
Abstract
Outbreak of Coronavirus (SARS-CoV-2) has thrown a big challenge to the globe by snatching millions of human lives from the world. In this study, inhibitory efficiency of ten anti-HIV compounds from different Indian medicinal plant parts have been virtually screened against Mpro, PLpro and RdRp proteins of SARS-CoV-2. The molecular docking study reflected that among these compounds, Proptine (PTP) has the highest binding affinity for the three cases. Introduction of PTP molecules within the binding pocket of these proteins showed a large structural and conformational changes on the structure of proteins which is revealed from molecular dynamics (MD) simulation studies. RMSD, RMSF and analysis of thermodynamic parameters also revealed that PTP makes a huge impact on the structures of the respective proteins which will pave an opportunity for doing advanced experimental research to evaluate the potential drug to combat COVID-19.
Graphic abstract

Similar content being viewed by others

1 Introduction
In late December 2019, a new strain of Coronavirus (SARS-CoV-2) has spread swiftly around the globe. WHO declared a pandemic situation within 3 months of its initial screening in Wuhan, China (Wang et al. 2020; Lu et al. 2020; Jiang et al. 2020). Hundreds of millions lives of approximately 188 countries are affected with this highly transmissible disease (Shekerdemian et al. 2020; Baildya et al. 2020). This pandemic efficiently strikes the national healthcare systems and global economy. While the year of 2020 was a challenging one, 2021 looks to be more difficult with the appearance of multiple variants SARS-CoV-2 (Fontanet et al. 2021). A new variant can emerge through numerous rounds of viral replication. The variant is co-termed as a strain if the sequence variation shows a distinctly different phenotypic characteristics (Mascola et al. 2021). In December, 2020, there was an unexpected rise of the COVID-19 cases due to the emergence of the new SARS-CoV-2 variants 501Y.V2 (B.1.351) in South Africa and 501Y.V1 (B.1.1.7) in the UK (Fontanet et al. 2021; Tegally et al. 2020; Volz et al. 2021; Davies et al. 2021; Xie et al. 2021). New strains are more transmissible than the previously one (Kirby 2021). Cold, flu, dihedra, headache, lymphopenia, etc. are the common symptoms of COVID-19 but it is not clear new strain-affected patients are sowing the same symptoms or not (Sahin et al. 2020; Graham et al. 2021). SARS-CoV-2, the single-stranded positive-sense RNA virus is included in β-coronavirus genus, closely related to the genomic organization of SARS-CoV identified in 2003 (Pal et al. 2020). With the interaction of angiotensin converting enzyme 2 of Human cell, spike protein of SARS-CoV-2 helps the virus to enter the human cell (Zhang et al. 2020). After entering into the cell, immediately viral replication and transcription are started with the functional proteins like main protease (Mpro), papain-like protease (PLpro), RNA-dependent RNA polymerase (RdRp) (Indwiani Astuti 2020). It is reported that in the new strains, mutation (N501Y) occurred in the receptor-binding domain of the spike protein, responsible for increased transmission (Davies et al. 2021).
With the heavy toll of the pandemic, scientists are actively checking various strategies either new or repurposed drugs as well as vaccines (Kandeel and Al-Nazawi 2020) but till now there is no potentially active drug available in the market which may effectively combat SARS-CoV-2. 2020 saw the development and testing of some COVID-19 vaccines (Fontanet et al. 2021; Baden et al. 2021; Polack et al. 2020). Till now, it is not clear that whether these COVID-19 vaccines can protect against the infection from these new SARS-CoV-2 variants or not (Fontanet et al. 2021; Mascola et al. 2021). Recently, name of the few drugs e.g., Remdesivir, Hydroxychloroquine, Chloroquine are the topic of discussion to researchers but none of these are potentially efficient against this deadly virus (Baildya et al. 2020, 2021a; Elfiky 2020; Khan et al. 2021; Mandal et al. 2021).
