
Abstract
Five undescribed alkaloids were isolated from the seeds of Cephalotaxus oliveri along with 27 known ones. The new structures were elucidated based on spectroscopic data including 1D and 2D NMR, MS and calculated ECD spectra. Among them, (+)-acetylcephalofortine C was an enantiomeric Cephalotaxine alkaloids. The performed bioassay revealed that those alkaloids were not cytotoxic against cancer cells and had no neuroprotective properties in the HEI-OC-1 cells model.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
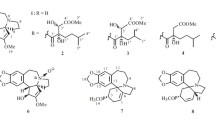
The cephalotaxine-type alkaloid was firstly found to possess inhibitory activities against several human tumor cell lines, especially the leukemia cell line [1, 2], consistent with Chinese folk usage. After immense effort of clinical researches, China first approved homoharringtonine as a drug in acute non-lymphocytic leukemia, myelodysplastic syndrome, chronic myelocytic leukemia and polycythemia vera in 1970’s [3, 4]. Recently, homoharringtonine was approved in 2012 by the Food and Drug Administration for the treatment of chronic myeloid leukemia [5]. Plants of the genus Cephalotaxus (Cephalotaxaceae) were the only resource of this type of natural products. So the alkaloids from Cephalotaxus species have attracted great attention from scientists [6, 7]. Till now, stems and leaves of Cephalotaxus have been carried out. Seeds of this genus were also important resources, for example, homoharringtonine was first found from seeds of C. harringtonia [1]. However, studies of alkaloids in seeds are virgin lands compared to that of stems and leaves. Over the past 60 years, more than 100 alkaloids in Cephalotaxus have been reported, which were mainly classified into homoerythrina and the cephalotaxine-types according to their skeleton features [8, 9]. In order to exploit the structure diversity and enrich the library of Cephalotaxus alkaloids, phytochemical investigations have been made on the seeds of C. oliveri. As a result, five new alkaloids, together with 27 known ones (Fig. 1) were isolated from the seeds of C. oliveri. Here we reported the isolation, structure elucidation of those alkaloids.

Structures of alkaloids from seeds of C. oliveri
2 Results and discussion
Newly isolates (1–5) probably belong to alkaloids as they exhibited a positive reaction with Dragendorff’s reagent. Alkaloid 1 was obtained as a white powder. Its molecular formula was determined as C17H19NO5 from HRESIMS m/z = 318.1338 [M+H]+ (Additional file 1: S5). The 1H and 13C NMR spectroscopic data (Table 1, Additional file 1: S1~S2) showed the occurrence of two aromatic protons (δH 6.71, 6.92, each s) at para-position, a methylenedioxy moiety (δH 5.88, 5.87), two O-bearing CH groups (δH 4.15, δC 76.2; δH 3.72, δC 83.0) and a ketal carbon (δC 98.7). The 1H and 13C NMR patterns of 1 were very similar to those of the known alkaloid cephalotine A (17) [10] with the exception for somewhat differences in chemical shifts values between both alkaloids. The same molecular formulas of both alkaloids assigned their isomeric relationship. H-3 (δH 3.72), H-10 (δH 2.70, 2.51), and H-17 (δH 6.92) showed the HMBC correlations to the ketal carbon (δC 98.7), allowing the ketal carbon at C-11 other than C-3 in 17 (Fig. 2, Additional file 1: S4). Configuration of H-2 was established as β-orientation on the basis of the coupling constant (d, J = 6.6 Hz) of H-2, which was same as that of 17. Consequently, the structure of 1 was confirmed as shown in Fig. 1, and named isocephalotine A.

Key HMBC correlations of alkaloids 1, 3 and 4
Alkaloid 2 was obtained as a white powder. Its molecular formula C17H19NO5 was same as 1 from its HRESIMS m/z 318.1342 [M+H]+ (Additional file 1: S11). The 1H and 13C NMR data of 2 and 1 (Table 1, Additional file 1: S7~S8) were almost same with exception for 1H and 13C NMR spectroscopic data of C-1/2/3 between both compounds, suggesting they shared the same planar structure. Obvious differences between both alkaloids were the highfield signals of C-1 (δC 34.5, ΔδC − 2.3), C-2 (δC 74.6, ΔδC − 1.6) and C-3 (δC 79.1, ΔδC − 3.9) in 2, indicating α-configuration of H-2 in 2 as in cephalotine B [10], which was confirmed by the ROESY correlation from H-2 (δH 4.09) to H-4 (δH 3.08) (Additional file 1: S10). Thus, 2 was established as shown in Fig. 1, and named isocephalotine B.
