
Abstract
Five new alkaloids (1–5) were isolated from the leaves and twigs of Cephalotaxus lanceolata and C. fortunei var. alpina along with 24 known alkaloids. The new structures were elucidated based on spectroscopic data including 1D and 2D NMR, FTIR, UV and MS. These new alkaloids showed no cytotoxicity to HeLa, SGC-7901 gastric cancer, and A-549 lung cancer cell lines.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
1 Introduction
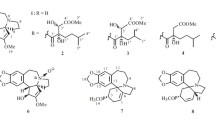
Various constituents of Cephalotaxus genus have been reported, including alkaloids [1–6], tropones [7–10], lignans [10, 11], diterpenes [9], flavonoids [6, 10]. Previous investigations led to approximate 100 Cephalotaxus alkaloids, which were mainly classified into two structural types, i.e., homoerythrina and cephalotaxine-type, and the latter demonstrated remarkable antitumor activities [12]. For example, homoharringtonine among cephalotaxine alkaloids was successfully used to treat acute leukemia. As for homoharringtonine, the side chains played an important role in the anticancer activity of these compounds which possessed H-3 α-configuration. So far only reported cephalezomines G possessed H-3 β-configuration. Both homoerythrina and cephalotaxine had same biogenetic origin. However, most of homoerythrinas almost with H-3 α-configuration reminded us that there were more cephalotaxines with same configuration. As a part of our continuous research for Cephalotaxus alkaloids, five new alkaloids, together with 24 known ones (Fig. 1) were isolated from leaves and twigs of C. lanceolata and C. fortunei var. alpina. The known alkaloids were identified as drupacine (6) [2], cephalotaxinone (7) [13], acetycephalotaxine (8) [14], cephalezomine J (9) [5], desmethylcephalotaxine (10) [15], isocephalotaxinone (11) [16], 1l-hydroxycephalotaxin (12) [2], cephalotaxine (13) [17], lucidinine (14) [18], comosidine (15) [18], schelhammeridine (16) [19], 3-epischelhammeridine (17) [20], comosine (18) [21], 3-epicomosine (19) [20], 3-epischelhammericine (20) [20], fortunine (21) [22], taxodine (22) [23], O-methylschlammericine (23) [13], cephalezomine M (24) [5], homoisoharringtonine (25) [24], homoharingtonine (26) [25], isoharringtonine (27) [25, 26], epidesoxyharringtonine (28) [27], desoxyharringtonine (29) [28] by comparison with literatures.

Structures of alkaloids from C. lanceolata and C. fortunei var. alpine
2 Results and Discussion
Newly isolates (1–5) probably belong to alkaloids as they exhibited a positive reaction with Dragendorff’s reagent. Alkaloid 1 was isolated as white powder. Its UV absorption bands at 203 and 291 nm and IR absorption bands at 3520, 3406, 1631, 1500, 1482, 1342 cm−1 were consistent with those of Cephalotaxus alkaloids [2]. Analysis of the 1H and 13C NMR data of 1 (Tables 1, 2) revealed several typical functionalities similar to those of the known alkaloid drupacine (6) [2], including a tetrasubstituted benzene ring with two para H-atoms (δ H 6.76, δ C 110.1; δ H 6.72, δ C 106.0; δ C 128.6, 132.4, 146.8, 147.4), a –OCH2O– moiety (δ H 5.97; δ C 101.5), a ketal carbon (δ C 106.7), two O-bearing CH groups (δ H 3.86, δ C 76.7; δ H 4.81, δ C 76.1), and two –OH groups (δ H 3.53 and 4.68). The molecular formula of 1 was established as C17H19NO5 with nine degrees of unsaturation by HRESIMS ([M+H]+ at m/z 318.1336), absence of a methyl than that of 6. The HMBC correlations (Fig. 2) of the methine signal (δ H 4.81) with C-12 (δ C 132.4), C-13 (δ C 128.6), and C-17 (δ C 106.0) allowed its position as C-11. Likewise, the other signal δ H 3.38 was assigned to CH-4 based on its HMBC correlations with δ C 110.1 (C-14), C-12 and δ C 39.2 (C-6). The obvious HMBC correlation between methylene protons (δ H 1.37 and 2.23) with C-6 and C-3 attributed it to C-1. The proton signal δ H 3.86 was assigned to H-2 based on its correlation with δ H 2.23 in the 1H–1H COSY (Fig. 2) spectrum. The ketal carbon (δ C 106.7) was located at C-3 by its HMBC correlations from H-1, 2 and 4. The HMBC crosspeak of H-11/C-3 showed an oxygen bridge between C-11/C-1 in 1 consistent with its degrees of unsaturation. H-2 was established as β-orientation on the basis of the coupling constant (d, J = 6.4 Hz) of H-2. Consequently, the structure of 1 was confirmed as shown in Fig. 1, and named cephalotine A.