Secondary metabolites from different tropical plants are greatly influenced by environmental factors. These secondary metabolites have already drawn the attention for developing medicine against different antiviral diseases (Guerriero et al. 2018; Yang et al. 2018). According to the previous reports, extracts from various medicinal plant parts or plant-derived natural products have shown anti-HIV activity and they are under different stages of clinical development in different parts of the globe (Bedoya et al. 2001; Min et al. 1999, 2001). Different attempts also have been made to search the natural products which can inhibit different proteins of SARS-CoV-2 (Dutta et al. 2021). Few scientists claimed that the leave extracts of Azadirachta indica and Ocimum sanctum showed significant inhibitory activity against SARS-CoV-2 (Baildya et al. 2021b; Varshney et al. 2020). In-silico-based screening is an effective tool to meet the challenges of antiviral drug discovery. Computational screening of natural compounds as docking followed by molecular dynamics saves resources in terms of money and time (Murgueitio et al. 2012). Generally, the active compounds from one plant were considered in the previous studies (Borkotoky and Banerjee 2020), but in our study, we have considered the most active anti-HIV compounds of different plants with the explanation of the effectiveness against new SARS-CoV-2 variants.
In this study, we have chosen ten highly active compounds (available in traditionally used medicinal plants of India) against HIV (Sabde et al. 2011) and virtually screened their inhibitory activity against Mpro, PLpro and RdRp protein of SARS-CoV-2. Molecular dynamics simulation also performed to check the stability of Mpro, PLpro and RdRp with the compound having the highest Molecular Docking score. These findings will provide valuable information to other researchers to identify the right drug to combat COVID-19.
2 Methodology
2.1 Docking of potentially active anti-HIV compounds with Mpro, PLpro and RdRp of SARS-CoV-2
Structures of Mpro (PDB ID: 6LU7), PLpro (PDB ID: 6W9C) and RdRp (PDB ID: 6M71) were downloaded from Protein Data Bank (http://www.rcsb.org). The “sdf” files of the ten anti-HIV compounds were downloaded from the PubChem (National Library of Medicine) and was converted to respective pdb files. All the structures of proteins were cleaned by removing hetero-atoms and water molecules using UCSF Chimera (Pettersen et al. 2004). Autodock Vina package (Trott and Olson 2010) was used for docking between the compounds and the best binding sites of the proteins. Autodock Tools was used for the preparation of pre-docking necessary files.
2.2 Molecular dynamics (MD) simulation of protein–protopine complex
MD studies using GROMACS (5.0) (Berendsen et al. 1995) were performed with the minimum energy conformer obtained after docking between proteins and protopine (PTP) by applying CHARMM36-mar2019 force-field (Lee et al. 2014) using TIP3P solvation model (Boonstra et al. 2016). CHARMM General Force Field server for was used the generation of topology and parameter files of the respective drug. Periodic boundary conditions were applied by generating a cubical box of 1-nm length by keeping 2-nm distance between two successive images. All the protein moieties were neutralized by adding adequate number of ions. Using steepest descent algorithm and conjugate gradient protocol, energy minimization was performed until the maximum force of at least 10 kJ mol−1 nm−1. Isochoric-isothermal (NVT) and isothermal-isobaric (NPT) ensembles were applied on the system for 100 ps for equilibration at 300 K by keeping 2-fs time step and 1-nm electrostatic and van der Waal cut-offs. Particle mesh Ewald (PME) (Abraham and Gready 2011) method was used for long range interaction calculations. Finally, 2000-ps MD simulation were subjected to the equilibrated ensembles with the same cut-offs. In all cases, snapshots of the trajectory were saved for each ns.
2.3 Binding free energy calculation
Binding energies between the proteins and PTP were calculated by molecular mechanics Poison–Boltzmann surface area (MM-PBSA) method, implemented on Gromacs tool (g_mmpbsa) (Baker et al. 2001; Kumari et al. 2014). The following formulae was used for binding energy calculation:
where Gw-complex is the total free energy of the proteins and PTP complex, Gw-protein, Gw-PTP are the free energies of the protein and PTP, respectively.
2.4 MD simulation result analysis
Using trjconv implemented on GROMACS, structural trajectories were calculated for protein and PTP complexes. To re-centre the proteins, PTP, ions and water molecules within the cubical box the same tool was used. gmx energy tool of GROMACS was applied for calculation of van der Waals interaction, electrostatic energy, interaction energy values, etc. With the help of xmgrace plotting tool RMSD plots, RMSF plot, Radius of gyration and SASA plots were generated.