Alkaloid 3 was obtained as a white powder. Its molecular formula was determined as C17H17NO5 from HRESIMS m/z = 316.1177 [M+H]+ (Additional file 1: S18). Alkaloid 3 displayed similar 1H and 13C NMR data (Table 1, Additional file 1: S13-S14) to the known alkaloid demethylcephalotaxinone (12), except that a methylene in 12 was substituted by a methine (δC 70.6, δH 4.90) in 3. In addition, the HMBC correlations from the methine (δH 4.90) to C-10 (δC 52.2), C-12 (δC 136.1), C-13 (δC 125.1), and C-17 (δC 111.5) located the methine at C-11 (Fig. 2, Additional file 1: S16). H-11 was assigned as α-orientated based on the ROESY correlation of H-11 with H-6 (δH 1.65) and H-8 (δH 2.76) (Fig. 3, Additional file 1: S17). Thus, the structure of 3 was established as shown in Fig. 1, and named as 11β-hydroxydemethylcephalotaxinone.

Key ROESY correlations of 3
Alkaloid 4 was obtained as a white powder. Its molecular formula was determined as C20H25NO5 from HRESIMS m/z = 360.1807 [M+H]+ (Additional file 1: S23). The 1H NMR data (Table 1, Additional file 1: S20) were very similar to those of the known alkaloid cephalofortine C [11], with exception for the downfield H-3 signal (δH 5.70, ΔδC + 0.97) and a methyl signal (δH 1.41). Additionally, compared to cephalofortine C, 4 had an acetyl group in its 13C NMR (δC 20.4, 171.9). The key HMBC correlations from both H-3 (δH 5.70) and CH3 (δH 1.41) to carbonyl carbon (δC 171.9) suggested the existence of the acetyl group (Additional file 1: S22). Thus the gross structure of alkaloid 4 was elucidated as shown in Fig. 2. The large coupling constant of H-3/4 (δH 5.70, d, J = 9.2 Hz) assigned the configurations of H-3 and H-4 as those of cephalotaxine [12]. However, like to (+)-acetylcephalotaxine ([α]D + 102) rather than (−)-acetylcephalotaxine (\([\alpha ]_{{\text{D}}}^{25}\) − 97) [13], 4 might be also assigned to (+)-form based on its specific rotation value (\([\alpha ]_{{\text{D}}}^{23.0}\) + 107.2). Finally, the absolute configuration of 4 was determined by the time-dependent density functional theory ECD calculation. The calculated ECD spectrum of (3R,4R,5S)-4 was consistent with the experimentally observed ECD spectrum (Fig. 4) (Additional file 1: S32). As a result, the absolute configuration of compound 4 was defined as 3R,4R,5S. Thus, the structure of alkaloid 4 was elucidated as shown in Fig. 1, and named as (+)-acetylcephalofortine C (4).

