo IV enzyme of K. pneumoniae (5EIX) and S. aureus gyrase (2XCT) indicate that they had stronger binding affinity in both enzymes than ciprofloxacin but have different binding interactions. The hybrid 6b could be considered a promising lead compound for finding new dual antibacterial/anticancer agents. Moreover, Compound 8a could be a lead for discovering new dual antibacterial/antifungal agents.
Graphical abstract

Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Fluoroquinolones represent the backbone stone among the highly potent antimicrobial agents many decades ago [1]. Fluoroquinolones can eventually result in bacterial cell death via the circumvention of DNA replication and transcription processes in bacterial cell lines. They hamper the topoisomerase II activity (DNA gyrase) or/and prevent the detachment of topoisomerase II from DNA [2,3,4]. Bacterial topoisomerase II is highly different than mammalian topoisomerase; therefore, fluoroquinolones show about 1000-fold specific selectivity against bacterial topoisomerase II rather than the same enzyme in human beings [5]. Fluoroquinolones have also been found to abolish the in vitro topoisomerase IV ligase activities that have a higher similarity in structure to the topoisomerase II. This enzyme possesses a critical function represented in partitioning the DNAchromosomes in bacterial strains through cell division. It probably can stand among the fundamental targets for affording the fluoroquinolones’ antibacterial activity in specifically Gram-positive bacteria [6]. This technique can be considered related to apoptosis rather than necrosis [6]. Nowadays, fluoroquinolones have enormous applications’ clinically, and a vast domain regarding their antimicrobial activity comprises Gram-negative, Gram-positive, aerobic and anaerobic pathogens [7, 8]. In relation, fluoroquinolones exhibited highly potent anti-tuberculosis activity with a promising ability to lessen the treatment duration and to decrease the availability of bacterial resistance’s existence [9, 10]. Moreover, fluoroquinolones are depicted by having a remarkable phagocytic cells’ entrance; and a higher comprehensive safety margin [11]. Aside from fluoroquinolones’ antibacterial activities [12,13,14], they exert other versatile non-classical biophysiological activities as instances; antifungal [15, 16], antitumor [17, 18], anti- HCV-NS3 helicase, HIV-1 integrase [19], anti-Alzheimer [20], and antimalarial activities [21]. As a result of the rapid emergence of highly resistant bacterial strains, it becomes a great necessity to ameliorate and develop new fluoroquinolones-based scaffolds that can withstand these virulent pathogens [22]. Brightly, a combination of one of the two well-known approved quinolones, trovafloxacin and ciprofloxacin with antifungal drugs such as fluconazole I or amphotericin B II afforded valuable antifungal activity against Candida albicans than using only quinolones themselves [23]. Currently approved antifungal agents are classified regarding their molecular targets. The main biochemical targets for antifungal drugs are enzymes and another additional bimolecular substances encompassed in biosynthesis of both cell wall (sorbitol), and plasma membrane (ergosterol); in addition to fungal DNA synthesis (metal chelation with DNA), and mitosis (microtubules) (Fig. 1) [24,25,26]. In relation, Metals play a fundamental role in various biological and infection processes in fungal pathogens as they act as adjuvants in a populace of enzymes—including plurality of direct and indirect roles in redox, catalytic, and malevolence, such as metal-dependent superoxide dismutases (SODs), and metalloproteases or melanin-producing laccases. Therefore, metals can affect the organizational, engulfing, and toxin removal systems in fungal strains [27]. As an example, 5‐chloro‐8‐hydroxyquinoline and 5‐nitro‐8‐hydroxyquinoline can serve as ligands forming complexes with metals (Cu, Zn, Al, and Ru) for potentiation of their antifungal activity via interfering cell membrane and DNA synthesis [28]. In addition, compounds III and IV (Fig. 2), thiol substituted quinolinium derivatives at 3‐ and 4‐positions were highly potent antifungal agents with 1 μg/mL MIC against C. albicans ATCC 60193 [29].

Biomolecular Targets for antifungals

Chemical structure of fluconazole I, amphotericin BII, compounds III, IV, and quinoline-1,3,4-oxadiazole hybrid V
On the other hand, quinoline derivatives stand among versatile heterocycles that can be considered as an efficient category for subsequent promising antifungal [30], antibacterial [31, 32], antimalarial [33, 34], anti-tuberculosis [35], anticancer agents [36,37,38]. Molecular hybridization represents a new promising trend utilizing two or more biologically active compounds to afford new scaffolds targeting multiple targets or specific drug targets [39, 40]. In literature data, it is reported that quinoline-1,3,4-oxadiazole hybrid V (Fig. 2) showed higher antibacterial activity against Staphylococcus aureus as a Gram-positive strain tested with a MIC value equal to 10 ng/mL, which is threefold lower than that of ciprofloxacin (30 ng/mL), and equal to 25 ng/mL against Escherichia coli Gram-negative bacteria in emulation to ciprofloxacin with MIC value equal to 60 ng/mL [41]. Upon depending on the hybridization strategy in design of this research work, the aim is to synthesize a new ciprofloxacin-linked quinoline hybrids via a quinoline-4-carbonyl linker (8a–c) or quinolin-4-yl-1,3,4-oxadiazole linker (6a–c); to study the impact of binding these groups to the C-7-piperazine moiety on antimicrobial and anticancer activities (Fig. 2).
Result and discussion
Chemistry
The starting appropriate acids 1a–d and the intermediates esters 2a–c and hydrazides 3a–c were elaborated in the same manner with reported literature procedures [42]. Hydrazides 3a–c converted to 1,3,4-oxadiazole-2-thione derivatives 4a–c were prepared as reported [43] by refluxing with CS2 in ethanolic solution aside with KOH as a strong base. The structures of oxadiazoles 4b and 4c were elucidated based on their 1H NMR, 13C NMR, and DEPT 13C NMR spectroscopy. The acylated ciprofloxacin 5 was prepared following the reported procedure via the treatment of ciprofloxacin with bromoacetyl bromide in an ice bath along with potassium carbonate as a base [44]. Alkylation of oxadiazole derivatives 4a–c with acylated ciprofloxacin 5 was accomplished in ACN with TEA to give the new hybrids 6a–c in an acceptable output [44] (Scheme 1).

