
Abstract
Two extraction procedures, namely dispersive liquid-liquid microextraction (DLLME) and dispersive liquid-liquid microextraction based on solidification of floating organic drop (DLLME-SFOD), have been compared for the spectrophotometric determination of Fe (III). In both procedures, Fe (III) was extracted after complexation with gallic acid in the presence of cetyltrimethylammonium bromide (CTAB). Tetrachloroethylene and 1-undecanol were used as extraction solvents in DLLME and DLLME-SFOD, respectively, while acetone was used as dispersing solvents. The effects of various experimental parameters (solution pH, the concentration of ligand and CTAB, as well as nature and amount of extraction and disperser solvents) on the extraction efficiency were investigated. Under optimum conditions, the calibration graphs were linear in the range of 50.0–650.0 and 8.0–800.0 μg L−1 and the detection limits were 15.0 and 5.0 μg L−1 for DLLME and DLLME-SFOD, respectively. The presence of NaCl, up to 1.0% (w/v) did not impact the extraction procedures. The analyte was good tolerated in the presence of most concomitant ions. The procedures were applied for the determination of Fe (III) in standard reference materials and real samples with good recoveries (95.5–99.0%) for DLLME-SFOD while poor recoveries (68.0–82.5%) were obtained when DLLME was applied. The analytical figures of the procedures were comparable with those listed in the literature and it could be concluded that DLLME-SFOD may be considered one of the best tools used for preconcentration of Fe (III), owing to its simplicity, time-saving and the possibility of using in conventional analytical laboratories.
Graphical abstract

Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Iron is an essential element not only for industries but also for all living organisms. In the human body, it is required for several biological processes including enzyme activities, erythropoiesis, as well as oxygen binding, transport, and storage [1, 2]. In the photosynthesis process of plants, chlorophyll formation requires iron [3]. Anemia is caused by a lack of iron, whereas its overdose may lead to hemochromatosis [4].
Thus, different techniques have been used to determine iron concentration in environmental and biological samples including inductively coupled plasma mass spectrometry (ICP-MS) [5], inductively coupled plasma optical emission spectrometry (ICP-OES) [6], flame atomic absorption spectrometry (FAAS) [7], and spectrophotometry [8]. Spectrophotometric techniques are simple, rapid, low-cost, accurate, and precise. However, it has lower sensitivity compared to the FAAS, ICP-OES, and ICP-MS [9, 10].
A preconcentration step is highly recommended to improve the sensitivity and selectivity of spectrophotometric tools [11]. This step can be performed using a variety of techniques, including liquid-liquid extraction (LLE) [12], solid phase extraction (SPE) [13] and chromatographic techniques [14, 15]. However, these techniques are time-consuming and they necessitate the use of harmful chemical solvents in relatively large quantities [16]. As a result, liquid phase microextraction techniques (LPME) have been developed to provide viable alternatives to classical extraction methods [17]. Herein, the volume of organic solvent utilized is typically in the microliter range. Examples of LPME procedures are solidified floating organic drop (SFOD) [18], hollow fiber liquid phase microextraction (HF-LPME) [19], and dispersive liquid-liquid microextraction (DLLME) [20].
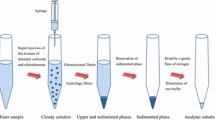
DLLME is a ternary solvent system in which a cloudy solution is generated by rapid injection of a mixture of the dispersing and the extractant solvents, into the aqueous sample solution. The target metal ion is extracted from its aqueous solution into a small volume of the organic phase upon centrifugation [21].
There are various modes of DLLME depending on the density and type of the extractant. Solvents with a density greater than that of water, such as chlorinated hydrocarbons, are utilized in the classical DLLME [22]. Low-density solvents such as hexane, heptane, and m-xylene are used in low-density solvent DLLME (LDS–DLLME) [23], but when solvents with low density have solidification temperature of 10–25 °C, such as 1-undecanol, are used the method is called dispersive liquid-liquid microextraction based on solidification of floating organic droplet (DLLME-SFOD) [24, 25]. The two procedures (DLLME and DLLME-SFOD) were extensively used for preconcentration of metal ions [26, 27], pesticides [28, 29], phenolic compounds [30, 31], pharmaceutical compounds [32, 33], and poly aromatic hydrocarbons [34, 35].