3 Results and discussion
The binding affinities of the ten anti-HIV compounds against Mpro, PLpro and RdRp are shown in Fig. 1. Highest binding affinity of − 7.9, − 7.4 and − 7.5 kcal/mol is achieved by protopine (PTP) against Mpro, PLpro and RdRp, respectively. This signifies that among these compounds PTP is strongly binds with these proteins.

Docking score of anti-HIV compounds against SARS-CoV-2 proteins
Moreover, to determine the level of toxicity of the selected anti-HIV compounds, we have examined out ADME toxicity analyzed by pkCSM online server (Pires et al. 2015).
3.1 Prediction of ADMET
Evaluation of pharmacokinetic properties of the studied anti-HIV compounds suggested that they were effectively absorbed by the gastro-intestinal part with low blood–brain-barrier (BBB) permeability value and these drugs do not affect CYP2D6, CYP2C9 and CYP2D6 cytochromes. Skin permeability for these compounds was found to be in between − 3.078 and 1.089 units. The toxicity table is shown in Table S1.
3.2 Prediction of toxicity
All compounds show negative hERGI inhibition activity. The LD50 values of the examined anti-HIV compounds fall in between 2.0 and 2.993 (mol/kg), while the chronic oral rat toxicity (LOAEL) values fall in between 1.112 and 2.49 (log mg/kg_bw/day). Most of the compounds do not show AMES toxicity and hepatotoxicity. None of the compounds show skin sensitization. T. pyriformis and minnow toxicity values are tabulated in Table 1.
From the above analysis, it is clear that the studied drugs showed a descent toxicity level. From the binding affinity and toxicity level analysis, we have selected PTP to carry out further analysis.
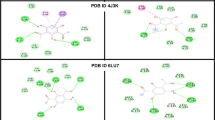
The binding interactions of PTP on the binding pockets of Mpro, PLpro and RdRp are shown in Fig. 2a–c. The major interactions that contributes to the binding of the corresponding proteins are electrostatic and Van der Waal types. The donor and acceptor sites that are engaged in H-bonding are shown 3D-contour in the left panel of Fig. 2a–c. The residues corresponding proteins that are involved to the binding of active compound PTP molecule are shown on the right panel of Fig. 2a–c.

Docked structure of PTP and SARS-CoV-2 proteins
To analyze the stability of the PTP–protein complexes, we have performed MD simulation. The root mean square deviation (RMSD) plots of corresponding complexes are shown in Fig. 3a–c. It is clear from Fig. 3 that Mpro–PTP, PLpro–PTP and RdRp achieved an equilibrium for about 500, 250 and 500 ps with an RMSD value around 2.2, 1.9 and 2.2 Å, respectively.

Root mean square deviation of docked a Mpro b PLpro and c RdRp during MD simulation
We found the RMSD fluctuation of the docked PLpro–PTP complex accounts lower RMSD fluctuation with respect to undocked one confirming introduction of PTP in the binding cavity of PLpro stabilizes the corresponding complex.
Figure 4a–c illustrates the radius of gyration (Rg) plot (left panel) and solvent accessible surface area (SASA) plot (right panel). Rg measures the compactness of a system. Except Mpro–PTP complex the docked PLpro and RdRp corresponds lower Rg value compare to undocked one revealing that these complexes are much compact. Higher value indicates lower fluctuation of proteins with time. Figure 4 (left panel) shows that all the docked structure has lower SASA value compare to undocked one which reveals the lower fluctuation of the docked structure. To further assess the flexibility and local motion of the drug–protein complexes, we have further analyzed root mean square fluctuation (RMSF) as shown in Fig. 5a–c. The lower fluctuation of the PTP-protein as compare to the protein clearly reveals that PTP moiety strongly interacts with the protein residues establishing stability of the complexes. We further noted that RMS fluctuation for the residue GLY143 for Mpro, LYS232 for PLpro and LYS545 and ARG858 for RdRp experienced lower fluctuation revealing that they are involved in interaction with the drug and located at the binding loop of the corresponding complexes. Furthermore, we have performed g_MMPBSA energy analysis. By applying the said tool, we have analyzed the contribution energy of each residue as shown in Fig. 6a–c.