Synthesis of quinolin-4-yl-1,3,4-oxadiazole linked-ciprofloxacin derivatives 6a–c. a 1Equ. H2SO4, Ethanol abs, Reflux, 10 h, then addition of 10% NaHCO3 solution, 88.60–89.10% yield; b NH2NH2.H2O, Ethanol abs, Reflux 5–8 h, 75.62–88.42% yield; c 1.5 equ KOH pellets, CS2, Reflux 12 h, then addition of 1 M HCl, 85.04–89.91% yield; d ACN, TEA, stirring 24 h, 60.29–72.71% yield
The chemical identity of compounds 6a–c was confirmed upon relying on their 1H NMRspectroscopy, mass spectrophotometry, and elemental analysis. 1H NMR spectra revealed the existence the protons of the parent ciprofloxacin scaffold in their expected chemical shifts. Additionally, the singlet signal of the linker CH2 appeared at δ 4.27–4.78 ppm. Trials to obtain reasonable 13C NMRspectrums of compounds 6a–c were failed due to the difficulty of solubility of these compounds even in DMSO-d6. On the other side, reaction of the appropriate 2-substituted quinoline-4-carboxylic acid derivatives 1a, 1b, or 1d with excess SOCl2 affording the corresponding acid chlorides 7a–c that were used in the following step without any purification. The reaction of acid chlorides 7a–c with ciprofloxacin in ACN using TEA afforded the targeted acylated ciprofloxacin derivatives 8a–c (Scheme 2). The structural identity of hybrids 8a–c was affirmed on the basis of their 1H NMRspectroscopy and elemental analysis. Trials to obtain reasonable 13C NMRspectrums of compounds 8a and 8b were failed due to the difficulty of solubility of these compounds except compound 8c that showed a good 13C NMRwhich revealed the presence of three carbonyl carbons appeared at δ: 176.83, 168.03, and 166.35 ppm related to C-4 of the quinolone nucleus, carbonyl group linker, and carboxylic group, respectively.

Synthesis of quinoline-4-carbonyl linked-ciprofloxacin derivatives 8a–c. a SOCl2, stirring at 60ºC, 4 h; b ACN, TEA, Reflux 24 h, 70.85–82.39% yield
Screening of the antimicrobial activity
The standard agar cup diffusion protocol [45] were used to determine the in vitro antibacterial activity against E. coli (ATCC 8739), Klebsiella pneumoniae (ATCC 10031), Pseudomonas aeruginosa (ATCC 10145), and S. aureus (ATCC 6538) of compounds, 6a–c and 8a–c compared with the reference ciprofloxacin as a positive control and DMSO as a negative control. Also, the antifungal activity of 6a–c and 8a–c was examined against C. albicans by standard agar cup diffusion protocol and using itraconazole as a positive antifungal reference and DMSO as a negative reference [46].
Upon relying on the antibacterial and antifungal in vitro results as shown in Table 1, it is clear that the ciprofloxacin-quinoline-4-yl-1,3,4-oxadiazole hybrid 6a and ciprofloxacin-quinoline-4-carbonyl hybrid 8b displayed remarkable antibacterial activity against Gram-positive S. aureus compared with ciprofloxacin with MICs values equal to 19.49, 18.24, 109.6 µg/mL, respectively. It was found that 6b, 8b, and 8c displayed comparable activity against S. aureus collated with ciprofloxacin with MIC of 50.92, 57.55, 56.73, 109.6 µg/mL, respectively. Meanwhile, some of the tested hybrids 6c and 8c showed an obvious antibacterial activity against Gram-negative E. coli collated with the ciprofloxacin with MIC values equal to 34.33, 46.01, and 85.11 µg/mL, respectively. Besides, compounds 6a, 6b, 8a, and 8b afforded weak activity against E. coli. Moreover, compound 6b revealed a high virility against K.pneumoniae compared with the ciprofloxacin with MIC values equal to 16.97 and 29.51 µg/mL, respectively, and other hybrids showed to some extent acceptable to weak activity. Thus, ciprofloxacin hybrids 6a, 6b, and 8a exhibited potent activity against ps. Aeruginosa higher than ciprofloxacin with MIC values equal to 30.29, 40.84, 46.33, and 72.44 µg/mL; respectively. On the basis of our investigated outcomes, it becomes clear that most of the tested ciprofloxacin-quinoline compounds showed more robust and broad-magnitude bactericidal activity than ciprofloxacin reference versus most of the tested Gram-positive and Gram-negative strains. It becomes clear that the existence of substitution on N4 of piperazinyl moiety enhances the bactericidal activity through improving their lipophilicity (expressed as logP values calculated via Swiss ADME Prediction web site; mentioned in Table 1) as increasing of logP values due to decrease zwitterion formation which led to increase penetration to bacterial cell [46].
In contrast to ciprofloxacin which exhibited negligible fungicidal activity against C. albicans with MIC equals 169.82 µg/mL, compounds 6b and 8a–8c showed good antifungal activity with MIC values equal to 28.87, 21.88, 32.43, and 27.94 µg/mL, respectively, but less than itraconazole. These promising antifungal activities of compounds 6b and 8a–8c may be due to enhancement of lipophilicity than the parent ciprofloxacin and subsequent increasing the penetration to fungal cell and/or synergistic antifungal activity as a result of gathering ciprofloxacin and quinoline moiety in one compact unit.
Screening of the anticancer activity
The NCI results of new heterocyclic compounds; 6a–c and 8a–c (Shown in Table 2) indicated that the hybrids 6a–c had better anti-proliferative activities than hybrids 8a–c.
For example, compounds 6a and 6b showed promising anti-proliferative activity against SR- leukemia cell lines with growth inhibition percentages; 33.25 and 52.62%, respectively. These results illustrated that electron-donating substitution such as p-methyl group on phenyl ring as in compound 6b was more favorable than being unsubstituted phenyl ring on this position like in compound 6a. On the other hand, replacing p-methyl group with p-methoxy group led to a lack of anti-proliferative activity as in compound 6c. Therefore, this results in a good impact on the anticancer activity of these compounds. Concerning CAKI-1 Renal Cancer, compound 6b had a positive effect on the anticancer activity compared to hybrids 6a and 6c with growth inhibition percentages; 39.81, 26.92 27.31%, respectively. Moreover, the hybrids 6a–c exhibited remarkable anti-proliferative activities against UO-31 renal cancer with growth inhibition percentages; 64.19, 55.49, 40.15%, respectively.
Furthermore, in relation to LOX IMVI Melanoma cancer cells, compounds 6a and 6b showed good anticancer activity with a percentage of growth inhibition of 39.14 and 36.64%, respectively. In contrast, compound 6c showed insignificant anticancer activity with a growth inhibition percentage, 8.95%. Therefore, by addition of a stronger electron-donating group such as methoxy group on phenyl ring in position 2 of quinoline ring in compound 6c had a negative effect on the anticancer activity of this compound compared to compounds 6a and 6b; so being unsubstituted phenyl ring on this position like in compound 6a was better for higher anti-proliferative against LOX IMVI Melanoma, and the same reason reflected on the growth inhibition percentages of compounds 6a–c on A498 renal cancer cell lines.
Notably, compounds 8a–c showed very week anti-proliferative activity versus most of the used cancer cell lines. On balance, the presence of 1,3,4 oxadiazole ring linkage in hybrids 6a–c may be had a striking positive effect on the anticancer activity of these hybrids rather than carbonyl linkage in hybrids 8a–c; this can be accredited to the well-known intrinsic anticancer activity of the 1,3,4 oxadiazole itself with multiple mechanisms published before in many literatures research works [47,48,49].
Docking studies of antibacterial activity of hybrids 6a–c and 8a–c
Firstly, we investigate both the mode of binding, and binding energies of the hybrids 6a–c, 8a–c, and the parent ciprofloxacin versus the active binding site of K.pneumoniae Topo IV enzyme (PDB: 5EIX) to stand on their efficient binding modes and evaluate their similarity to the standard ligand binding modes [50]. The deprotonated forms of hybrids 6a–c & 8a–c and the zwitterionic forms of ciprofloxacin & levofloxacin were used in this molecular modeling study using Molecular docking software (MOE® version 2014.01). The K. pneumoniae Topo IV X-ray crystallographic structure (PDB ID: 5EIX) was taken from the protein data bank [51]. The binding free energy (ΔG) values of all the new compounds range from − 8.234 to − 7.17 kcal/mol, comparable to the parent reference drug ciprofloxacin (ΔG = − 6.05 kcal/mol) and the reference levofloxacin downloaded as ligand with enzyme (ΔG = − 5.55 kcal/mol) which means powerful binding, as outlined in Table 3.
From the docking results in K. pneumoniae Topo IV active site as shown in Figs. 3, 4, 5, 6, 7 and 8 and Table 3, we can claim that all the new tested compounds make different interactions to some extent to that of ciprofloxacin or levofloxacin except compound 8c which forms one metal chelation with magnesium ion Mg 4:1501 through the carboxylic group at C-3 and one pi-H bond with His A1077 residue as ciprofloxacin does. Moreover, levofloxacin which is the native ligand with the enzyme, it forms one metal chelation of the carboxylic group at C-3 with magnesium ion Mg 4:1501 as ciprofloxacin does and two hydrogen bonds with DA B5 and Gly A420 residues.