Gallic acid (GA), 3,4,5-trihydroxybenzoic acid, is a potent complexing agent that forms stable compounds with Fe(III) [36]. It coordinates to Fe(III) through two adjacent OH [37]. Gallic acid-based sorbents were used in to preconcentrate Fe(III) by SPE [38, 39]. It was also used for spectrophotometric determination of Fe(II) and Fe(III) in environmental samples [40]. To the best of our knowledge, GA was not used before as a complexing agent to extract Fe(III) by liquid-liquid based extraction procedure.
The primary goal of this work is to compare the extraction efficiency of DLLME and DLLME-SFOD for preconcentration and extraction of Fe (III) in water and food samples prior to detection by spectrophotometric technique. The analyte was extracted as a ternary complex after complexation with GA in the presence of cetyltrimethylammonium bromide (CTAB). The ternary complex GA-Fe-CTAB was extracted into an organic phase which was subjected to spectrophotometric analysis.
Experimental
Chemicals
Analytical grade chemicals were received from Merck (Darmstadt, Germany), Sigma-Aldrich Corporation (St. Louis, MO, USA), or Alpha Chemika (Mumbai, Maharashtra, India). All aqueous solutions were prepared using double-distilled water. The stock solution of Fe (III) (1000 mg L−1) was prepared by dissolving the appropriate amount of FeCl3 in 0.1 mol L−1 HCl. The working solutions of Fe (III) were freshly prepared from the stock solution by dilution with double-distilled water. An appropriate amount of GA was dissolved in 10 mL ethanol then diluted to 100 mL using double-distilled water to prepare 1.0 × 10–2 mol L−1 solution. An aqueous solution of CTAB (1.0 × 10–2 mol L−1) was prepared by dissolving the appropriate amount in double-distilled water. Buffer solutions were prepared using sodium acetate/acetic acid (pH 3.0–6.0), Hexamine (pH 7.0–8.0).
Instrumentation
All spectrophotometric measurements were taken using UNICO 1200 UV–VIS spectrophotometer (UNICO Instruments Co., NJ, USA). The pH was measured using AD1030 pH meter (Mettler-Toledo, China). Centrifugation was performed with a HERMLE Z 306 centrifuge (HERMLE Labortechnik, Germany). A microwave digestion system (Speedwave®four, Berghof Products, Germany) with polytetrafluoroethylene (PTFE) vessels was used for the digestion of real samples.
Real sample
Water samples were gathered in polyethylene jars that had been acid cleaned. To eliminate any suspended particles, all collected samples were filtered using a cellulose membrane filter (Millipore) with a 0.45 µm pore size and kept frozen until analysis. Food samples were prepared according to previous work in our scientific group [41]. Briefly, samples of food (spinach leaf, apple, and tomato) were rinsed with double-distilled water, dried at 90 °C until constant weight, and then crushed into fine powder in a porcelain mortar. The sample (0.3–0.4 g) was mixed with 4 of HNO3 and 2 mL of H2O2 and digested using a microwave digestion technique with the following parameters: "power, 1600 W; ramp time, 15 min; temperature, 200 °C; hold time, 15; and cooling time, 15 min". After cooling to ambient temperature, the liquid was filtered and diluted to 10 mL.