Experimental and calculated ECD spectra of 4
Alkaloid 5 was obtained as a colorless crystal. Analysis of the 1H and 13C NMR spectra (Table 1, Additional file 1: S25~S26) indicated the presence of following signals, a 1,2,4,5-tetra substituted benzenoid ring with a methylenedioxy substitution (δH 6.70, 6.66, 5.83, 5.81), five methylenes (δC 40.9, 19.9, 53.7, 48.9, 29.7), three methines (δC 93.7, 73.9, 55.8), two quaternary carbons (δC 77.6, 168.2) and one methoxy (δC 58.3), which were agreed well with signals of cephalotaxine. Its molecular formula C18H21NO4 from HRESIMS m/z 316.1549 [M+H]+ (Additional file 1: S29) was also same with that of cephalotaxine. The rest of C-1′ (δC 128.9), C-2′ (δC 132.4, δH 7.71), C-3′ (δC 115.3, δH 6.65), C-4′ (δC 161.2) and a carbonyl group (δC 174.6) signals revealed a p-hydroxybenzoic acid structure, which was supported by the HMBC correlations from H-2′ to C-3′, C-4′, the carbonyl group and from H-3′ to C-4′ (Fig. 2, Additional file 1: S28). Its molecular formula C7H6O3 was revealed by the HRESIMS m/z = 137.0238 [M−H]− (Additional file 1: S30). Carefully analyzing the 13C NMR spectra data of 5, revealed the upfield signals of C-1 (δC 93.7), C-4 (δC 55.8), C-6 (δC 40.9) and the downfield signal of C-5 (δC 77.6). These changes were consistent with the 13C NMR signals of cephalotaxine ester salts [14], which means compound 5 was a p-hydroxybenzoate salt of cephalotaxine. The specific rotation (\([\alpha ]_{{\text{D}}}^{23.5}\) − 216.4) of 5 was close to that of (−)-cephalotaxine (\([\alpha ]_{{\text{D}}}^{25}\) − 204). Thus, the structure of 5 was established as (−)-cephalotaxine p-hydroxybenzoate.
The known alkaloids were identified as 11-hydroxycephalotaxine (6) [15], cephalotaxine (7) [16], acetylcephalotaxine (8) [17], drupacine (9) [17], 3-deoxy-3,11-epoxy- cephalotaxine (10) [18], drupacinamide (11) [19], demethylcephalotaxinone (12) [20], cephalotaxinone (13) [21], 11β-hydroxycephalotaxine N-oxide (14) [22], cephalotine D (15) [10], cephalancetine C (16) [23], cephalotine A (17) [10], cephalotine B (18) [10], cephalocyclidin A (19) [24], cephalofortunone (20) [25], deoxyharringtonine (21) [2], isoharringtonine (22) [1, 26], 3'-R-isoharringtonine (23) [26], harringtonine (24) [1], deoxyharringtonic acid (25) [13], 5'-des-O-methylisoharringtonine (26) [13], 5'-des-O-methylharringtonine (27) [27], nordeoxyharringtonine (28) [28], epischellhammericine B (29) [29], 3-epicomosine (30) [30], 3-epischellhammericine (31) [29], taxodinea (32) [31] by comparison with literatures.
In biological activity research, none of these new compounds showed cytotoxic activity against HeLa, SGC-7901 gastric cancer, and A-549 lung cancer cell lines (IC50 > 20 µM). Additionally, unlike homologous Erythrina alkaloids [32], all of them also had no neuroprotective effects on HEI-OC-1 cells model.
Alkaloids 1–5 were undescribed compounds. Interestingly, the 4 was assigned as rare (+)-form enantiomer. This was the first to report alkaloids from the seeds of C. oliveri.
3 Experimental section
3.1 General experimental procedures
Optical rotations were measured with a RUDOLPH APVI-6 automatic polarimeter. UV spectra were recorded on a Shimadzu 2401A spectrophotometer. 1D and 2D NMR spectra were acquired Bruker AVANCE III-600 MHz spectrometers with SiMe4 as an internal standard. MS data were obtained using a Shimadzu UPLC-IT-TOF (Shimadzu Corp., Kyoto, Japan). Column chromatography (CC) was performed on either silica gel (200–300 mesh, Qing-dao Haiyang Chemical Co., Ltd., Qingdao, China) or RP-18 silica gel (20–45 um, Fuji Silysia Chemical Ltd., Japan). Fractions were monitored by TLC on silica gel plates (GF254, Qingdao Haiyang Chemical Co., Ltd., Qingdao, China), and spots were visualized with Dragendorff’s reagent spray. MPLC was performed using a Buchi pump system coupled with RP-18 silica gel-packed glass columns (19 × 480, 40 × 480, 45 × 480, and 55 × 480 mm, respectively). HPLC was performed using Waters 1525EF pumps coupled with analytical semi-preparative or preparative Xbridge C18 columns (4.6 × 150 and 19 × 250 mm, respectively). The HPLC system employed a Waters 2998 photodiode array detector and a Waters fraction collector III.
