
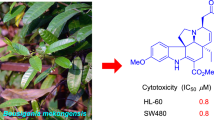
Abstract
Three new monoterpene indole alkaloids, 3α-hydroxymethyl-ibogamine (1), 3α-acetatemethoxyl-ibogamine (2), 16α-hydroxyl-ibogamine (3) together with six known alkaloids were isolated from the branches and leaves of Tabernaemontana divaricata (Apocynaceae). The structures of these alkaloids were determined by spectroscopic analyses. All isolated compounds showed no significant cytotoxicity against SGC-7901 gastric cancer, HeLa, and A-549 lung cancer cell lines (IC50 > 20 µM).
Graphical Abstract

Similar content being viewed by others
Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
1 Introduction
Alkaloids are unique natural products denoted by the presence of a nitrogen atom as part of a heterocyclic ring, constituting a highly diverse group of compounds. At present, more than 12,000 different alkaloids, all of them plant-derived, are known. To deal with this enormous diversity, these compounds are organized into dozens of structural related groups [1, 2]. Among this class of compounds, monoterpene indole alkaloids (MIAs) may account for a quarter of naturally occurring alkaloids. Both, their highly intricate chemical structures and pronounced pharmacological activities attracted many people from the research community to illuminate the structural diversity, bioactivities and biosynthetic pathways [3]. Species of the genus Tabernaemontana L. (Apocynaceae) are widely distributed in tropical and subtropical regions of Africa, Asia, North America, Pacific Islands, South America, including five species in China. This species are recognized as a rich source of MIAs [4], whereas the Iboga-type alkaloids ibogamine [5], voacangine [6], and coronaridine [7] are common in these species, hence, they can be regarded as chemical markers for the genus. These compounds feature a nitrogen-containing seven-membered ring which is linked to the indole system. As part of continuing search for bioactive alkaloids, three new alkaloids named as 3α-hydroxymethyl-ibogamine (1), 3α-acetatemethoxyl-ibogamine (2), 16α-hydroxyl-ibogamine (3), as well as six known alkaloids 4–9, were isolated from the branches and leaves of T. divaricata (L.) R. Br. ex Roem. & Schult. The known alkaloids were identified as coronaridine (4) [8], isovoacangine (5) [9], taberdivarine G (6) [10], voacangine (7) [11], heyneanine (8) [12], coronaridine hydroxyindolenine (9) [13]. All of them belong to the iboga-type alkaloids. In the assessment of their bioactivities, the isolated alkaloids did not show significant activities against SGC-7901 gastric cancer, HeLa, and A-549 lung cancer cell lines (IC50 > 20 µM).
2 Results and discussion
The alkaloid fraction of T. divaricata was separated as described in experimental section to yield a total of nine compounds, including three new alkaloids 1–3 (Fig. 1). All compounds showed a positive response to Dragendorff’s reagent on TLC.

Structures of compounds 1–9
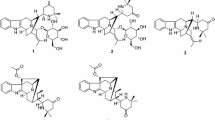
Compound 1 was obtained as pale yellow amorphous powder. The UV absorption bands at 284, 227, and 214 nm suggest the presence of an indole chromophore [14]. The molecular formula of compound 1 was established as C20H26N2O with nine degrees of unsaturation by HRESIMS ([M + H]+ at m/z 311.2118). The 1H NMR data (Table 1) indicated an unsubstituted indole ring with signals at δH 7.38 (1H, d, J = 7.7 Hz), 6.94 (1H, t, J = 7.7 Hz), 6.99 (1H, t, J = 7.7 Hz), 7.22 (1H, d, J = 7.7 Hz), and one methyl signal at δH 0.87 (3H, t, J = 7.3 Hz). The 13C NMR and DEPT data (Table 1) revealed the presence of four quaternary carbons (δC 143.1, 136.2, 130.7, 108.6), nine methines (δC 121.1, 119.1, 118.3, 111.0, 59.8, 59.7, 42.3, 41.9, 27.9), six methylenes (δC 62.4, 53.8, 37.2, 27.9, 27.4, 21.3), and one methyl group (δC 12.2). The 1D NMR spectroscopic data of 1 were similar to those of ibogamine, except the presence of a hydroxymethyl group at C-3 and a methine carbon (δC 59.8) of C-3 in 1 instead of a methylene (δC 49.9) in ibogamine [5]. The HMBC correlations from δH 3.20 (H-3) to δC 27.4 (C-15), δC 37.2 (C-17), δC 53.8 (C-5), and from δH 3.48 (H-22) to δC 27.9 (C-14), δC 59.8 (C-3) confirmed this result (Fig. 2). Thus, the planar structure of compound 1 was determined. The relative configuration of 1 was determined on the basis of the ROESY spectrum, the correlation of H-17 (δH 2.17)/H-16, H-3/H-17 (δH 1.61), suggested H-3 was β-oriented (Fig. 3). Consequently, the structure of 1 was confirmed as shown in Fig. 1, and named 3α-hydroxymethyl-ibogamine (1).