Extraction procedures
Dispersive liquid–liquid microextraction procedure
In DLLME, 10.0 mL of the standard solution or analyte aqueous solution (pH 5.0) was mixed with 0.1 mL of 1.0 × 10–2 mol L−1 GA and 0.1 mL of 1.0 × 10–2 mol L−1 CTAB. A cloud solution was formed after rapid injection of a mixture of 200 μL of tetrachloroethylene (extraction solvent) and 200 μL of acetone (disperser solvent). The dispersion of fine droplets of extraction solvent in the aqueous sample resulted in a cloud solution. The complex of Fe with GA was separated into fine tetrachloroethylene droplets in this step. The small droplets of tetrachloroethylene dispersed throughout the test tube were settled to the bottom after centrifugation at 4000 rpm for 5 min. The upper phase was carefully eliminated, and the volume of the sediment phase was made up to 0.5 mL by the addition of ethanol, and the absorbance was spectrophotometrically measured at 560 nm against a reagent blank prepared by the same manner in absence of Fe (III).
Dispersive liquid–liquid microextraction based on solidification of floating organic droplet
An aqueous sample (10.0 mL) containing Fe (III) at pH 5.0 was mixed in a conical tube with 0.1 mL of 1.0 × 10–2 mol L−1 GA and 0.1 mL 1.0 × 10–2 mol L−1 CTAB. After injecting a mixture of 100 µL 1-undecanol and 200 µL acetone with a gastight syringe, the solution became cloudy. Upon centrifugation (4000 rpm, 5 min), an organic droplet of the extraction solvent containing the analyte was floated on the solution surface and was solidified by cooling in an ice bath for 5 min. A spatula was used to transfer the solidified floated organic drop to an Eppendorf® tube, and the total volume was increased to 0.5 mL by adding ethanol. The absorbance was measured as illustrated in the previous subsection.
Calculations
All trials were carried out in triplicate, and the findings were given as mean ± standard deviation (S.D.). The extraction efficiency was described in terms of recovery (R) during the optimization study:
where V and C stand for volume and concentration, where f and i represent final and initial states, respectively.
Results and discussion
Preliminary study
On mixing Fe (III) with GA a violet color is spontaneously formed at pH 5.0 (acetate buffer) and the maximum absorbance is achieved at 560 nm (Fig. 1). According to Masoud et al., gallic acid forms a stable complexes with iron in different metal:ligand ratio [42]. Therefore, the extraction and subsequent spectrophotometric determination of Fe (III) as GA complex is possible either by DLLME or DLLME-SFOD. The preliminary investigations also indicated that the extraction process could not be completed in the absence of CTAB, i.e.; the analyte was extracted as a ternary complex (Fe-GA-CTAB). To acquire the best efficiency, the experimental conditions that may affect the extraction procedure were optimized.

Visible spectra of gallic acid and its complex with Fe (III)
Optimization of the extraction procedures
Effect of pH
The initial pH of the aqueous phase may have a great impact on the complex's formation, stability, and subsequent extraction. The effect of pH on the microextraction of Fe (III) using DLLME and DLLME-SFOD procedures was investigated in the pH range of 3.0–8.0. According to the findings in Fig. 2a, the absorbance increased with increasing pH until attained a plateau at pH 5.0–7.0 then decreased after that may be interpreted in the view of hydrolysis of Fe (III) ions. At pH < 5.0, hydronium ions compete with Fe (III) and hinder metal ion binding to the ligand, reducing the possibility of complex formation [43]. Based on these findings, pH 5.0 was selected for the subsequent extraction procedures.
Effect of gallic acid concentration:
Gallic acid was used as a chelating agent in our work because of its ability to form a hydrophobic and colored complex with Fe(III) that can be easily extracted into the organic phase and analyzed using spectrophotometry. In the range of 0.01–0.3 mmol L−1, the effect of GA concentration on the microextraction of Fe (III) using DLLME and SFOD-DLLME was investigated. According to the results shown in Fig. 2b, the extraction increased as the ligand concentration increased up to 0.1 mmol L−1. As a result, the concentration of GA in the following studies was kept constant at 0.1 mmol L−1.