3.2 Plant materials
Seeds of Cephalotaxus oliveri Mast. (Cephalotaxaceae) were collected in October 2013 in Yunnan Province, P. R. China and identified by Xiang-hai Cai. The voucher specimen (cai20131002) was preserved in the State Key Laboratory of Phytochemistry and Plant Resources in West China, Kunming Institute of Botany, Chinese Academy of Sciences.
3.3 Extraction and isolation
The air-dried and powdered seeds of C. oliveri (14 kg) were extracted with MeOH at room temperature, and the solvent was evaporated in vacuo. The crude extract was suspended in HCl (1%) and partitioned with Petroleum ether and EtOAc. Then the acidic layer was adjusted to pH 7–8 with 5% ammonia solution and subsequently extracted with EtOAc to obtain crude alkaloid extract (45 g). The crude alkaloids was subjected to CC over silica gel (400 g) and eluted with a CHCl3–MeOH gradient (1:0 to 0:1, v/v) to give five fractions (I–V) based on TLC analysis.
Fraction I (10 g) was subjected to C18 MPLC with MeOH–H2O (1:9 to 100:0, v/v) as the eluent to obtain eight fractions (I-1 ~ 8). Subfraction I-1 ~ 2 were identified as 9 (4.5 g) by 1H-NMR. Subfraction I-3 was purified by a Sephadex LH-20 column and eluted with MeOH to obtain two fractions (I-3-1 ~ 2). Subfraction I-3-1 was then separated on a preparative C18 column with a gradient CH3CN–H2O (1:4 to 7:13, v/v) to afford 25 (3.6 mg), 26 (11.5 mg). Subfraction 1-3-2 was separated on a preparative C18 column with a gradient CH3CN–H2O (3:7 to 9:11, v/v) to afford 20 (1 mg); Subfraction I-4 was purified by a Sephadex LH-20 column and eluted with MeOH to obtain 22 (23 mg). Subfraction I-5 was purified by a Sephadex LH-20 column (eluted with MeOH) and then separated on a preparative C18 column with a gradient CH3CN–H2O (3:7 to 9:11, v/v) to afford 8 (41.3 mg). Subfraction 1–6 was identified as 22 (1 g) by 1H-NMR. Subfraction I-7 was purified by a Sephadex LH-20 column (eluted with MeOH) and then separated on a preparative C18 column with a gradient CH3CN–H2O (9:11 to 3:2, v/v) to afford 28 (21 mg) and 31 (66.3 mg). Subfraction I-8 was identified as 21 (2.5 g) by 1H-NMR.
Fraction II (2 g) was subjected to CC over silica gel (20 g) and eluted with a CHCl3–MeOH gradient (1:0 to 0:1, v/v) to give two fractions (II-1 ~ 2). Subfraction II-1 was purified by a Sephadex LH-20 column and eluted with MeOH to obtain two fractions (II-1-1 ~ 2). Subfraction II-1-1 was identified as 22 (0.6 g) by 1H-NMR. Subfraction II-1-2 was subjected to C18 MPLC with MeOH–H2O (1:9 to 100:0, v/v) as the eluent and then separated on a preparative C18 column with a gradient CH3CN–H2O (1:4 to 7:13, v/v) to afford 11 (1.4 mg), 16 (2 mg), 29 (24.6 mg), 30 (15.7 mg). Subfraction II-2 was purified by a Sephadex LH-20 column and eluted with MeOH to obtain 7 (93.8 mg), the rest part was then separated on a preparative C18 column with a gradient CH3CN–H2O (1:4 to 7:13, v/v) to afford 26 (11.5 mg).
Fraction III (1 g) was subjected to C18 MPLC with MeOH–H2O (1:9 to 100:0, v/v) as the eluent to obtain three fractions (III-1 ~ 3). Subfraction III-1 was identified as 7 (90.0 mg) by 1H-NMR. Subfraction III-2 was identified as 22 (162 mg) by 1H-NMR. Subfraction III-3 was purified by a Sephadex LH-20 column and eluted with MeOH to obtain two fractions (III-3-1 ~ 2). Subfraction III-3-1 was then separated on a preparative C18 column with a gradient CH3CN–H2O (1:4 to 7:13, v/v) to afford 26 (8 mg). Subfraction III-3-2 was then separated on a preparative C18 column with a gradient CH3CN–H2O (9:11 to 3:2, v/v) to afford 23 (23.1 mg).