2D Diagram of ciprofloxacin as a reference complexed with the active site of Topo IV enzyme (PDB: 5EIX)

3D Diagram of ciprofloxacin as a reference complexed with the active site of Topo IV enzyme (PDB: 5EIX)

2D Diagram of levofloxacin as a reference complexed with the active site of Topo IV enzyme (PDB: 5EIX)

2D Diagram of hybrid 6a complexed with the active site of Topo IV enzyme (PDB: 5EIX)

2D Diagram of hybrid 6b complexed with the active site of Topo IV enzyme (PDB: 5EIX)

3D Diagram of hybrid 6b complexed with the active site of Topo IV enzyme (PDB: 5EIX)
Regarding compound 6b, which had higher antibacterial activity against K. pneumonia, it interacts with 5EIX binding site in different binding mode with various amino acid residues than that of ciprofloxacin Hybrid 6b constructs one pi-H bond with amino acid residue His A1077, and three hydrogen bonds with DT B7, DA B7, and Arg A1029 amino acid residues. Also, the unsubstituted quinoline hybrid 6a forms two pi-cationic interactions with His A1077, and Pro A1076 amino acid residues and one hydrogen bond with amino acid residue DA B7. On the other hand, Compounds 6c, 8a have interactions with different residues than that interacted with ciprofloxacin or compounds 6a or 6b.
Depending on the data obtained from this docking study hybrids 6a–c, and 8a–c in comparison with reference ciprofloxacin (Table 2 supporting information), it becomes clear that formation of metal chelation with Mg 4:1501 as in compound 8c or formation of at least one hydrogen bond with one of the following amino acids; DT B7, DA B7, and Arg A1029 amino acid residues is essential for those hybrids to show good antibacterial activity against K.pneumoniae as in 6a, and 6b.
Secondly, the tested compounds 6a, 6b, 8b, and 8c were docked into the binding pocket of with the active binding site of S. aureus gyrase enzyme (PDB ID: 2XCT) to highlight on their effective binding styles and evaluate their resemblance to the positive control ciprofloxacin;so it is enabled the investigation of both binding style and energies of the final hybrids 6a, and 8b that afford higher bactericidal effects versus S. Aureus comparing with ciprofloxacin with MIC values 19.49, 18.24, and 109.6 µg/mL, respectively. The S. aureus gyrase enzyme X-ray crystallographic structure (PDB ID: 2XCT) was obtained from the protein data bank [51].
Docking outcomes showed robust binding affinity to the S. aureus gyrase which appears on the values of their binding free energy (ΔG); extent from − 7.83 to − 6.88 kcal/mol, collated to the ciprofloxacin (ΔG = − 6.03 kcal/mol), as shown in Table 4.
On the basis of docking outcomes mentioned in Table 4, Figs. 9, 10, 11 and 12, it can be noted that the new tested compounds afford different binding interactions’ styles than that of ciprofloxacin which forms two metal chelation bonds with Mn atom, a hydrogen bond with Ser A1084, two pi–pi interactions with DG9 residue of DNA nucleotides, and two H-pi bonds with DA H13 and DC H12. Concerning hybrids 6a and 8b, which afford MIC equal to 19.49 μg/mL and 18.24 μg/mL; respectively, they form interactions with the 2XCT binding site in a different mode with various amino acid residues than that of ciprofloxacin. Docking studies are in a good endorsment with the obtained in vitro antibacterial results. In relation, Compound 6a forms five hydrogen bonds with Lys A1043, Arg A1092, His A1046, DG B9, and DC C14; in addition to one pi-H bond with DNA nucleotides residues DA C13 (Fig. 11). Besides, compound 8b has an ionic bond with Arg A1092, one H–pi bond with DNA nucleotides residues DG B9, and two hydrogen bonds with Lys A1043, and Ser A1085 amino acid residues (Fig. 12). On contrast, the least active compounds 6c and 8a exert weaker interactions with the 2XCT binding site than the parent, and these interactions do not comprise any ionic bonds. Therefore, the more efficient anti-bactericidal effect of the hybrids 6a, and 8b may be due to improvement of the lipophilicity, and subsequently increasing the passage into microbial cells and respectable fitting binding modes on the predictable target site. Additionally, compounds 6a and 8b may had another antibacterial technique in addition to their activity against DNA gyrase of gram positive S. aureus bacteria; that is reflected in more stronger antibacterial activity of these compounds comparing with parent positive control ciprofloxacin.