Effect of (a) pH (b) amount of gallic acid (c) amount of CTAB on the extraction of Fe (III) by DLLME-SFOD and DLLME
Effect of CTAB concentration:
The effect of CTAB concentration on the extraction of Fe (III) by the two extraction procedures was studied in the range of 0.01 – 0.3 mmol L−1. Figure 2c shows that as the CTAB concentration increased, the extraction efficiency increased until reached a constant limit at 0.1 mmol L−1. The CTAB concentration of 0.1 mmol L−1, which yielded the maximum extraction efficiency, was thus chosen for further investigations.
Effect of extracting solvent
The extraction solvent used in both DLLME and DLLME-SFOD is a crucial factor in achieving efficient performance. Three extractant solvents (chloroform, carbon tetrachloride, and tetrachloroethylene) with densities larger than water and poor miscibility in water were studied in DLLME. Other two low-density solvents (m-xylene and 1-undecanol) were used in DLLME-SFOD. Figure 3 shows that the highest extraction efficiency was obtained when using tetrachloroethylene in DLLME and 1-undecanol in DLLME-SFOD. The extraction efficiency increases with an increasing volume of the extracting solvent and reaches a plateau at 100 µL of 1-undecanol and 200 µL of tetrachloroethylene. Therefore, these volumes were selected as optimum extractant volumes.

Effect of type and volume of extraction agent on extraction efficiency of Fe (III) by (a) DLLME-SFOD and (b) DLLME
Effect of disperser solvent
The miscibility of the disperser solvent in the extraction solvent and the aqueous solution is the most important factor in selecting it. Furthermore, the type of disperser has a direct impact on the binary solvent's viscosity. As a result, this solvent can regulate droplet production and extraction efficiency [44]. Three solvents, acetone, methanol, and ethanol were used to investigate this effect. The results in Table 1 show that the highest extraction efficiency was obtained when using 200 µL acetone as a dispersive solvent in both procedures.
Effect of ionic strength
In most modes of liquid–liquid extraction, adding salt to the aqueous sample can considerably increase the extraction of various analytes “salting-out effect”. In this study, several tests were carried out with varying NaCl concentrations (0.1–1.0%) to evaluate the possibility of a salting-out effect. According to these findings, adding NaCl in this concentration range does not affect the efficiency of Fe(III) extraction. These findings are most likely the outcome of two opposing salt addition factors [18]. One of these is the salting-out phenomenon, which promotes extraction by lowering the aqueous medium's analyte solubility and thereby increasing analyte extraction into the organic drop. The other effect is a reduction in the preconcentration factor as the organic drop volume increases. As a result, all the extraction trials were conducted without the use of salt.
Effect of centrifugation time
To obtain two distinct phases in the tubes, centrifugation is required. The effect of centrifugation time on Fe (III) extraction was investigated at 4000 rpm for 2–10 min. The results show that centrifugation at 4000 rpm for 5 min is sufficient to obtain maximum recovery by the two procedures (R = 98.5 ± 2.0% and 80.5 ± 2.9% for DLLME-SFOD and DLLME, respectively).
Interference
To study the selectivity of the two extraction procedures for preconcentration and determination of Fe (III), aliquots containing 100 µg L−1 and different concentrations of the interfering ions were prepared and then extracted by the optimized procedure. The tolerance limit is defined as the highest amount of the concomitant ion (in mg L−1) that produces ± 5% change in the extraction efficiency. The results (Table 2) showed that most of the cations and anions tested did not affect Fe (III) extraction and determination. Thus, the two procedures have high selectivity for the analyte.