Key 1H–1H COSY ( ) and HMBC (
) and HMBC ( ) correlations of compound 1. (Color figure online)
) correlations of compound 1. (Color figure online)
Alkaloid 2 had the same molecular formula (HRESIMS m/z 318.1335 [M+H]+) and very similar UV and IR spectra as 1. Comparison of the 13C NMR data of 2 and 1 (Table 2) suggested that both compounds shared the same planar structure. In the 1H NMR spectrum (Table 1), obvious difference between both alkaloids was that a proton signal δ H 3.86 (d, J = 6.4 Hz, H-2) in 1 was replaced by δ H 4.05 (t, J = 8.9 Hz) in 2. This indicated α-configuration of H-2 in 2, and confirmed by a ROESY correlation from H-2 to H-4. Thus, 2 was established as 3-epicephalotine A and named cephalotine B.
Alkaloid 3 displayed similar 1H and 13C NMR data (Tables 1, 2) to the known alkaloid cephalotaxinone (7) [13] except that a quaternary carbon (δ C 81.9) in 3 substituted a methine in 7. In addition, the HMBC correlations of both H-1 and H-14 with δ C 81.9 located the quaternary carbon to C-4. The molecular formula C18H19NO5 of 3 from HRESIMS m/z at 330.1337 [M+H]+, 16 mass units higher than that of 7, further indicated that 3 was an 4-hydroxy cephalotaxinone. Alkaloid 4 showed the similar 13C NMR data to the known alkaloid acetycephalotaxine (8) [14], except that a methine signal of 8 was substituted by a quaternary carbon δ C 86.1 (s) in 4. Like in 3, the additional hydroxyl of 4 was also located at C-4 by its molecular formula C20H23NO6 by HRESIMS at m/z 374.1604 [M+H]+), 16 mass units higher than that of 8. Further, this was supported by the HMBCs of δ H 5.21 (H-1) and δ H 7.15 (H-14) with δ C 86.1 (C-4). The hydroxyl of 3 and 4 adopted α-orientation by the molecular model. The configuration of H-3 in both alkaloids was α-oriented by ROESY correlation between H-3 and H-11. Therefore, 3 and 4 were named cephalotines C and D, respectively.
Six methylenes, 3 methines, a methyoxyl and 5 quaternary carbons in the 13C NMR spectrum of alkaloid 5 revealed that 5 belongs to homoerythrina-type alkaloids rather than cephalotaxine-type alkaloids [2]. The 13C NMR and DEPT data of alkaloid 5 were similar to those of comosine (18) [21] with exception for three downfielded signals [87.0 (s), 67.6 (t), 63.3 (t)], suggesting a N-oxide moiety. Additionally, its molecular formula C20H23NO4 by HRESIMS (m/z 330.1717 [M+H]+) could support this presumption. The H-3 was allowed at β-configuration through ROESY correlations of H-3 with H-10 and H-12. Thus 5 was named as cephalotine E.
None of these compounds showed any significant activity against HeLa, SGC-7901 gastric cancer, and A-549 lung cancer cell lines (IC50 > 20 μM).