2D Diagram of ciprofloxacin as a reference complexed with the active site of Topo IV enzyme (PDB: 2XCT)

3D Diagram of ciprofloxacin as a reference complexed with the active site of DNA gyrase enzyme (PDB: 2XCT)

2D Diagram of hybrid 6a complexed with the active site of DNA gyrase enzyme (PDB: 2XCT)

2D Diagram of hybrid 8b complexed with the active site of DNA gyrase enzyme (PDB: 2XCT)
Conclusion
Ciprofloxacin-quinoline hybrids 6a–c and 8a–c were chemically elaborated, characterized and investigated for their antimicrobial and anticancer activities. Ciprofloxacin-linked quinoline-4-yl-1,3,4 oxadiazoles 6a and 6b showed promising anticancer activity against SR-leukemia and UO-31 renal cancer cell lines and greater bactericidal effect versus S. aureus, highly resistant K. pneumoniae and Ps. aeruginosa than the reference ciprofloxacin. On the other side, the gathering of ciprofloxacin with a quinolone-4-yl nucleus in one compact unit through carbonyl group led to synergistic antifungal activities as shown by compounds 8a–c against C. Albicans. Docking studies on the S. aureus gyrase and K. pneumonia binding sites indicated that the tested compounds interacted in a different manner than that of ciprofloxacin. Therefore, the more efficient antibacterial activity of the tested compounds may be because of improvement of the lipophilicity, and subsequently increasing the passage into microbial cells and respectable fitting binding modes on the predictable target site. Additionally, these compounds may have another antibacterial mechanization in addition to their activity against DNA gyrase or Topo IV enzymes; that is reflected in more efficient antibacterial activity of these compounds comparing with parent positive control ciprofloxacin. Finally, Ciprofloxacin-quinoline-4-yl-1.3.4 oxadiazole hybrids 6a and 6b may be considered promising new dual antibacterial/anticancer lead compound that require further modification and investigation for their mechanism of action. Additionally, compound 8a could be a suitable lead compound for innovating new dual antibacterial/antifungal drugs.
Experimental
Chemistry
Materials and reagents
-
The chemicals used in this study were purchased from Sigma Aldrich.
-
The reaction progress was monitored using TLC (Kieselgel 60 F254 pre-coated plates, E. Merck, Darmstadt, Germany) and the spots were visualized by UV lamp at λ 254 and 365 nm.
-
Melting points detection of the new compounds was done using Stuart electro-thermal melting point apparatus and they were uncorrected.
-
Bruker Advance III 400 MHz records 1H NMR spectra of tested compounds. Chemical shift (δ) in ppm relative to TMS (δ = 0 ppm) as internal standard using DMSO-d6 as solvent, and 13C NMR spectra were recorded on Bruker Advance III 100 MHz; at faculty of Pharmacy, Beni-Sweif University, Egypt.
-
Shimadzu GC/MS-QP5050A records the elemental microanalysis values of the new hybrids, at regional center for mycology and biotechnology, Al-Azhar University, Cairo, Egypt.
-
LC/MS/MS (Agilent 1260 Infinity II (USA) with 6420 Triple Quad LC/MS detector was utilized to make Mass spectrophotometry for new compounds, at Nawah, Mokatam, Egypt.
Chemistry
2-Phenylquinoline-4-carboxylic acid (1a)
Yellowish white powder; 0.48 g; 68.38%yield; mp: 210–211 °C (reported mp: 209–210 °C) [52].
2-(p-Tolyl)quinolone-4-carboxylic acid (1b)
Orange powder; 0.61 g; 81.22%yield; mp: 243–244 °C (reported mp: 242 °C) [52].
2-(4-Methoxyphenyl)quinolone-4-carboxylic acid (1c)
Buff powder; 0.68 g; 85.11%yield; mp: 196 °C (reported mp: 195–196 °C) [52].
2-(4-Chlorophenyl)quinolone-4-carboxylic acid (1d)
Yellowish white powder; 0.49 g; 60.33%yield; mp: 144 °C (reported mp: 142–144 °C) [52].
Ethyl-2-(phenyl) quinolone-4-carboxylate (2a)
Yellowish white powder; 0.38 g; 88.60%yield; mp: 57–58 °C (reported mp: 56–57 °C) [53].
Ethyl-2-(p-tolyl)quinolone-4-carboxylate (2b)
Orange powder; 0.49 g; 89.10%yield; mp: 54–55 °C (reported mp: 54 °C) [53].
Ethyl-2-((4-methoxy)phenyl)quinolone-4-carboxylate (2c)
Buff powder; 0.48 g; 88.90%yield; mp: 79–80 °C (reported mp: 78–79 °C) [53].
2-(Phenyl)quinolone-4-carbohydrazide (3a)
Yellowish white powder; 0.28 g; 88.42%yield; mp: 222–223 °C (reported mp: 222 °C) [44].
2-(p-Tolyl)quinolone-4-carbohydrazide (3b)
Orange powder; 0.38 g; 80.57%yield; mp: 251 °C (reported mp: 249–251 °C) [54].
2-(p-Methoxy-phenyl)quinolone-4-carbohydrazide (3c)
Buff powder; 0.31 g; 75.62%yield; mp: 236–237 °C (reported mp: 233–236 °C) [44].
5-(2-Phenylquinolin-4-yl)-1,3,4-oxadiazole-2-thione (4a)
Yellowish white powder; 0.32 g; 85.04%yield; mp: 246–247 °C (reported m.p:244–246 °C) [55].
5-(2-(p-Tolyl)quinolin-4-yl)-1,3,4-oxadiazole-2-thione (4b)