Analytical figures of merit
Under the optimum experimental conditions, the calibration curves were linear over concentration ranges of 50.0–650.0 and 8.0–800.0 µg L−1 for Fe (III) with enrichment factors (EF = the ratio between the slopes of the calibration graphs after and before preconcentration) of 19.5 and 20.2 for DLLME and DLLME-SFOD, respectively. The equations of the calibration curves after the preconcentration procedure were Y = 0.008X + 0.002 (R = 0.997) and Y = 0.011X + 0.001 (R = 0.999) for DLLME and DLLME-SFOD, respectively. The limits of detection (LOD = 3 × Standard deviations of 5 blanks/slope of calibration graph) and limits of quantification (LOQ = 10 × Standard deviations of 5 blanks/slope of calibration graph) were 15.0 and 50.0 for DLLME and 5.0 and 8.0 µg L−1 for DLLME-SFOD. The relative standard deviations (RSD) were 4.9% and 2.1% for ten independent measurements of 100 µg L−1 of Fe (III) by DLLME and DLLME-SFOD, respectively.
Accuracy and recovery experiments
To validate the under-investigated procedures, they were used for the determination of Fe in two standard reference materials (SRMs) (SRM 1573a for tomato leaves and SRM 3255 for green tea extract). Table 3 displays the analytical results. As can be shown, the measured values by DLLME-SFOD were in good agreement with the certified values, whereas the results obtained by DLLME are significantly lower than the certified values (t > t critical). Moreover, the proposed approaches were used to determine the concentrations of Fe in food samples. In addition, the recovery tests of different concentrations of Fe were performed. The results in Table 4 show that the recoveries for trace analysis are reasonable (95.5 – 99.0%) when the DLLME-SFOD procedure was applied. On the other hand, non-quantitative recoveries (68.0 – 82.5%) were achieved by the DLLME procedure. The poor recoveries obtained for DLLME may result from the inadequate separation of the analyte into the organic phase.
Comparison of the investigated approaches with one another
The two microextraction approaches are simple, rapid, and reproducible, and require only a few amounts (in µL) of organic solvents. Both procedures extract Fe (III) as gallate complex at the same pH range (5.0–7.0) and the addition of CTAB is required to form a ternary complex. 1-Undecnol and tetrachloroethylene are used as extraction solvents in DLLME-SFOD and DLLME, respectively, while acetone is employed as a disperser solvent in both procedures. Excellent analytical features, i.e., lower LOD and wider linear range, were achieved in the case of DLLME-SFOD which also shows good accuracy and quantitative recoveries in the analysis of SRMs and real samples, respectively.
Comparison with other procedures
Table 5 displays a comparison between the presented procedures and other preconcentration approaches, coupled with the spectrophotometric determination of Fe (III), in terms of the analytical features. As clearly shown, the developed procedures, especially DLLME-SFOD, have lower LOD and a wider dynamic analytical range than those of most published procedures.
Conclusions
The efficiency of two microextraction procedures, DLLME and DLLME-SFOD, was examined in this work for the extraction and preconcentration of Fe (III) in aqueous samples prior to spectrophotometric measurement using GA as a complexing agent. To the best of our knowledge, this is the first report to employ GA as a complexing agent to extract Fe(III) using a liquid–liquid extraction process. Based on the results of the present study, the two procedures are quick and simple for extracting the target analyte. It is worth mentioning that the DLLME-SFOD approach has major advantages over the DLLME, including accuracy, repeatability, lower LODs and LOQs, and a wider analytical range. Also, quantitative recoveries were achieved by DLLME-SFOD in the analysis of water and food samples. The developed procedures, particularly DLLME-SFOD, have lower LOD and a wider dynamic analytical range than most published spectrophotometric procedures for the determination of iron.
Data availability
The datasets used or analyzed during the current study are available from the corresponding author upon reasonable request.