Fraction IV (0.7 g) was subjected to C18 MPLC with MeOH–H2O (1:9 to 100:0, v/v) as the eluent to obtain three fractions (IV-1 ~ 3). Subfraction IV-1 was purified by a Sephadex LH-20 column and eluted with MeOH to obtain 7 (550 mg). Subfraction IV-2 was purified by a Sephadex LH-20 column (eluted with MeOH) and then separated on a preparative C18 column with a gradient CH3CN–H2O (2:3 to 11:9, v/v) to afford 24 (51.5 mg). Subfraction IV-3 was purified by a Sephadex LH-20 column to gain 7 (40 mg).
Fraction V (25 g) was subjected to C18 MPLC with MeOH–H2O (1:19 to 100:0, v/v) as the eluent to obtain four fractions (V-1 ~ 4). Subfraction V-1 was purified by C18 MPLC with MeOH–H2O (1:19 to 100:0, v/v) as the eluent to obtain three fractions (V-1-1 ~ 3). Subfraction V-1-1 separated on a preparative C18 column with a gradient CH3CN–H2O (1:9 to 1:3, v/v) to afford 19 (4.7 mg), 18 (1 mg) and 17 (2.2 mg); Subfraction V-1-2 was then purified by a Sephadex LH-20 column and eluted with MeOH to obtain 5 (1.5 g). Subfraction V-1-3 was then purified by a Sephadex LH-20 column and eluted with MeOH to obtain 7 (7.3 g). Subfraction V-2 was purified by C18 MPLC with MeOH–H2O (1:19 to 100:0, v/v) as the eluent and then separated on a preparative C18 column with a gradient CH3CN–H2O (3:7 to 9:11, v/v) to afford 10 (1.1 mg). Subfraction V-3 was subjected to C18 MPLC with MeOH–H2O (1:19 to 100:0, v/v) as the eluent to obtain five fractions (V-3-1 ~ 5). Subfraction V-3-1 was purified by a Sephadex LH-20 column and eluted with MeOH to obtain two fractions (V-3-1-1 ~ 2). Subfraction V-3-1-1 was identified as 10 (560.9 mg). Subfraction V-3-1-2 was then separated on a preparative C18 column with a gradient CH3CN–H2O (3:17 to 3:7, v/v) to afford 1 (1.9 mg), 2 (5.5 mg) and 13 (0.5 mg). Subfraction V-3-2 was purified by a Sephadex LH-20 column and eluted with MeOH to obtain five fractions (V-3-2-1 ~ 5). Subfraction V-3-2-1 was then separated on a preparative C18 column with a gradient CH3CN–H2O (1:3 to 2:3, v/v) to afford 4 (4.6 mg). Subfraction V-3-2-2 was then separated on a preparative C18 column with a gradient CH3CN–H2O (3:7 to 9:11, v/v) to afford 32 (3.9 mg), 15 (10.4 mg). Subfraction V-3-2-3 was identified as 12 (55.1 mg) by 1H-NMR. Subfraction V-3-2-4 was separated on a preparative C18 column with a gradient CH3CN–H2O (1:3 to 2:3, v/v) to afford 3 (7.6 mg), 12 (13.0 mg). Subfraction V-3-3 was purified by a Sephadex LH-20 column and eluted with MeOH and separated on a preparative C18 column with a gradient CH3CN–H2O (1:3 to 2:3, v/v) to obtain 14 (2.5 mg) and 32 (5.6 mg). Subfraction V-3-4 was purified by a Sephadex LH-20 column and eluted with MeOH and separated on a preparative C18 column with a gradient CH3CN–H2O (7:13 to 1:1, v/v) to obtain 6 (302 mg). Subfraction V-3-5 was purified by a Sephadex LH-20 column and eluted with MeOH to obtain 26 (68.8 mg). Subfraction V-4 was purified by C18 MPLC with MeOH–H2O (1:19 to 100:0, v/v) as the eluent to obtain two fractions (V-4 ~ 1–2). Subfraction V-4-1 was separated on a preparative C18 column with a gradient CH3CN–H2O (1:3 to 2:3, v/v) to obtain 27 (6.3 mg). Subfraction V-4-2 was purified by a Sephadex LH-20 column and eluted with MeOH to obtain 24 (98.4 mg).