Orange powder; 0.35 g; 89.91%yield; mp: 239-240 °C; 1H NMR(500 MHz, DMSO-d6) δ: (ppm) 2.42 (3H, s, CH3), 7.41 (2H, d, J = 8 Hz, ArH), 7.77–7.82 (1H, m, ArH), 7.91–8.01 (1H, m, ArH), 8.18–8.23 (3H, m, ArH), 8.42 (1H, s, H-3 of quinolone ring), 8.81 (1H, d, J = 8 Hz, ArH); 13C NMR(125 MHz, DMSO-d6) δ: (ppm) 21.37, 118.28, 122.03, 125.55, 127.64, 127.95, 128.66, 130.15, 130.50, 131.19, 135.26, 140.50, 148.92, 156.09, 159.17, 178.02.; DEPT13C NMR (125 MHz, DMSO-d6) δ: (ppm) 21.65, 118.27, 125.69, 128.02, 129.02, 130.15, 130.42, 131.19.
5-(2-(p-Methoxy phenyl)quinolin-4-yl)-1,3,4-oxadiazole-2-thione (4c)
Buff powder; 0.31 g; 88.48%yield; mp: 245–247 ºC; 1H NMR(500 MHz, DMSO-d6) δ: (ppm) 3.83 (3H, s, OCH3), 7.16 (2H, d, J = 8 Hz, ArH), 7.73–7.77 (1H, m, ArH), 7.91–7.96 (1H, m, ArH), 8.18–8.23 (2H, m, ArH), 8.28–8.32 (1H, m, ArH), 8.45 (1H, s, H-3 of quinolone ring), 8.79 (1H, d, J = 8 Hz, ArH); 13C NMR(125 MHz, DMSO-d6) δ: (ppm) 55.88, 114.97, 117.71, 118.41, 121.75, 125.88, 128.29, 128.65, 129.50, 129.83, 129.98, 131.44, 147.84, 155.59, 159.99, 165.89.; DEPT13C NMR (125 MHz, DMSO-d6) δ: (ppm) 56.23, 114.73, 118.52, 125.70, 128.22, 129.03, 130.09, 131.80.
1-Cyclopropyl-6-fluoro-4-oxo-7-(4-(2-((5-(2-phenylquinolin-4-yl)-1,3,4-oxadiazol-2-yl)thio)acetyl)piperazin-1-yl)-1,4-dihydroquinoline-3-carboxylic acid (6a)
Yellowish white powder; 0.37 g; 67.30%yield; mp: 285 ºC;1H NMR (400 MHz, DMSO-d6) δ: (ppm) 1.12–1.24 (2H, m, cyclopropyl-H), 1.25–1.39 (2H, m, cyclopropyl-H), 3.41–3.52 (4H, m, piperazinyl-H), 3.72–3.84 (5H, m, 4 piperazinyl-H and cyclopropyl-H), 4.23 (2H, s, CH2–C=O), 7.53–7.56 (1H, m, ArH), 7.59 (2H, d, J = 7.6 Hz, ArH), 7.8–7.85 (1H, m, ArH), 7.93 (3H, d, J = 12 Hz, H-5 and 2 ArH), 8.22 (1H, d, J = 7.3 Hz, ArH), 8.36 (1H, d, J = 7.6 Hz, ArH), 8.59–8.71 (3H, m, H-2 and 2ArH), 9.03–9.44 (1H, m, ArH), 15.18 (1H, s, COOH); MS (ESI+) m/z 677.3 [M + H]+; Anal. Calcd. For C36H29FN6O5S (676.19); C, 63.90; H, 4.32; N, 12.42; S, 4.74; Found C, 64.13; H, 4.50; N, 12.69; S, 4.82.
1-Cyclopropyl-6-fluoro-4-oxo-7-(4-(2-((5-(2-(p-tolyl)quinolin-4-yl)-1,3,4-oxadiazol-2-yl)thio)acetyl)piperazin-1-yl)-1,4-dihydroquinoline-3-carboxylic acid (6b)
Orange powder; 0.38 g; 60.29%yield; mp: 293–294 ºC;1H NMR (400 MHz, DMSO-d6) δ: (ppm) 1.03–1.17 (2H, m, cyclopropyl-H), 1.24–1.32 (2H, m, cyclopropyl-H), 2.38 (3H, s, CH3), 3.17–3.23 (6H, m, piperazinyl-H), 3.48–3.85 (2H, m, piperazinyl-H), 4.09–4.11 (1H, m, cyclopropyl-H), 4.78 (2H, s, CH2–C=O), 7.36 (2H, d, J = 7.7 Hz, ArH), 7.62–7.82 (2H, m, ArH), 7.91–7.96 (2H, m, H-8 and ArH), 8.21 (1H, d, J = 8.9 Hz, ArH), 8.26–8.29 (2H, m, H-5, ArH), 8.62 (1H, s, H-2), 8.69 (1H, s, ArH), 9.03 (1H, d, J = 8.9 Hz, ArH), 15.23 (1H, s, COOH)); MS (ESI+) m/z 713.3 [M + Na]+; Anal. Calcd. For C37H31FN6O5S (690.21); C, 64.34; H, 4.52; N, 12.17; S, 4.64; Found C, 64.59; H, 4.61; N, 12.43; S, 4.84.
1-Cyclopropyl-6-fluoro-7-(4-(2-((5-(2-(4-methoxyphenyl)quinolin-4-yl)-1,3,4-oxadiazol-2-yl)thio)acetyl)piperazin-1-yl)-4-oxo-1,4-dihydroquinoline-3-carboxylic acid (6c)
Buff powder; 0.45 g; 72.71%yield; mp: 296ºC;1H NMR (400 MHz, DMSO-d6) δ: (ppm) 1.15–1.21 (2H, m, cyclopropyl-H), 1.23–1.31 (2H, m, cyclopropyl-H), 3.33–3.50 (4H, m, piperazinyl-H), 3.73–3.79 (4H, m, piperazinyl-H), 3.84 (3H, s, OCH3), 3.87–3.89 (1H, m, cyclopropyl-H), 4.78 (2H, s, CH2–C=O), 7.09 (2H, d, J = 8.8 Hz, ArH), 7.49–7.55 (1H, m, ArH), 7.75–7.77 (1H, m, ArH), 7.89–7.95 (2H, m, H-8 and ArH), 8.18 (1H, d, J = 12.0 Hz, H-5), 8.33 (2H, d, J = 8.0 Hz, ArH), 8.58 (1H, s, ArH), 8.68 (1H, s, H-2), 9.01 (1H, d, J = 8.8 Hz, ArH), 15.27 (1H, s, COOH); MS (ESI+) m/z 729.3 [M + Na]+; Anal. Calcd. For C37H31FN6O6S (706.20); C, 62.88; H, 4.42; N, 11.89; S, 4.54; Found C, 63.12; H, 4.57; N, 12.05; S, 4.80.
1-Cyclopropyl-6-fluoro-4-oxo-7-(4-(2-phenylquinoline-4-carbonyl)piperazin-1-yl)-1,4-dihydroquinoline-3-carboxylic acid (8a)
Yellowish white powder; 0.36 g; 70.85%yield; mp: < 300 ºC; 1H NMR (400 MHz, DMSO-d6) δ: (ppm) 1.09–1.20 (2H, m, cyclopropyl-H), 1.25–1.45 (2H, m, cyclopropyl-H), 3.18–3.26 (4H, m, piperazinyl-H), 3.43–3.47 (4H, m, piperazinyl-H), 4.07–4.15 (1H, m, cyclopropyl-H), 7.52 (2H, d, J = 6.6 Hz, ArH), 7.55 (2H, d, J = 6.6 Hz, ArH), 7.62–7.74 (2H, m, H-8 and ArH), 7.79–7.86 (2H, m, H-5 and ArH), 7.95 (1H, d, J = 7.0 Hz, ArH), 8.10–8.19 (2H, m, H-2 and ArH), 8.34–8.39 (1H, m, ArH), 8.65–8.72 (1H, m, ArH), 15.18 (1H, s, COOH); MS (ESI+) m/z 585.5 [M + Na]+; Anal. Calcd. For C33H27FN4O4 (562.20); C, 70.45; H, 4.84; N, 9.96; Found C, 70.63; H, 5.01; N, 10.13.
1-Cyclopropyl-6-fluoro-4-oxo-7-(4-(2-(p-tolyl)quinoline-4-carbonyl)piperazin-1-yl)-1,4-dihydroquinoline-3-carboxylic acid (8b)
Orange powder; 0.51 g; 82.39%yield; mp: > 300 ºC;1H NMR(400 MHz, DMSO-d6) δ: (ppm) 1.16–1.32 (4H, m, cyclopropyl-H), 2.40 (3H, s, CH3), 3.00–3.05 (6H, m, piperazinyl-H), 3.13–3.20 (2H, m, piperazinyl-H), 3.37–3.47 (1H, m, cyclopropyl-H), 7.36 (2H, d, J = 7.9 Hz, H-8 and ArH), 7.57 (1H, d, J = 9.3 Hz, ArH), 7.73 (1H, d, J = 6.6 Hz, ArH), 7.94 (2H, d, J = 13.1 Hz, H-5 and ArH), 8.05 (1H, d, J = 8.4 Hz, ArH), 8.15 (1H, d, J = 7.9 Hz, ArH), 8.20 (1H, s, H-2), 8.69 (3H, d, J = 8.4 Hz, ArH); MS (ESI+) m/z 593.3 [M + Na]+; Anal. Calcd. For C34H29FN4O4 (570.22); C, 70.82; H, 5.07; N, 9.72; Found C, 70.61; H, 5.23; N, 10.01.