Key HMBC correlations of compounds 1–3

Key ROESY correlations of compounds 1 and 2
Compound 2 was isolated as pale yellow amorphous powder. The 1H and 13C NMR data (Table 1) of 2 was very similar to those of 1, except the presence of an additional –COOCH3 signal (δH 3.60, δC 51.5, δC 173.2) and the 13C NMR chemical shift at C-22 (δC 38.5) was different from that in compound 1 (δC 62.4). Moreover, compound 2 was assigned the molecular formula of C22H28N2O2, differing from 1 by addition of 42 daltons (da) and consistent with substitution of an OH by a COOCH3 group based on the analysis of HRESIMS ([M + H]+ at m/z 353.2223). This assumption was supported by HMBC correlations from δH 2.39 (H-22) to δC 30.7 (C-14), δC 55.4 (C-3), δC 173.2 (C-23), and from δH 3.60 (H-24) to δC 173.2 (C-23) (Fig. 2). Similar to 1, the ROESY correlation of H-17 (δH 2.15)/H-16, H-3/H-17 (δH 1.59), H-15 (δH 1.34)/H-22 (δH 2.39) suggested that H-3 was β-oriented (Fig. 3). Hence, compound 2 was identified as 3α-acetatemethoxyl-ibogamine (2).
Compound 3 showed a molecular ion peak at m/z 297.1964 [M + H]+ (calcd. 297.1961) by HRESIMS, corresponding to the molecular formula C19H24N2O, which is 16 da more than that of the known alkaloid ibogamine [5]. Comparison of its 1D NMR data (Table 1) with those of ibogamine showed that the major differences were the signal due to C-17, C-21 were found to resonate at lower field (δC 44.0, 63.4) and a quaternary carbon (δC 74.7) in 3 substituted a methine in ibogamine. In addition, the HMBC correlations from the cross peaks of H-17 (δH 1.93)/C-16 (δC 74.7), C-2 (δC 145.6), and of H-21 (δH 2.79) /C-20 (δC 35.1), C-17 (δC 44.0), C-5 (δC 54.4), C-16 (δC 74.7), C-2 (δC 145.6) located the quaternary carbon of C-16 (Fig. 2), indicating that 3 was 16-hydroxy ibogamine. According to the biogenetic pathway of iboga-type monoterpenoid indole alkaloids and molecular model, the substituent of C-16 was α-oriented. Therefore, the structure of 3, 16α-hydroxyl-ibogamine (3), was defined as shown in Fig. 2.
Compounds 1–9 were evaluated for their cytotoxicity activity based on the MTT method (camptothecin was used as a positive control). However, none of these compounds showed any significant activity against SGC-7901 gastric cancer, HeLa, and A-549 lung cancer cell lines (IC50 > 20 µM).