Radius of gyration (left) and SASA plot (right) of a Mpro, b PLpro and c RdRp during MD simulation

Root mean square fluctuation of before and after docking of a Mpro, b PLpro and c RdRp

Contribution energy of different residues of a Mpro, b PLpro and c RdRp
Both the RMSF and the contribution energy graph showed the residues which directly interacts with the PTP moiety expose their highest energy contributions as indicated by the residue numbers in corresponding RMSF graphs (left panel Fig. 5). Different free energy parameters that contribute to the binding energy are plotted in Fig. 7. It is clear from the figure that none of the parameter has its contribution to the overall binding. Highest Van der Waal interaction is achieved by RdRp, while least is seen by Mpro. Same trend is followed in case of non-polar energy term. Gibb’s free energy change for binding is highest with PLpro, while least is experienced by RdRp.

Different types of free energies of the PTP docked proteins
Furthermore, to analyze the effect of entry of drug to the binding pocket of the corresponding complexes, we have plotted the snapshots after every 1000 ps. We note that there is a huge conformational change experienced by the corresponding proteins at 2000 ps, 1000 ps and 2000 ps for Mpro, PLpro and RdRp, respectively. The snapshots at 0 ps and 2000 ps clearly raised the profound structural changes for stronger binding interactions between drug and proteins during the trajectory.
As per the report, the new variants of SARS-CoV-2 [i.e., 501Y.V1 (B.1.1.7), 501Y.V2 (B.1.351)] carry a mutation in spike protein and for that reason, the transmission rate is increased upto 70% (Xie et al. 2021; Gu et al. 2020; Tang et al. 2020). Our study shows that PTP has potential inhibitory activities against Mpro, PLpro and RdRp proteins of SARS-CoV-2. Hence, there is a chance of PTP to show the effectiveness against the new variants of SARS-CoV-2 (Fig. 8).

Structural changes of Mpro, PLpro and RdRp during MD simulation
4 Conclusions
The present work deals with the virtual screening of ten anti-HIV compounds which are extracted from various Indian medicinal plants to retard the overall replication and transmission process associated with SARS-CoV-2. Molecular docking studies revealed that out of ten compounds highest binding affinity is found with PTP against Mpro, PLpro and RdRp. Furthermore, molecular dynamics simulation and evaluation of different parameters such as RMSD, RMSF, Rg, SASA and thermodynamic parameters revealed that introduction PTP moiety to the binding loop of the corresponding proteins has a huge impact on overall structural and conformational changes. Analysis of the results revealed that PTP could be a potential therapeutic drug to combat the replication and transcription process of SARS-CoV-2. It is expected that PTP will be effective against the new strains of SARS-CoV-2 but more research is required for a proper understanding of the continuous mutation process of the SRAS-CoV-2 such that PTP can be converted from an experimental level to a conclusion.