7-(4-(2-(4-Chlorophenyl)quinoline-4-carbonyl)piperazin-1-yl)-1-cyclopropyl-6-fluoro-4-oxo-1,4-dihydroquinoline-3-carboxylic acid (8c)
Yellowish white powder; 0.38 g; 77.22%yield; mp: > 300 ºC; 1H NMR(400 MHz, DMSO-d6) δ: (ppm) 1.18–1.12 (2H, m, cyclopropyl-H), 1.29–1.36 (2H, m, cyclopropyl-H), 3.26–3.58 (8H, m, piperazinyl-H), 3.83–3.90 (1H, m, cyclopropyl-H), 7.62–7.74 (4H, m, H-8 and 3 ArH), 7.85–7.91 (2H, m, H-5 and ArH), 7.93–8.03 (1H, m, ArH), 8.16 (1H, d, J = 8.1 Hz, ArH), 8.34 (2H, d, J = 8.1 Hz, ArH), 8.47 (1H, s, H-2), 8.65 (1H, d, J = 8.1 Hz, ArH), 15.17 (1H, s, COOH)); 13C NMR(100 MHz, DMSO-d6) δ: (ppm) 8.07, 9.02, 36.37, 37.35, 46.06, 49.37, 107.26, 108.58, 112.83, 119.38, 124.00, 125.91, 128.44, 128.85, 129.48, 129.90, 130.24, 130.82, 131.87, 134.62, 134.72, 135.39, 137.16, 138.46, 148.79, 150.05, 155.04, 166.35, 168.03, 176.83; MS (ESI−) m/z 595.2 [M + H]−; Anal. Calcd. For C33H26ClFN4O4 (596.16); C, 66.39; H, 4.39; N, 9.38; Found C, 66.52; H,4.57; N,9.61.
Screening of the antimicrobial acti vity
The antibacterial activity of compounds 6a–c, and 8a–c were tested versus Staphylococcus aureus (ATCC 6538), Klebsiella pneumoniae (ATCC 10031), Pseudomonas aeruginosa (ATCC 10145), and Escherichia coli (ATCC 8739) as an in vitro experiment compared to ciprofloxacin as a reference using standard agar cup protocol [45]. The antifungal activity of hybrids 6a–c and 8a–c were evaluated in vitro against Candida albicans (ATCC 10231) using Muller Hinton media for fungal strain growth.
Screening of the anticancer activity
All the synthesized hybrids 6a–c (Ciprofloxacin- Quinoline hybrids linked via 1,3,4 oxadiazole linkage) and 8a–c (Ciprofloxacin- Quinoline hybrids linked via carbonyl linkage) were selected by National Cancer Institution (NCI), Bethesda, USA, for in vitro one-dose antitumor assay. The methodology protocol for the NCI anticancer screening is reported in detail elsewhere (http://www.dtp.nci.nih.gov). The anticancer assay was performed in full NCI 60 cell lines extracted from nine tumor cell lines, including leukemia, melanoma, lung, colon, CNS, ovarian, renal, prostate, and breast cancer cell lines.
Docking studies of antibacterial activity of hybrids 6a–c and 8a–c
A molecular modeling study was performed for 6a–c, and 8a–c compared to ciprofloxacin at the active sites of K.pneumoniae. Topo IV enzyme (PDB: 5EIX) [50], and S. aureus gyrase enzyme (PDB ID: 2XCT) [51] using Molecular docking software (MOE® version 2014.09)_ using levofloxacin as method’s validation reference; showed good fitting of these compounds with the active sites of K.pneumoniae. Topo IV enzyme (PDB: 5EIX), and S. aureus gyrase enzyme (PDB ID: 2XCT) that agreed with the antibacterial screening results obtained against K.pneumoniae and S. aureus. Each compound was confirmed depending basically on its docking score value.
References
H. Koga, A. Itoh, S. Murayama, S. Suzue, T. Irikura, Structure-activity relationships of antibacterial 6,7- and 7,8-disubstituted 1–a1kyl-1,4-dihydro-4-oxoquinoline-3-carboxyliaccids’. J. Med. Chem. 12(23), 1358–1363 (1980)
K. Mizuuchi, L.M. Fisher, M.H. O’Dea, M. Gellert, DNA gyrase action involves the introduction of transient double-strand breaks into DNA. Proc. Natl. Acad. Sci. 77, 1847–1851 (1980)
J.D. Walters, F. Zhang, R.J. Nakkula, Mechanisms of fluoroquinolone transport by human neutrophils. Antimicrob. Agents Chemother. 11(43), 2710–2715 (1999)
F. Schmitz, Relationship between ciprofloxacin, ofloxacin, levofloxacin, sparfloxacin, and moxifloxacin (BAY 12–8039) MICs and mutations in grlA, grlB, gyrA and gyrB in 116 unrelated clinical isolates of Staphylococcus aureus. J. Antimicrob. Chemother. 4(41), 481–484 (1998)
S. Nawaz, R. Bodla, R. Kant, S.P. Singh, R. Bhutani, G. Kapoor, Fluoroquinolone as antimicrobial agent: a review. Int. J. Pharm. Sci. Res. 2(3), 57–63 (2017)
N.H. Nasser, M. Abdulbary, S.H. Shaalan, E.S. Hadi, Synthesis, characterization, and antibacterial assessment of new gatifloxacin analogues. World J. Pharm. Pharm. Sci. 2(7), 175–188 (2018)
R.J. O’Brien, M. Spigelman, New drugs for tuberculosis: current status and future prospects. Clin. Chest Med. 2(26), 327–340 (2005)
Y.-L. Fan, J.-B. Wu, X.-W. Cheng, F.-Z. Zhang, L.-S. Feng, Fluoroquinolone derivatives and their anti-tubercular activities. Eur. J. Med. Chem. 146, 554–563 (2018)
M. Asif, A review on potent antitubercuolar agent isiniazid and its analoges. Int. J. Pharm. Chem. 4(2), 110–120 (2012)
P.C. Drobac, G. Anca, J.K. Joseph, J. Furin, S. Shin, Treatment of multidrug-resistant tuberculosis during pregnancy: long-term follow-up of 6 children with intrauterine exposure to second-line agents. Clin. Infect. Dis. 11(40), 1689–1692 (2005)
K. Soni, Fluoroquinolones: chemistry & action—a review. Indo Glob. J. Pharm. Sci. 1(2), 43–53 (2012)