3 Experimental
3.1 General Experimental Procedures
Optical rotations were measured with a JASCO P-1020 digital polari-meter (Horiba, Kyoto, Japan). UV spectra were recorded on a Shimadzu UV2401PC spectrometer (Shimadzu, Kyoto, Japan). NMR spectra were performed on a Bruker AVANCE III 400 MHz spectrometers (Bruker Biospin GmbH, Karlsruhe, Germany) and a Bruker AVANCE III 500 MHz spectrometers (Bruker Biospin GmbH, Karlsruhe, Germany). HRESIMS data were acquired on a Shimadzu UPLC-IT-TOF spectrometer (Shimadzu, Kyoto, Japan). HPLC analyses were performed on a Waters instrument consisting of a Waters 1525EF pump coupled with a Waters 2998 photodiode array detector and a Waters fraction collector III. The analytical separations were performed on a Sunfire C18 column (5 μm, 150 mm × 4.6 mm). Preparative separations were done a Sunfire C18 column (5 μm, 250 mm × 19 mm). The following stationary phases were used for column chromatography (CC): silica gel (200–300 mesh, Qingdao Marine Chemical Ltd., Qingdao, China), C18 silica gel (50 μm, YMC Co. Ltd., Japan), and Sephadex LH-20 (Mitsubishi Co., Ltd., Japan). Each separation step was monitored by TLC on silica gel plates (GF254, Qingdao Marine Chemical Co., Ltd., Qingdao, China), and spots were visualized using Dragendorff’s reagent. MPLC was performed using a Buchi pump system coupled with RP-18 silica gel-packed glass columns (15 × 230 and 26 × 460 mm, respectively).
3.2 Plant Material
Leaves and branches of Tabernaemontana divaricata were collected in Hainan Province, P. R. China, and identified by Dr. Sheng-Zhou Huang. A voucher specimen (Cai20150424) was deposited in the State Key Laboratory of Phytochemistry and Plant Resources in West China, Kunming Institute of Botany, Chinese Academy of Sciences.
3.3 Extraction and Isolation
Air-dried branches and leaves of T. divaricata (46 kg) were powdered and extracted with MeOH (96 h × 3) at room temperature. The extract (2.5 kg) was partitioned between 0.5% HCl solution and EtOAc, and the acidic water layer was adjusted to pH 8–9 with 15% ammonia solution and subsequently extracted with EtOAc. This yielded 325 g crude alkaloid extract. This extract was subjected to column chromatography (CC) over silica gel and eluted with gradient CHCl3–Me2CO (1:0–1:1, v/v) to afford five fractions (I–V).
Fraction II (20.3 g) was further chromatographed on a C18 MPLC column eluted with a gradient of MeOH–H2O (30:70–100:0, v/v). Six subfractions II-1 to II-6 were collected from this procedure. Subfraction II-2 was subjected to C18 MPLC column once again using MeOH–H2O (20:80–70:30, v/v) as eluent which yielded seven subfractions (II-2-1 to II-2-7). Subfraction II-2-5 was refined by a preparative C18 HPLC column using a gradient of MeCN–H2O (45:55–60:40, v/v). This afforded 4.0 mg of 1. Fraction II-2-7 was further purified by preparative HPLC with a gradient from 50 to 65% aqueous acetonitrile. This afforded 6.7 mg of 8. Fraction II-3 was separated by reversed phase MPLC column with a gradient of MeOH–H2O (40:60–85:15, v/v). This step afforded five subfractions (II-3-1 to II-3-5). Fraction II-3-5 was separated by prep. HPLC with a gradient of MeOH–H2O (65:35–80:20, v/v). This step yielded 8.2 mg of compound 9. Fraction II-4 was separated using a Sephadex LH-20 column eluted with MeOH. Four subfractions (II-4-1 to II-4-4) were collected. Fraction II-4-2 was separated on a prep. C18 HPLC using a gradient of MeCN–H2O (55:45–70:30, v/v) which afforded 6.7 mg of 6. Fraction II-4-3 was subjected to a prep. C18 HPLC eluted with a gradient of MeOH–H2O (75:25–90:10, v/v). This step generated 2.2 mg of 4. Compound 7 (1.5 g) was crystalized from fraction II-5. Subfraction II-6 was subjected to C18 MPLC eluted with mixtures of MeOH and water (60:40–90:10, v/v). Five subfractions (II-6-1 to II-6-5) were collected. Fraction II-6-2 was further separated using reversed phase MPLC with a gradient of MeOH (from 60 to 85%) in H2O, affording one subfraction (II-6-2-4). Refining of this subfraction by prep. HPLC with a gradient of MeCN–H2O (65:35–80:20, v/v) gave 30.9 mg of 5. Fraction II-6-5 was chromatographed over Sephadex LH-20 column eluted with MeOH. This generated five subfractions (II-6-5-1 to II-6-5-5). Fraction II-6-5-1 was separated by prep. HPLC using a gradient of H2O (from 70 to 85%) in MeCN. This step yielded 7.1 mg of 2 and 5.7 mg of 3.