3 Experimental Section
3.1 General Experimental Procedures
Optical rotations were carried out using a Horiba SEPA-300 polarimeter and JASCO DIP-370 digital polarimeter. UV spectra were recorded on Shimadzu 2401Aspectrophotometer. IR Spectra were obtained on Brucker Tensor 27 infrared spectrophotometer with KBr pellets. 1H, 13C and 2D NMR spectral data were measured on a Bruker Avance III-600, DRX-500, and AM-400 MHz spectrometers with SiMe4 as an internal standard. HRESIMS data were recorded on an Agilent G6230 TOF MS. Column chromatography (CC) was performed with silica gel (200–300 mesh, Qing-dao Haiyang Chemical Co., Ltd., Qingdao, China). RP-18 silica gel (20–45 μm, Fuji Silysia Chemical Ltd., Japan). Fractions were monitored by TLC on silica gel plates (GF254, Qingdao Haiyang Chemical Co., Ltd.) and spots visualized with Dragendorff’s reagent spray. MPLC was employed using a Buchi pump system coupled with RP-18 silica gel packed glass columns(15 × 230 and 26 × 460 mm, respectively). HPLC system was carried out on a Waters HPLC system (Waters 1525E pumps, Waters 2996 photodiode array detector, Waters fraction collector II) using a analytical semi-preparative or preparative Sunfire C18 column (4.6 × 150, 10 × 150, and 19 × 250 mm, respectively).
3.2 Plant Materials
Leaves and stems of C. lanceolata and C. fortunei var. alpina were collected from Yunnan Province, P. R. China and identified by Dr. Jie Cai, respectively. Two voucher specimen (cai20131002 and cai20140501) was preserved in the State Key Laboratory of Phytochemistry and Plant Resources in West China, Kunming Institute of Botany, Chinese Academy of Sciences.
3.3 Extraction and Isolation of C. lanceolata and C. fortunei var. alpina
The air-dried and powdered leaves and stems of C. lanceolata (19 kg) and C. fortunei var. alpina (39 kg) was extracted with MeOH (3 × 50 L, 3 × 100 L, 2 days each) at room temperature, respectively, and the solvent was evaporated in vacuo. The extract was dissolved in 1 % HCl solution (v/v) to pH 2–3, basified with 10 % ammonia solution (v/v) to pH 7–8, and partitioned with EtOAc to afford the crude alkaloids (39 and 198 g).
The alkaloidal extract of C. lanceolata (39 g) was subjected to CC over silica gel (400 g) and eluted with a CHCl3–MeOH gradient (1:0 to 0:1, v/v) to give four fractions (I-IV) based on TLC analysis. Fraction I (7.5 g) was subjected to C18 MPLC with MeOH–H2O (20:80 to 100:0, V/V) as the eluent to obtain four fractions (I-1–I-4). I-1 (800 mg) was further separated on a C18 MPLC with a gradient of MeOH–H2O (20:80 to 40:60, v/v) and then separated on a preparative C18 column with a gradient MeOH–H2O (30:70 to 40:60, v/v) to afford 6 (30 mg). I-2 (3 g) was purified on a C18 MPLC with a gradient of MeOH–H2O (30:20 to 60:40, v/v) to afford the alkaloid 7 (8 mg). 11 (33 mg) was crystallized from I-3 (1 g), and the mother liquid of this fraction was separated on a C18 MPLC with a gradient of MeOH–H2O (40:60 to 70:30, v/v) to afford the alkaloids 16 (18 mg) and 18 (14 mg). I-4 (2 g) was applied to a C18 HPLC with a gradient of MeOH–H2O (50:40 to 80:10, v/v) then separated on a preparative C18 column with a gradient MeOH–H2O (55:45 to 65:35) to obtain 17 (20 mg), 20 (12 mg) and 21 (5.5 mg). Fraction II (15 g) was applied to a C18 MPLC with a gradient of MeOH–H2O (20:80–100:0, v/v) to obtain four subfractions II-1–II-4. II-1 (5 g) was further applied to a C18 MPLC with a gradient of MeOH–H2O (10:90 to 70:30, v/v) to give four fractions II-1-1–II-1-4. II-1-1 (0.8 g) was separated on a C18 MPLC with a gradient of MeOH–H2O (10:90 to 30:70, v/v) and then separated on a preparative C18 column with a gradient MeOH–H2O (25:75 to 35:65, v/v) to give 1 (8 mg) and 2 (12.5 mg). II-1-3 (2 g) was subjected to a C18 MPLC with a gradient of MeOH–H2O (30:70 to 60:40, v/v) and then separated on a preparative C18 column with a gradient MeOH–H2O (48: 52 to 58:42, v/v) to give 12 (55 mg). II-3(4 g) was applied to a C18 MPLC with a gradient of MeOH–H2O (20:80 to 50:50, v/v) to obtain 27 (9.5 mg), and then separated on a preparative C18 column with a gradient MeOH–H2O (38:62 to 48:52, v/v) to give 14 (11 mg). II-4 (3.0 g) was subjected to CC over silica gel (30 g) and eluted with a CHCl3–MeOH gradient (25:1 to 15:1, v/v) and further purified on a preparative C18 column with a gradient MeOH–H2O (50:50 to 60:40, v/v) to give 9 (14 mg). III (12 g) was applied to C18 MPLC with a gradient of MeOH–H2O (20:80 to 60:40, v/v) to obtain four subfractions III-1-III-4. III-1 (4 g) was separated on a C18 MPLC with a gradient of MeOH–H2O (10:90 to 40:60, v/v) to give 13 (10 mg). III-3 (2.5 g) was separated on a C18 MPLC with a gradient of MeOH–H2O (30:70 to 60:40, v/v) to give 5 (10 mg) and 22 (22 mg).