Isocephalotine A (1): White powder; C17H19NO5; \([\alpha ]_{{\text{D}}}^{23.2}\) − 30.67 (c 0.03, CH3OH); UV (CH3OH) λmax (logε): 203.6 (3.77), 228.4 (3.06), 289.2 (2.95) (Additional file 1: S6); 1H (600 MHz) and 13C (150 MHz) NMR data (methanol-d4), see Table 1; positive HRESIMS m/z 318.1338 [M+H]+ (calcd. for C17H20NO5, 318.1342).
Isocephalotine B (2): White powder; C17H19NO5; \([\alpha ]_{{\text{D}}}^{23.4}\) − 26.0 (c 0.11, CH3OH); UV (CH3OH) λmax (logε): 203.6 (3.08), 231.2 (3.30), 290.0 (3.08) (Additional file 1: S12); 1H (500 MHz) 13C (125 MHz) NMR data (methanol-d4), see Table 1; positive HRESIMS m/z 318.1342 [M+H]+ (calcd. for C17H20NO5, 318.1342).
11β-Hydroxydemethylcephalotaxinone (3): White powder; C17H17NO5; \([\alpha ]_{{\text{D}}}^{22.7}\) 250.5 (c 0.04, CH3OH); UV (CH3OH) λmax (logε): 202.8 (3.48), 251.4 (3.03), 298.8 (3.15) (Additional file 1: S19); 1H (600 MHz) and 13C (150 MHz) NMR data (methanol-d4), see Table 1; positive HRESIMS m/z 316.1177 [M+H]+ (calcd. for C17H18NO5, 316.1179).
(+)-Acetylcephalofortine C (4): White powder; C20H25NO5; \([\alpha ]_{{\text{D}}}^{23.0}\) 107.2 (c 0.05, CH3OH); UV (CH3OH) λmax (logε): 204.2 (3.94), 226.4 (3.23), 283.8 (2.79) (Additional file 1: S24); 1H (600 MHz) and 13C (150 MHz) NMR data (methanol-d4), see Table 1; positive HRESIMS m/z 360.1807 [M+H]+ (calcd. for C20H26NO5, 360.1811).
Cephalotaxine P-hydroxybenzoate (5): \([\alpha ]_{{\text{D}}}^{23.5}\) − 216.4 (c 0.05, CH3OH); UV (CH3OH) λmax (log ε): 203.6 (4.17), 246.2 (3.45), 286.2 (3.08) (Additional file 1: S31); 1H (500 MHz) and 13C (125 MHz) NMR data (methanol-d4) δC: 128.9 (s, C-1′), 132.4 (d, C-2′/6′), 115.3 (d, C-3′/5′), 161.2 (d, C-4′), 174.6 (s, C-7′), other data see Table 1; positive HRESIMS m/z 316.1549 [M+H]+ (calcd. for C18H22NO4, 316.1549); negative HRESIMS m/z 137.0238 [M+H]+ (calcd. for C7H5NO3, 137.0239).
3.4 Cytotoxicity assay
The cytotoxicity of compounds 1–5 was tested by the MTS assay. The cells were seeded into 96-well tissue culture dishes at 5 × 103 cells/well for SMMC-7721, HT-29 and A-549 and cultured overnight at 37 °C in a 5% CO2 incubator for cell adhesion. Cells were then incubated in culture medium with each compound for 48 h. The MTS-reducing activity was evaluated by measuring the absorbance at 490 nm using the CellTiter 96 Aqueous One Solution Cell Proliferation Assay kit (Promega, USA) and an Infinite M200 Pro (Tecan, Austria) microplate reader. IC50 values were calculated using Reed–Muench method.