References
D. Stoyanovsky, Y. Tyurina, I. Shrivastava, I. Bahar, V. Tyurin, O. Protchenko, S. Jadhav, S. Bolevich, A. Kozlov, Y. Vladimirov, Free. Radic. Biol. Med 133, 153–161 (2019)
Y. Bi, A. Ajoolabady, L.J. Demillard, W. Yu, M.L. Hilaire, Y. Zhang, J. Ren, Biochem. Pharmacol. 190, 114661 (2021)
G.R. Rout, S. Sahoo, Rev. Agric. Sci. 3, 1 (2015)
H. Tapiero, L. Gate, K. Tew, Biomed. Pharmacother 55, 324 (2001)
T.S. Alomar, M.A. Habila, N. AlMasoud, Z.A. Alothman, M. Sheikh, M. Soylak, Appl. Sci. 11, 7792 (2021)
M.A. Gab-Allah, A.B. Shehata, Chem. Pap. 75, 4239 (2021)
D. Sulthoniyah, R. Primaharinastiti, F. Annuryanti, Res. J. Pharm. Technol. 11, 2569 (2018)
Y.G. Abou,El-Reash, H.A. Tantawy, E. Abdel-Latif, W.I. Mortada, Microchem J 158, 105280 (2020)
A. Bazmandegan Shamili, A. Haji Shabani, S. Dadfarnia, M. Saeidi, M. Rohani Moghadam, Turk. J. Chem 39, 1059 (2015)
M. Soylak, M. Agirbas, E. Yilmaz, Food Chem. 338, 128068 (2021)
R. Gürkan, N. Altunay, N. Gürkan, J. Iran. Chem. Soc. 14, 1033 (2017)
T. Vander Hoogerstraete, B. Onghena, K. Binnemans, J. Phys. Chem. Letter 4, 1659 (2013)
W.I. Mortada, A.M. Abdelghany, Biol. Trace Elem. Res. 193, 100 (2020)
S. Chen, N. Li, X. Zhang, D. Yang, H. Jiang, Spectrochim. Acta. Part. A. Mol. Biomol. Spectrosc 138, 375 (2015)
S. Zareba, K. Szarwiło, A. Pomykalski, Farmaco. Soc. Chim. Ital. 1989. 60, 459 (2005)
Z. Temerdashev, T. Musorina, T. Chervonnaya, Z.V. Arutyunyan, J. Anal. Chem. 76, 1357 (2021)
A. Quigley, W. Cummins, D. Connolly, J. Chem. 2016, (2016).
G.G. El-Gamal, W.I. Mortada, M.M. Hassanien, A.A. Ibrahim, Y.G. Abou El-Reash, J. Anal. Atomic Spectrom. 36, 1306 (2021)
M. Ghambarian, Y. Yamini, A. Esrafili, Microchim. Acta. 177, 271 (2012)
A. Zgoła-Grześkowiak, T. Grześkowiak, TrAC. Trend. Anal. Chem. 30, 1382 (2011)
S.Z. Mohammadi, Y.M. Baghelani, F. Mansori, T. Shamspur, D. Afzali, Química. Nova. 35, 198 (2012)
L. Kocúrová, I.S. Balogh, J. Šandrejová, V. Andruch, Microchem. J. 102, 11 (2012)
X. Li, A. Xue, H. Chen, S. Li, J. Chromatograph. A 1280, 9 (2013)
W. I. Mortada, E. A. Azooz, Trend. Environ. Anal. Chem e00163 (2022).
M.S. Jagirani, M. Soylak, Crit. Rev. Anal. Chem. 52, 968 (2022)
W. I. Mortada, G. G. El-Gamal, M. M. Hassanien, A. A. Ibrahim, Y. G. Abou El-Reash, Int. J. Environ. Anal. Chem. 1 (2022).