The alkaloidal extract of C. fortunei var. alpina (198 g) was subjected to CC over silica gel (2.0 kg), eluted with CHCl3–MeOH gradient (1:0 to 0:1, v/v) to yield six fractions (I-VI). Fraction II (43 g) was gradually purified C18 MPLC with MeOH–H2O (30:70 to 100:0, V/V), to afford subfractions II-1–II-6. 6 (200 mg) was crystallized from II-1 (7 g), and the mother liquid of this fraction was separated on a C18 MPLC with a gradient of MeOH–H2O (30:70 to 50:50, v/v) to afford 7 (5 mg). II-3 (11 g) was subjected to CC over silica gel (120 g) with CHCl3–Me2CO(20:1 to 5:1, v/v) as the eluent and then further purified on a C18 MPLC with a gradient of MeOH–H2O (30:70 to 50:50, v/v) to afford 3 (5 mg). II-4 (8 g) was gradually purified on a C18 MPLC (MeOH–H2O, 40:60 to 60:40, v/v) to afford 20 (98 mg) and then further purified on a preparative C18 column with a gradient MeOH–H2O (48: 52 to 58:42, v/v) to give 21 (17 mg). II-5 (7 g) was separated by C18 MPLC with a gradient of MeOH–H2O (50:50 to 70:30, v/v) to give 23 (39 mg). Fraction III (41 g) was separated on a C18 MPLC with a gradient of MeOH–H2O (20:80 to 100:0, v/v) to afford subfractions (III-1–III-5). Subfraction III-3 (12 g) was gradually separated on a C18 MPLC, eluted with MeOH–H2O (30:70 to 50:50, v/v) to afford 14 (141 mg). 12 (133 mg) was crystallized from III-5 (13 g), and the mother liquid of this fraction was separated on a C18 MPLC with a gradient of MeOH–H2O (20:80 to 40:60, v/v) to afford 10 (32 mg). IV (31 g) was separated on a C18 MPLC with a gradient of MeOH–H2O (10:90 to 100:0, v/v) to yield subfractions IV-1–IV-9. IV-2(8 g) was further purified on a C18 MPLC with CH3CN–H2O (5:95 to 15:85, v/v) as the eluent to give 7 (200 mg). IV-3 (3 g) was subjected to a C18 MPLC with MeOH–H2O (20:80 to 50:50, v/v), then further purified on a preparative C18 column with a gradient MeOH–H2O (35:65 to 45:55, v/v) to give 26 (46) and 27 (18 mg). 25 (54 mg) was crystallized from IV-5 (13 g). IV-9 (5 g) was gradually separated on a C18 MPLC, eluted with MeOH–H2O (35:65 to 55:45, v/v) to afford 28 (100 mg) and 29 (380 mg). V (25 g) was subjected to a C18 MPLC with a gradient of MeOH–H2O (10:90 to 100:0, v/v) to give five subfractions (V-1–V-5). V-2 (4 g) was separated on a C18 MPLC with a gradient of MeOH–H2O (10:90 to 30:70, v/v) to afford 4 (600 mg). VI (17 g) was purified on C18 MPLC with a gradient of MeOH–H2O (10:90 to 100:0, v/v), and VI-3 (3 g) was gradually purified on a C18 MPLC (MeOH–H2O, 10:90 to 30:70, v/v) and further purified on a preparative C18 column with a gradient MeOH–H2O (15: 85 to 25:75, v/v) to yield 15 (4 mg), 24 (5 mg) and 19 (18 mg).