M. LeBel, Ciprofloxacin: chemistry, mechanism of action, resistance, antimicrobial spectrum, pharmacokinetics, clinical trials, and adverse reactions. Pharmacother. J. Hum. Pharmacol. Drug Ther. 1(8), 3–30 (1988)
R. Bartzatt, S.L.G. Cirillo, J.D. Cirillo, Design of ciprofloxacin derivatives that inhibit growth of methicillin resistant Staphylococcus aureus (MRSA) and methicillin susceptible Staphylococcus aureus (MSSA). Med. Chem. 2(6), 51–56 (2010)
B. Marquez, V. Pourcelle, C.M. Vallet et al., Pharmacological characterization of 7-(4-(piperazin-1-yl)) ciprofloxacin derivatives: antibacterial activity, cellular accumulation, susceptibility to efflux transporters, and intracellular activity. Pharm. Res. 5(31), 1290–1301 (2014)
S. Srinivasan, R.M. Beema Shafreen, P. Nithyanand, P. Manisankar, S.K. Pandian, Synthesis and in vitro antimicrobial evaluation of novel fluoroquinolone derivatives. Eur. J. Med. Chem. 12(45), 6101–6105 (2010)
A.M. Sugar, X.-P. Liu, Combination antifungal therapy in treatment of murine pulmonary mucormycosis: roles of quinolones and azoles. Antimicrob. Agents Chemother. 7(44), 2004–2006 (2000)
T. Gumbo, A. Louie, M.R. Deziel, G.L. Drusano, Pharmacodynamic evidence that ciprofloxacin failure against tuberculosis is not due to poor microbial kill but to rapid emergence of resistance. Antimicrob. Agents Chemother. 8(49), 3178–3181 (2005)
R.K. Shandil, R. Jayaram, P. Kaur et al., Moxifloxacin, ofloxacin, sparfloxacin, and ciprofloxacin against mycobacterium tuberculosis: evaluation of in vitro and pharmacodynamic indices that best predict in vivo efficacy. Antimicrob. Agents. Chemother. 2(51), 576–582 (2007)
M. Abdel-Aziz, S.-E. Park, G.E.-D.A.A. Abuo-Rahma, M.A. Sayed, Y. Kwon, Novel N-4-piperazinyl-ciprofloxacin-chalcone hybrids: Synthesis, physicochemical properties, anticancer and topoisomerase I and II inhibitory activity. Eur. J. Med. Chem. 69, 427–438 (2013)
P.C. Sharma, A. Jain, S. Jain, R. Pahwa, M.S. Yar, Ciprofloxacin: review on developments in synthetic, analytical, and medicinal aspects. J. Enzyme Inhib. Med. Chem. 4(25), 577–589 (2010)
M. Daneshtalab, A. Ahmed, Nonclassical biological activities of quinolone derivatives. J. Pharm. Pharm. Sci. 1(15), 52–72 (2011)
M. Pudlo, V. Luzet, L. Ismaïli, I. Tomassoli, A. Iutzeler, B. Refouvelet, Quinolone–benzylpiperidine derivatives as novel acetylcholinesterase inhibitor and antioxidant hybrids for Alzheimer disease. Bioorg. Med. Chem. 8(22), 2496–2507 (2014)
J. Li, S. Li, C. Bai, H. Liu, P. Gramatica, Structural requirements of 3-carboxyl-4(1H)-quinolones as potential antimalarials from 2D and 3D QSAR analysis. J. Mol. Graph. Model. 44, 266–277 (2013)
J.M.A. Blair, M.A. Webber, A.J. Baylay, D.O. Ogbolu, L.J.V. Piddock, Molecular mechanisms of antibiotic resistance. Nat. Rev. Microbiol. 13, 42–51 (2015)
E. Sasaki, S. Maesaki, Y. Miyazaki, K. Yanagihara, K. Tomono, T. Tashiro, S. Kohno, Synergistic effect of ofloxacin and fluconazole against azole-resistant Candida albicans. J. Infect. Chemother. 3(6), 151–154 (2000)
F.C. Odds, A.J.P. Brown, N.A.R. Gow, Antifungal agents: mechanisms of action. Trends Microbiol. 6(11), 272–279 (2003)
R. Prasad, A.H. Shah, M.K. Rawal, Antifungals: mechanism of action and drug resistance, in Yeast Membrane Transport, Advances in Experimental medicine and Biology, New Delhi, Inida, vol. 892, ed. by J. Ramos, H. Sychrová, M. Kschischo (Springer, Cham, 2016), pp.327–349
B. Marcel, Mechanism of action of antifungal drugs, with special reference to the imidazole derivatives. Rev. Infect. Dis. 4(2), 520–534 (1980)
F. Gerwien, V. Skrahina, L. Kasper, B. Hube, S. Brunke, Metals in fungal virulence. FEMS Microbiol. Rev. 1(42), 1–21 (2018)
C. dos Santos Chagas, F.L.A. Fonseca, I.A. Bagatin, Quinoline-derivative coordination compounds as potential applications to antibacterial and antineoplasic drugs. Mater. Sci. Eng. C 98, 1043–1052 (2019)
A. Empel, E. Kisiel, D.R. Wojtyczka, M.K. Epa, D. Idzik, A. Sochanik, T.J. Wasik, A. Zieba, Synthesis and antimicrobial activity of sulfur derivatives of quinolinium salts. Molecules 23, 218 (2018)
A. Dorababu, Recent update on antibacterial and antifungal activity of quinoline scaffolds. Arch. Pharm. 3(354), e2000232 (2021)
R.S. Upadhayaya, J.K. Vandavasi, N.R. Vasireddy, V. Sharma, S.S. Dixit, J. Chattopadhyaya, Design, synthesis, biological evaluation and molecular modelling studies of novel quinoline derivatives against Mycobacterium tuberculosis. Bioorg. Med. Chem. 17, 2830–2841 (2009)
R. Wise, J.M. Andrews, L.J. Edwards, In vitro activity of bay 09867, a new quinoline derivative, compared with those of other antimicrobial agents. Antimicrob. Agents Chemother. 4(23), 559–564 (1983)
H. Shiraki, M.P. Kozar, V. Melendez et al., Antimalarial activity of novel 5-aryl-8-aminoquinoline derivatives. J. Med. Chem. 1(54), 131–142 (2011)
B.N. Acharya, D. Thavaselvam, M.P. Kaushik, Synthesis and antimalarial evaluation of novel pyridine quinoline hybrids. Med Chem Res. 17, 487–494 (2008)