References
Abraham MJ, Gready JE (2011) Optimization of parameters for molecular dynamics simulation using smooth particle-mesh Ewald in GROMACS 4.5. J Comput Chem 32(9):2031–2040. https://doi.org/10.1002/jcc.21773
Baden LR, El Sahly HM, Essink B, Kotloff K, Frey S, Novak R, Diemert D, Spector SA, Rouphael N, Creech CB (2021) Efficacy and safety of the mRNA-1273 SARS-CoV-2 vaccine. N Engl J Med 384(5):403–416. https://doi.org/10.1056/NEJMoa2035389
Baildya N, Ghosh NN, Chattopadhyay AP (2020) Inhibitory activity of hydroxychloroquine on COVID-19 main protease: an insight from MD-simulation studies. J Mol Struct 1219:128595. https://doi.org/10.1016/j.molstruc.2020.128595
Baildya N, Ghosh NN, Chattopadhyay AP (2021a) Inhibitory capacity of Chloroquine against SARS-COV-2 by effective binding with Angiotensin converting enzyme-2 receptor: an insight from molecular docking and MD-simulation studies. J Mol Struct 1230:129891. https://doi.org/10.1016/j.molstruc.2021.129891
Baildya N, Khan AA, Ghosh NN, Dutta T, Chattopadhyay AP (2021b) Screening of potential drug from Azadirachta Indica (Neem) extracts for SARS-CoV-2: an insight from molecular docking and MD-simulation studies. J Mol Struct 1227:129390. https://doi.org/10.1016/j.molstruc.2020.129390
Baker NA, Sept D, Joseph S, Holst MJ, McCammon JA (2001) Electrostatics of nanosystems: application to microtubules and the ribosome. Proc Natl Acad Sci 98(18):10037–10041. https://doi.org/10.1073/pnas.181342398
Bedoya L, Sanchez-Palomino S, Abad M, Bermejo P, Alcami J (2001) Anti-HIV activity of medicinal plant extracts. J Ethnopharmacol 77(1):113–116. https://doi.org/10.1016/S0378-8741(01)00265-3
Berendsen HJ, van der Spoel D, van Drunen R (1995) GROMACS: a message-passing parallel molecular dynamics implementation. Comput PhysCommun 91(1–3):43–56. https://doi.org/10.1016/0010-4655(95)00042-E
Boonstra S, Onck PR, van der Giessen E (2016) CHARMM TIP3P water model suppresses peptide folding by solvating the unfolded state. J Phys Chem B 120(15):3692–3698. https://doi.org/10.1021/acs.jpcb.6b01316
Borkotoky S, Banerjee M (2020) A computational prediction of SARS-CoV-2 structural protein inhibitors from Azadirachta indica (Neem). J BiomolStruct Dyn. https://doi.org/10.1080/07391102.2020.1774419
Davies NG, Abbott S, Barnard RC, Jarvis CI, Kucharski AJ, Munday JD, Pearson CA, Russell TW, Tully DC, Washburne AD (2021) Estimated transmissibility and impact of SARS-CoV-2 lineage B. 1.1. 7 in England. Science. https://doi.org/10.1126/science.abg3055
Dutta T, Ghorai S, Khan AA, Baildya N, Ghosh NN (2021) Screening of potential anti-HIV compounds from Achyranthes aspera extracts for SARS-CoV-2: an insight from molecular docking study. J PhysConfSer. https://doi.org/10.1088/1742-6596/1797/1/012042
Elfiky AA (2020) Ribavirin, remdesivir, sofosbuvir, galidesivir, and tenofovir against SARS-CoV-2 RNA dependent RNA polymerase (RdRp): a molecular docking study. Life Sci. https://doi.org/10.1016/j.lfs.2020.117592
Fontanet A, Autran B, Lina B, Kieny MP, Karim SSA, Sridhar D (2021) SARS-CoV-2 variants and ending the COVID-19 pandemic. Lancet. https://doi.org/10.1016/S0140-6736(21)00370-6
Graham MS, Sudre CH, May A, Antonelli M, Murray B, Varsavsky T, Klaser K, Canas LDS, Molteni E, Modat M (2021) The effect of SARS-CoV-2 variant B. 1.1. 7 on symptomatology, re-infection and transmissibility. medRxiv. https://doi.org/10.1101/2021.01.28.21250680
Gu H, Chen Q, Yang G, He L, Fan H, Deng Y-Q, Wang Y, Teng Y, Zhao Z, Cui Y (2020) Adaptation of SARS-CoV-2 in BALB/c mice for testing vaccine efficacy. Science 369(6511):1603–1607. https://doi.org/10.1126/science.abc4730