Cephalotine A (1): white powder; [α] 25D -31.5 (c 0.09, MeOH); UV (MeOH) λ max (log ε) 203 (3.01), 291 (3.91) nm; IR (KBr) ν max 3520, 3406, 1631, 1500, 1482, 1342 cm−1; 1H (400 MHz) and 13C NMR (100 MHz) data (acetone-d 6), see Tables 1 and 2; positive HRESIMS m/z 318.1336 (calcd for C17H20NO5 [M+H]+, 318.1342).
Cephalotine B (2): white powder; \( [\alpha]_{\rm D}^{25}\) −35.8 (c 0.12, MeOH); UV (MeOH) λ max (log ε) 204 (2.95), 291 (3.81) nm; IR (KBr) ν max 3450, 3430, 1631, 1484, 1342 cm−1; 1H (400 MHz) and 13C NMR (100 MHz) data (acetone-d 6), see Tables 1 and 2; positive HRESIMS m/z 318.1335 (calcd for C17H20NO5 [M+H]+, 318.1342).
Cephalotine C (3): brown oil; \( [\alpha ]_{\text{D}}^{25} \) +9.0 (c 0.13, MeOH); UV (MeOH) λ max (log ε) 237(3.67), 280 (3.80) nm; IR (KBr) ν max 3437, 2954, 1752, 1735, 1654, 1223 cm−1; 1H (600 MHz) and 13C NMR (150 MHz) data (DMSO-d 6 ), see Tables 1 and 2; positive HRESIMS m/z 330.1337 (calcd for C18H20NO5 [M+H]+, 330.1336).
Cephalotine D (4): colorless powder; \( [\alpha ]_{\text{D}}^{25} \) +138.0 (c 0.41, MeOH); UV (MeOH) λ max (log ε) 240 (3.84), 279 (3.89) nm; IR (KBr) ν max 3437, 2922, 1659, 1590, 1130 cm−1; 1H (400 MHz) and 13C NMR (100 MHz) data (acetone-d 6), see Tables 1 and 2; positive HRESIMS m/z 374.1604 (calcd for C20H24NO6 [M+H]+, 374.1598).
Cephalotine E (5): white powder; \( [\alpha ]_{\text{D}}^{25} \) +46.3 (c 0.10, MeOH); UV (MeOH) λ max (log ε) 204 (3.66), 243 (2.73), 288 (2.67)nm; IR (KBr) ν max 3419, 2934, 1623, 1507, 1490 cm−1; 1H (400 MHz) and 13C NMR (100 MHz) data (acetone-d 6 ), see Tables 1 and 2; positive HRESIMS m/z 330.1717 (calcd for C20H24NO4 [M+H]+, 330.1705).
3.4 Cytotoxicity Assay
Three human cancer cell lines, HeLa, SGC-7901 gastric cancer, and A-549 lung cancer, were used in the cytotoxicity assay. All the cells were cultured in RPMI-1640 or DMEM media (Hyclone, USA), supplemented with 10 % fetal bovine serum (Hyclone, USA) in 5 % CO2 at 37 °C. The cytotoxicity assay was performed according to the MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide) method in 96-well microplates. Briefly, 100 µL adherent cells were seeded into each well of 96-well cell culture plates and allowed to adhere for 12 h before addition of the test compound/drug. Meanwhile suspended cells were seeded with initial density of 1 × 105 cells/mL just before addition of the test compound/drug. Each tumor cell line was exposed to the test compound at concentrations of 0.06, 0.32, 1.60, 8.0, and 40 μM for 48 h. Each of these tests was conducted in triplicate, with cisplatin (sigma, USA) as the positive control. After the end of the treatment period, cell viability was measured and cell growth curve was plotted.