Z.A. Alothman, N.H. Al-Shaalan, M.A. Habila, Y.E. Unsal, M. Tuzen, M. Soylak, Environ. Monitor. Assess. 187, 1 (2015)
H. Musarurwa, N.T. Tavengwa, Food Chem. 342, 127943 (2021)
A. Jouyban, M.A. Farajzadeh, M.R.A. Mogaddam, Talanta 206, 120169 (2020)
E. Nalewajko-Sieliwoniuk, M. Hryniewicka, D. Jankowska, A. Kojło, M. Kamianowska, M. Szczepański, Food Chem. 327, 126996 (2020)
F. Hou, T. Deng, X. Jiang, Microchimica Acta 180, 341 (2013)
A.V. Herrera-Herrera, J. Hernández-Borges, T.M. Borges-Miquel, M.Á. Rodríguez-Delgado, J. Pharm. Biomed. Anal. 75, 130 (2013)
X. Sun, X. Xing, Z. Du, Talanta 209, 120540 (2020)
G. Leng, G. Lui, Y. Chen, H. Yin, D. Dan, J. Separat. Sci. 35, 2796 (2012)
M. Guiñez, L.D. Martinez, L. Fernandez, S. Cerutti, Microchem. J. 131, 1 (2017)
S.A. Kazmi, M.S. Qureshi, Z. Maqsood, Inorg. Chimica. Acta 137, 151 (1987)
M.J. Hynes, M.N.Ó. Coinceanainn, J. Inorg. Biochem 85, 131 (2001)
R. Sharma, P. Pant, J. Hazard. Mater. 163, 295 (2009)
X. Pu, B. Hu, Z. Jiang, C. Huang, Analyst 130, 1175 (2005)
J. Zolgharnein, H. Abdollahi, D. Jaefarifar, G. Azimi, Talanta 57, 1067 (2002)
W.I. Mortada, M.M. El-Defrawy, E. Erfan, H.A. El-Asmy, J. Food Compos. Anal. 108, 104445 (2022)
M.S. Masoud, A.E. Ali, S.S. Haggag, N.M. Nasr, Spectrochim. Acta. Part. A. Mol. Biomol. Spectrosc 120, 505 (2014)
A. Abdallah, A.M. Youins, M.R. El-Kholany, RSC Adv. 12, 8520 (2022)
M. Gharehbaghi, F. Shemirani, M. Baghdadi, Int. J. Environ. Anal. Chem 88, 513 (2008)
J.V. Maciel, B.M. Soares, J.S. Mandlate, R.S. Picoloto, C.A. Bizzi, E.M. Flores, F.A. Duarte, J. Agric. Food Chem. 62, 8340 (2014)
M. Borzoei, M.A. Zanjanchi, H. Sadeghi-Aliabadi, L. Saghaie, Biol. Trace Elem. Res. 192, 319 (2019)
M. Borzoei, M.A. Zanjanchi, H. Sadeghi-Aliabadi, L. Saghaie, Food Chem 264, 9 (2018)
M.R. Moghadam, A.M.H. Shabani, S. Dadfarnia, J Hazard Mater 197, 176 (2011)
E.R. Pereira, B.M. Soares, J.V. Maciel, S.S. Caldas, C.F. Andrade, E.G. Primel, F.A. Duarte, Anal. Method 5, 2273 (2013)
D.L. Giokas, E.K. Paleologos, M.I. Karayannis, Anal. Bioanal. Chem. 373, 237 (2002)
M.J. Almendral, A. Alonso, M.J. Porras, M.A. García, Y. Curto, Microchimica. Acta 147(1), 117 (2004)
E. Alian, A. Semnani, A. Firooz, M. Shirani, B. Azmoon, Arab. J. Sci. Eng. 43(1), 229–240 (2018)
Funding
Open access funding provided by The Science, Technology & Innovation Funding Authority (STDF) in cooperation with The Egyptian Knowledge Bank (EKB). Not applicable.
Author information
Authors and Affiliations
Contributions
HEZ contributed to data curation, investigation, methodology, and writing—original draft. WIM contributed to conceptualization, data curation, investigation, methodology, supervision, and writing—review and editing. MEK contributed to conceptualization, data curation, investigation, methodology, supervision, and writing—review and editing.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Consent for publication
The authors declare that they have not applicable for that section of “Ethics approval and consent to participate” and “Consent for publication”.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zedan, H.E., Mortada, W.I. & Khalifa, M.E. Microextraction procedures for preconcentration of Fe (III) in water and food samples prior to colorimetric detection: a comparative study. J IRAN CHEM SOC 20, 645–653 (2023). https://doi.org/10.1007/s13738-022-02697-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13738-022-02697-3