Guerriero G, Berni R, Muñoz-Sanchez JA, Apone F, Abdel-Salam EM, Qahtan AA, Alatar AA, Cantini C, Cai G, Hausman J-F (2018) Production of plant secondary metabolites: Examples, tips and suggestions for biotechnologists. Genes 9(6):309. https://doi.org/10.3390/genes9060309
Indwiani Astuti Y (2020) Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2): an overview of viral structure and host response. Diabetes MetabSyndr 14(4):407–412. https://doi.org/10.1016/j.dsx.2020.04.020
Jiang F, Deng L, Zhang L, Cai Y, Cheung CW, Xia Z (2020) Review of the clinical characteristics of coronavirus disease 2019 (COVID-19). J Gen Intern Med. https://doi.org/10.1007/s11606-020-05762-w
Kandeel M, Al-Nazawi M (2020) Virtual screening and repurposing of FDA approved drugs against COVID-19 main protease. Life Sci 251:117627. https://doi.org/10.1016/j.lfs.2020.117627
Khan AA, Baildya N, Dutta T, Ghosh NN (2021) Inhibitory efficiency of potential drugs against SARS-CoV-2 by blocking human angiotensin converting enzyme-2: Virtual screening and molecular dynamics study. MicrobPathog 152:104762. https://doi.org/10.1016/j.micpath.2021.104762
Kirby T (2021) New variant of SARS-CoV-2 in UK causes surge of COVID-19. Lancet Respir Med 9(2):e20–e21. https://doi.org/10.1016/S2213-2600(21)00005-9
Kumari R, Kumar R, Lynn A, Consortium O S D D (2014) g_mmpbsa-A GROMACS tool for high-throughput MM-PBSA calculations. J Chem Inf Model 54(7):1951–1962. https://doi.org/10.1021/ci500020m
Lee S, Tran A, Allsopp M, Lim JB, Hénin JRM, Klauda JB (2014) CHARMM36 united atom chain model for lipids and surfactants. J Phys Chem B 118(2):547–556. https://doi.org/10.1021/jp410344g
Lu H, Stratton CW, Tang YW (2020) Outbreak of pneumonia of unknown etiology in Wuhan, China: the mystery and the miracle. J Med Virol 92(4):401–402. https://doi.org/10.1002/jmv.25678
Mandal M, Chowdhury SK, Khan AA, Baildya N, Dutta T, Misra D, Ghosh NN (2021) Inhibitory efficacy of RNA virus drugs against SARS-CoV-2 proteins: an extensive study. J Mol Struct 1234:130152. https://doi.org/10.1016/j.molstruc.2021.130152
Mascola JR, Graham BS, Fauci AS (2021) SARS-CoV-2 viral variants—tackling a moving target. JAMA. https://doi.org/10.1001/jama.2021.2088
Min BS, Bae KH, Kim YH, Miyashiro H, Hattori M, Shimotohno K (1999) Screening of Korean plants against human immunodeficiency virus type 1 protease. Phytother Res 13(8):680–682. https://doi.org/10.1002/(SICI)1099-1573(199912)13:8%3c680::AID-PTR501%3e3.0.CO;2-H
Min BS, Kim YH, Tomiyama M, Nakamura N, Miyashiro H, Otake T, Hattori M (2001) Inhibitory effects of Korean plants on HIV-1 activities. Phytother Res 15(6):481–486. https://doi.org/10.1002/ptr.751
Murgueitio MS, Bermudez M, Mortier J, Wolber G (2012) In silico virtual screening approaches for anti-viral drug discovery. Drug Discov Today Technol 9(3):e219–e225. https://doi.org/10.1016/j.ddtec.2012.07.009
Pal M, Berhanu G, Desalegn C, Kandi V (2020) Severe acute respiratory syndrome Coronavirus-2 (SARS-CoV-2): an update. Cureus. https://doi.org/10.7759/cureus.7423
Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE (2004) UCSF Chimera—a visualization system for exploratory research and analysis. J Comput Chem 25(13):1605–1612. https://doi.org/10.1002/jcc.20084
Pires DE, Blundell TL, Ascher DB (2015) pkCSM: predicting small-molecule pharmacokinetic and toxicity properties using graph-based signatures. J Med Chem 58(9):4066–4072. https://doi.org/10.1021/acs.jmedchem.5b00104