3.5 Protecting hearing loss assay
HEI-OC1 cells were seeded into 96-well tissue culture dishes at 5 × 103 cells/well and cultured overnight in DMEM medium supplemented with 10% FBS and 50 µg/mL ampicillin. The cells were then incubated with different doses of compounds in DMEM without FBS for 12 h before neomycin exposure. After this pre-treatment, the experimental group was treated with 20 mM neomycin together with compounds, the neomycin-only group was treated with 20 mM neomycin and an equivalent volume of DMSO, and the control group was treated with equivalent volume of DMSO without neomycin or compounds. After another 24 h of culture, the cells were thoroughly washed with PBS and cultured in DMEM with ampicillin for an additional 12 h recovery. The cell viability was then measured using the CellTiter 96 AQueous One Solution Cell Proliferation Assay kit (Promega, USA).
References
Powell RG, Weisleder D Jr, Smith CR, Rohwedder WK. Structures of harringtonine, isoharringtonine, and homoharringtonine. Tetrahedron Lett. 1970;11:815.
Mikolajczak KL, Powell RG Jr, Smith CR. Deoxyharringtonine, a new antitumor alkaloid from Cephalotaxus: structure and synthetic studies. Tetrahedron. 1972;28:1995–2001.
[No authors listed]. Cephalotaxine esters in the treatment of acute leukemia. A preliminary clinical assessment. Chin Med J (Engl). 1976;2:263–72.
Zhang ZY, Hou CH, Zhu YF. A preliminary therapeutic analysis of 82 cases of chronic granulocytic leukemia treated with harringtonine. Chin J Intern Med. 1986;25:156–7.
Kantarjian HM, O’Brien S, Cortes J. Homoharringtonine/omacetaxine mepesuccinate: the long and winding road to Food and Drug Administration Approval. Clin Lymph Myelom Leuk. 2013;13:530.
Mei WL, Wu J, Dai HF. Advances in studies on chemical constituents in plants of Cephalotaxus Sieb. et Zucc. and their pharmacological activities. Chin Tradit Herb Drugs. 2006;37:452–8.
Perard-Viret J, Quteishat L, Alsalim R, Royer J, Dumas F. Cephalotaxus alkaloids. Alkaloids Chem Biol. 2017;78:205.
Abdelkafi H, Nay B. Natural products from Cephalotaxus sp.: chemical diversity and synthetic aspects. Nat Prod Rep. 2012;29:845.
Kobayashi J, Yoshinaga M, Yoshida N, Shiro M, Morita H, Cephalocyclidin A. a novel pentacyclic alkaloid from Cephalotaxus harringtonia var nana. J Org Chem. 2002;67:2283.
Ni L, Zhong XH, Cai J, Bao MF, Zhang BJ, Wu J, Cai XH. Five new alkaloids from Cephalotaxus lanceolata and C. fortunei var alpina. Nat Prod Bioprospect. 2016;6:149–54.
Zhu L, Gong LJ, Zhu DR, Zhu JM, Li Y, Kong LY, Luo JG. Cephalotaxine-type alkaloids from the seeds of Cephalotaxus fortunei and their cytotoxic activities. Phytochemistry. 2021;191:112903.
Planas L, Perard-Viret J, Royer J. Tereoselective synthesis of (−)-cephalotaxine and C-7 alkylated analogues. J Org Chem. 2004;69:3087–92.
Xue Z, Xu LZ, Chen DH, Huang L. Studies on the minor alkaloids of Cephalotaxus Hainanensis Li. Acta Pharm Sin. 1981;16:755.
Robin JP, Radosevic N, Blan-chard J, Roisnel T, Bataille T. Water soluble crystalline salts of harringtonines protonated on alkaloid nitrogen and use as chemotherapeutics. U.S., Patent PCT/EP/2014/079456 July 2015.
Powell RG, Madrigal RV Jr, Smith CR, Mikolajczak KL. Alkaloids of Cephalotaxus harringtonia var drupacea: 11-hydroxycephalotaxine and drupacine. J Org Chem. 1974;39:676.