References
W.W. Paudler, G.I. Kerley, J. McKay, J. Org. Chem. 28, 2194 (1963)
R.G. Powell, R.V. Madrigal, C.R. Smith Jr, K.L. Mikolajczak, J. Org. Chem. 39, 676 (1974)
I. Takano, I. Yasuda, M. Nishijima, Y. Hitotsuyanagi, K. Takeya, H. Itokawa, Bioorg. Med. Chem. Lett. 6, 1689 (1996)
H. Morita, M. Arisaka, N. Yoshida, Tetrahedron 56, 2929 (2000)
H. Morita, M. Yoshinaga, J.I. Kobayashi, Tetrahedron 58, 5489 (2002)
L.W. Wang, H.J. Su, S.Z. Yang, S.J. Won, C.N. Lin, J. Nat. Prod. 67, 1182 (2004)
J.G. Buta, J.L. Flippen, W.R. Lusby, J. Org. Chem. 43, 1002 (1978)
J. Du, M.H. Chiu, R.L. Nie, J. Nat. Prod. 62, 1664 (1999)
Y.R. He, Y.H. Shen, L. Shan, X. Yang, B. Wen, J. Ye, X. Yuan, H.L. Li, X.K. Xu, W.D. Zhang, RSC Adv. 5, 4126 (2015)
K.D. Yoon, D.G. Jeong, Y.H. Hwang, J.M. Ryu, J. Kim, J. Nat. Prod. 70, 2029 (2007)
K.D. Yoon, Y.W. Chin, J.W. Kim, Bull. Korean Chem. Soc. 31, 495 (2010)
H. Abdelkafi, B. Nay, Nat. Prod. Rep. 29, 845 (2012)
R. Powell, Phytochemistry 11, 1467 (1972)
R. Powell, D. Weisleder, C. Smith, J. Pharm. Sci. 61, 1227 (1972)
R. Powell, K. Mikolajczak, Phytochemistry 12, 2987 (1973)
S. Asada, Yakugaku Zasshi 93, 916 (1973)
R. Powell, D. Weisleder, C. Smith, I. Wolff, Tetrahedron Lett. 10, 4081 (1969)
N. Langlois, J. Razafimbelo, J. Nat. Prod. 51, 499 (1988)
J.S. Fitzgerald, S. Johns, J. Lamberton, A. Sioumis, Aust. J. Chem. 22, 2187 (1969)
S. Johns, J. Lamberton, A. Sioumis, Aust. J. Chem. 22, 2219 (1969)
L. Lacombe, N. Langlois, B. Das, P. Potier, Bull. Soc. Chim. Fr. 10, 3535 (1970)
M.H. Qiu, B.P. Lu, X. Ma, R.L. Nie, Acta Botanica Yunnanica 19, 99 (1997)
F. Chang, C. Wang, W. Pan, Y. Li, L. Mai, C. Sun, K. Ma, J. Integr. Plant Biol. 20, 129 (1978)
S. Li, Y. Cui, Y. Li, X. Pan, Y. Wang, W. Hung, Acta Chim. Sinica 45, 687 (1987)
R. Powell, D. Weisleder, C. Smith, W. Rohwedder, Tetrahedron Lett. 11, 815 (1970)
S.B. Li, Y.X. Cui, X.Y. Nie, X.X. Pan, Y.L. Li, Sci. Bull. 33, 1436 (1988)
W.K. Huang, Y.L. Li, X.X. Pan, Sci. China Ser. A 23, 835 (1980)
K. Mikolajczak, R. Powell, C. Smith, Tetrahedron 28, 1995 (1972)
Acknowledgments
This project was financially supported by the Young Academic and Technical Leader Raising Foundation of Yunnan Province (No. 2010CI049).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare no conflict of interest.
Additional information
Ling Ni and Xiu-Hong Zhong have contributed equally to this work.
Electronic Supplementary Material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Ni, L., Zhong, XH., Cai, J. et al. Five New Alkaloids from Cephalotaxus lanceolata and C. fortunei var. alpina . Nat. Prod. Bioprospect. 6, 149–154 (2016). https://doi.org/10.1007/s13659-016-0093-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13659-016-0093-7