Polack FP, Thomas SJ, Kitchin N, Absalon J, Gurtman A, Lockhart S, Perez JL, Pérez Marc G, Moreira ED, Zerbini C (2020) Safety and efficacy of the BNT162b2 mRNA Covid-19 vaccine. N Engl J Med 383(27):2603–2615. https://doi.org/10.1056/NEJMoa2034577
Sabde S, Bodiwala HS, Karmase A, Deshpande PJ, Kaur A, Ahmed N, Chauthe SK, Brahmbhatt KG, Phadke RU, Mitra D (2011) Anti-HIV activity of Indian medicinal plants. J Nat Med 65(3–4):662–669. https://doi.org/10.1007/s11418-011-0513-2
Sahin AR, Erdogan A, Agaoglu PM, Dineri Y, Cakirci AY, Senel ME, Okyay RA, Tasdogan AM (2020) 2019 novel coronavirus (COVID-19) outbreak: a review of the current literature. EJMO 4(1):1–7. https://doi.org/10.14744/ejmo.2020.12220
Shekerdemian LS, Mahmood NR, Wolfe KK, Riggs BJ, Ross CE, McKiernan CA, Heidemann SM, Kleinman LC, Sen AI, Hall MW (2020) Characteristics and outcomes of children with coronavirus disease 2019 (COVID-19) infection admitted to US and Canadian pediatric intensive care units. JAMAPediatr 174(9):868–873. https://doi.org/10.1001/jamapediatrics.2020.1948
Tang JW, Tambyah PA, Hui DS (2020) Emergence of a new SARS-CoV-2 variant in the UK. J Infect. https://doi.org/10.1016/j.jinf.2020.12.024
Tegally H, Wilkinson E, Giovanetti M, Iranzadeh A, Fonseca V, Giandhari J, Doolabh D, Pillay S, San EJ, Msomi N (2020) Emergence and rapid spread of a new severe acute respiratory syndrome-related coronavirus 2 (SARS-CoV-2) lineage with multiple spike mutations in South Africa. medRxiv. https://doi.org/10.1101/2020.12.21.20248640
Trott O, Olson AJ (2010) AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem 31(2):455–461. https://doi.org/10.1002/jcc.21334
Varshney KK, Varshney M, Nath B (2020) Molecular Modeling of Isolated Phytochemicals from Ocimum sanctum Towards Exploring Potential Inhibitors of SARS Coronavirus Main Protease and Papain-Like Protease to Treat COVID-19. SSRN 3554371. https://ssrn.com/abstract=3554371
Volz E, Mishra S, Chand M, Barrett JC, Johnson R, Geidelberg L, Hinsley WR, Laydon DJ, Dabrera G, O’Toole Á (2021) Transmission of SARS-CoV-2 Lineage B. 1.1. 7 in England: Insights from linking epidemiological and genetic data. medRxiv. https://doi.org/10.1101/2020.12.30.20249034
Wang C, Horby PW, Hayden FG, Gao GF (2020) A novel coronavirus outbreak of global health concern. Lancet 395(10223):470–473. https://doi.org/10.1016/S0140-6736(20)30185-9
Xie X, Liu Y, Liu J, Zhang X, Zou J, Fontes-Garfias CR, Xia H, Swanson KA, Cutler M, Cooper D (2021) Neutralization of SARS-CoV-2 spike 69/70 deletion, E484K and N501Y variants by BNT162b2 vaccine-elicited sera. Nat Med. https://doi.org/10.1038/s41591-021-01270-4
Yang L, Wen K-S, Ruan X, Zhao Y-X, Wei F, Wang Q (2018) Response of plant secondary metabolites to environmental factors. Molecules 23(4):762. https://doi.org/10.3390/molecules23040762
Zhang H, Penninger JM, Li Y, Zhong N, Slutsky AS (2020) Angiotensin-converting enzyme 2 (ACE2) as a SARS-CoV-2 receptor: molecular mechanisms and potential therapeutic target. Intensive Care Med 46(4):586–590. https://doi.org/10.1007/s00134-020-05985-9
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Dutta, T., Baildya, N., Khan, A.A. et al. Inhibitory effect of anti-HIV compounds extracted from Indian medicinal plants to retard the replication and transcription process of SARS-CoV-2: an insight from molecular docking and MD-simulation studies. Netw Model Anal Health Inform Bioinforma 10, 32 (2021). https://doi.org/10.1007/s13721-021-00309-3
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s13721-021-00309-3