Powell RG, Weisleder D, Smith C, Wolff I. Structure of cephalotaxine and related alkaloids. Tetrahedron Lett. 1969;10:4081.
Powell RG, Weisleder D, Smith C. Antitumor alkaloids from Cephalotaxus–Harringtonia-structure and activity. J Pharm Sci. 1972;61:1227.
Manivannan R. Isolation and characterizations of new alkaloid 3-deoxy-3, 11-epoxy cephalotaxine from Clitoria ternatea. J Drug Deliv Ther. 2019;9:458–62.
Wu KM, Ji XJ, Ye XR. Preparation of new harringtonine derivative and its application as medical composition. China, patent CN1463975, 2003.
Wang LW, Su HJ, Yang SZ, Won SJ, Lin CL. New alkaloids and a tetraflavonoid from Cephalotaxus wilsoniana. J Nat Prod. 2004;67:1182–5.
Eckelbarger JD, Wilmot JT, Gin DY. Strain-release rearrangement of N-vinyl-2-arylaziridines. Total synthesis of the anti-leukemia alkaloid (-)-deoxyharringtonine. J Am Chem Soc. 2006;128:10370–1.
Bocar M, Jossang A, Bodo B. New alkaloids from Cephalotaxus fortune. J Nat Prod. 2003;66:152–4.
He YR, Shen YH, Li B, Li B, Lu L, Tian JM, Zhang WD. Alkaloids from Cephalotaxus lanceolata and their cytotoxicities. Chem Biodivers. 2013;10:584–95.
Kobayashi J, Yoshinaga M, Yoshida N, Shiro M, Morita H, Cephalocyclidin A. Cephalocyclidin A, a novel pentacyclic alkaloid from Cephalotaxus harringtonia var. nana. J Org Chem. 2002;67:2283–6.
Li HX, Wen YH, Wang FF, Wu P, Wei XY. Cephalofortunone, a structurally unique Cephalotaxus alkaloid from Cephalotaxus fortune Hook f. Tetrahedron Lett. 2015;56:5735–7.
Li YN, Wu KM, Huang L. Synthesis of isoharringtonine and separation of its isomers. Acta Pharm Sin. 1984;19:582–9.
Takano I, Yasuda I, Nishijima M, Hitotsuyanagi Y, Takeya K, Itokawa H. Alkaloids from Cephalotaxus harringtonia. Phytochemistry. 1996;43:299–303.
Takano I, Yasuda I, Nishijima M, Hitotsuyanagi Y, Takeya K, Itokawa H. New Cephalotaxus alkaloids from Cephalotaxus harringtonia var drupacea. J Nat Prod. 1996;59:965–7.
Powell RG. Structures of homoerythrina alkaloids from Cephalotaxus harringtonia. Phytochemistry. 1972;11:1467–72.
Aladsesanmi AJ, Snyder JK, Kelley CJ, Hoffmann JJ. Homoerythrina alkaloids of Phelline comosa. Phytochemistry. 1991;30:3497–8.
Langlois N. New homoerythrinane alkaloids from Phelline species. Heterocycles. 1990;30:659–64.
Tang YT, Wu J, Yu Y, Bao MF, Tan QG, Schinnerl J, Cai XH. Colored dimeric alkaloids from the barks of Erythrina variegata and their neuroprotective effects. J Org Chem. 2021;86:13381–7.
Acknowledgements
This project was funded in part by the National Natural Science Foundation of China (No. 31872677).
Author information
Authors and Affiliations
Contributions
GY carried out the isolation and identification experiment and drafted the original. JW performed bioassay experiment. BS completed the ECD calculation. MB help to provide various experimental reagents. XC contributed to the conception, methodology, review and funding acquisition. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1:
Enantiomeric Cephalotaxus Alkaloids from Seeds of C. oliveri. General NMR, HRESIMS, UV and ECD spectra of compound 1-5 and computational methods for the ECD of compound 4.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Yu, GX., Wu, J., Shi, BB. et al. Enantiomeric Cephalotaxus alkaloids from seeds of Cephalotaxus oliveri. Nat. Prod. Bioprospect. 12, 24 (2022). https://doi.org/10.1007/s13659-022-00344-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s13659-022-00344-1