M. Shiradkar, G.V.S. Kumar, V. Dasari, S. Tatikonda, K.C. Akula, R. Shah, Clubbed triazoles: a novel approach to antitubercular drugs. Eur. J. Med. Chem. 42, 807–816 (2007)
R. Lin, P.J. Connolly, S. Huang et al., 1-Acyl-1H-[1,2,4]triazole-3,5-diamine analogues as novel and potent anticancer cyclin-dependent kinase inhibitors: synthesis and evaluation of biological activities. J. Med. Chem. 48, 4208–4211 (2005)
N. Demirbas, S.A. Karaoglu, A. Demirbas, K. Sancak, Synthesis and antimicrobial activities of some new 1-(5-phenylamino-[1,3,4]thiadiazol-2-yl)methyl-5-oxo-[1,2,4]triazole and 1-(4-phenyl-5-thioxo-[1,2,4]triazol-3-yl)methyl-5-oxo- [1,2,4]triazole derivatives. Eur. J. Med. Chem. 39, 793–804 (2004)
A. Pachuta-Stec, J. Rzymowska, L. Mazur, E. Mendyk, M. Pitucha, Z. RzaogonczyNska, Synthesis, structure elucidation and antitumour activity of N-substituted amides of 3-(3-ethylthio-1,2,4-triazol-5-yl)propenoic acid. Eur. J. Med. Chem. 44, 3788–3793 (2009)
M.S. Abdelbaset, Novel pyrrol-2(3H)-ones and pyridazin-3(2H)-ones carrying quinoline scaffold as anti-proliferative tubulin polymerization inhibitors. Bioorg. Chem. 38, 151–163 (2018)
T.S. Kaoud, A.M. Mohassab, H.A. Hassan, C. Yan, S.X. Van Ravenstein, D. Abdelhamid, K.N. Dalby, M. Abdel-Aziz, NO-releasing STAT3 inhibitors suppress BRAF-mutant melanoma growth. Eur. J. Med. Chem. (2019). https://doi.org/10.1016/j.ejmech.2019.111885
H.A. Hofny, Design, synthesis, and antibacterial evaluation of new quinoline-1,3,4-oxadiazole and quinoline-1,2,4-triazole hybrids as potential inhibitors of DNA gyrase and topoisomerase IV. Bioorg. Chem. 112, 104920 (2021)
A.M. Mohassab, H.A. Hassan, D. Abdelhamid, M. Abdel-Aziz, K.N. Dalby, T.S. Kaoud, Novel quinoline incorporating 1,2,4-triazole/oxime hybrids: synthesis, molecular docking, anti-inflammatory, COX inhibition, ulceroginicity and histopathological investigations. Bioorg. Chem. 75, 242–259 (2017)
B. Bonev, J. Hooper, J. Parisot, Principles of assessing bacterial susceptibility to antibiotics using the agar diffusion method. J. Antimicrob. Chemother. 6(61), 1295–1301 (2008)
G.E.-D.A.A. Abuo-Rahma, A. Sarhan Hatem, G.F.M. Gad, Design, synthesis, antibacterial activity and physicochemical parameters of novel N-4-piperazinyl derivatives of norfloxacin. Bioorg. Med. Chem. 11(17), 3879–3886 (2009)
L.M. Bagella, V. Nieddu, Biological evaluation of 1,3,4-oxadiazoles bis-substitute derivatives as potential anticancer agents, Ph.D. thesis, International Ph.D. School in Biomolecular and Biotechnological Sciences (POR) University of Sassari (2013–2014)
S. Bondock, S. Adel, H.A. Etman, F.A. Badria, Synthesis and antitumor evaluation of some new 1,3,4-oxadiazole-based heterocycles. Eur. J. Med. Chem. 48, 192–199 (2012)
I. Hatti, R. Sreenivasulu, S.S. Jadav, M.J. Ahsan, R.R. Raju, Synthesis and biological evaluation of 1,3,4-oxadiazole-linked bisindole derivatives as anticancer agents. Monatsh Chem. 146, 1699–1705 (2015)
D.A. Veselkov, I. Laponogov, X.-S. Pan et al., Structure of a quinolone-stabilized cleavage complex of topoisomerase IV from Klebsiella pneumoniae and comparison with a related Streptococcus pneumoniae complex. Acta Crystallogr D Struct Biol. 72, 488–496 (2016)
B.D. Bax, P.F. Chan, D.S. Eggleston, A. Fosberry, D.R. Gentry, F. Gorrec et al., Type IIA topoisomerase inhibition by a new class of antibacterial agents. Nature 466, 935–940 (2010)
G.C. Muscia, J.P. Carnevale, M. Bollini, S.E. Asís, Microwave-assisted Döbner synthesis of 2-phenylquinoline-4-carboxylic acids and their antiparasitic activities. J. Heterocycl. Chem. 45, 611–614 (2008)
R. Leardini, Aromatic annelation by reaction of arylimidoyl radicals with alkynes: a new synthesis of chinolines. J. Chem. Soc. Chem. Commun. 20, 1320–1321 (1984)
M.N. Zemtsova, A.V. Zimichev, P.L. Trakhtenberg et al., Synthesis and antiviral activity of several quinoline derivatives. Pharm. Chem. J. 45, 267–269 (2011)
H.A.K. Abd, Efficient synthesis, characterization and biological evaluation of some new atophan carbohydrazide derivatives. J. Chem. Pharm. Res. 12(6), 90–99 (2014)
Acknowledgements
Authors are thankful for Dr. Rehab Mahmoud Abdel-Baky, professor of Microbiology, Faculty of Pharmacy, Minia University for her great collaboration in assessment antibacterial and antifungal screening studies.
Funding
Open access funding provided by The Science, Technology & Innovation Funding Authority (STDF) in cooperation with The Egyptian Knowledge Bank (EKB).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that there is no conflict of interest.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Ezelarab, H.A.A., Hassan, H.A., Abuo-Rahma, G.ED.A. et al. Design, synthesis, and biological investigation of quinoline/ciprofloxacin hybrids as antimicrobial and anti-proliferative agents. J IRAN CHEM SOC 20, 683–700 (2023). https://doi.org/10.1007/s13738-022-02704-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13738-022-02704-7