3.3.1 3α-Hydroxymethyl-ibogamine (1)
Pale yellow amorphous powder, [α] 17.5D −29.9 (c 0.13, MeOH); UV (MeOH) λmax (log ε) 196 (4.10), 214 (4.30), 227 (4.41), 284 (3.75) nm; 1H and 13C NMR data, see Table 1. HRESIMS m/z 311.2118 [M + H]+ (calcd. for C20H26N2O, 311.2118).
3.3.2 3α-acetatemethoxyl-ibogamine (2)
Pale yellow amorphous powder, [α] 17.5D −15.0 (c 0.11, MeOH); UV (MeOH) λmax (log ε) 211 (4.26), 227 (4.40), 285 (3.74) nm; 1H and 13C NMR data, see Table 1. HRESIMS m/z 353.2223 [M + H]+ (calcd. for C22H28N2O2, 353.2224).
3.3.3 16α-hydroxyl-ibogamine (3)
Pale yellow amorphous powder, [α] 17.5D −19.2 (c 0.07, MeOH); UV (MeOH) λmax (log ε) 211 (4.09), 223 (4.10), 285 (3.49) nm; 1H and 13C NMR data, see Table 1. HRESIMS m/z 297.1964 [M + H]+ (calcd. for C19H24N2O, 297.1961).
3.4 Cytotoxicity
The human A-549 lung cancer, SGC-7901 gastric cancer, and HeLa cell lines were used in the performed cytotoxic assay. These cells were grown in DMEM media (HyClone, USA) supplemented with 10% fetal bovine serum (HyClone, USA) at 37 °C in 5% CO2. The cytotoxicity of all alkaloids was determined based on the MTT method in 96-well microplates. In short, 100 µL adherent cells were seeded into each well and incubated for 12 h before the addition of the test alkaloids/drug. At the same time, the suspended cells were seeded at an initial density of 1 × 105 cells/mL just before addition of the purified compounds. Each tumor cell line was exposed to a single test compound at concentrations of 0.8, 4 and 20 μM in DMSO. Camptothecin was used as positive control. Each test was performed in triplicate. After treatment, cell viability was assessed, cell growth graphed and IC50 values were calculated using Reed and Muench’s method [15].
References
S.E. O’Connor, J.J. Maresh, Nat. Prod. Rep. 23, 532–547 (2006)
H. Abdelkafi, B. Nay, Nat. Prod. Rep. 29, 845–869 (2012)
K. Folkers, R.T. Major, J. Am. Chem. Soc. 59, 1580–1581 (1937)
Editorial Board of the Flora of China of Chinese Academy of Sciences, Flora of China, vol 16 (Science Press, Beijing, 1996), p. 153
M. Damak, C. Poupat, A. Ahon, Tetrahedron Lett. 39, 3531–3534 (1976)
D.W. Thomas, K. Biemann, Tetrahedron 24, 4223–4231 (1968)
M. Gorman, N. Neuss, N.J. Cone, J.A. Deyrup, J. Am. Chem. Soc. 82, 1142–1145 (1960)
P.S. Pereira, S.D.C. Franca, P.V.A.D. Oliveira, C.M.D.S. Breves, S.I.V. Pereira, Quim. Nova 31, 20–24 (2008)
J.P. Kutney, A. Horinaka, R.S. Ward, B.R. Worth, Can. J. Chem. 58, 1829–1838 (1980)
B.J. Zhang, X.F. Teng, M.F. Bao, X.H. Zhong, L. Ni, X.H. Cai, Phytochemistry 120, 46–52 (2015)
E. Wenkert, D.W. Cochran, H.E. Gottlied, E.W. Hagaman, R. Brazfilho, F.J.D.A. Matos, M.I.L.M. Madruga, Helv. Chim. Acta 59, 2437–2442 (1976)
Z.J. Zhan, L.W. Zhang, W.G. Shan, J. Chem. Res. 7, 416–417 (2009)
H. Zhang, X.N. Wang, L.P. Lin, J. Ding, J.M. Yue, J. Nat. Prod. 70, 54–59 (2007)
B. Albinsson, B. Norden, J. Phys. Chem. 96, 6204–6212 (1992)
L. Reed, H. Muench, L.J. Reed, Am. J. Hyg. 27, 493–497 (1938)
Acknowledgements
This project was financially supported by the Applied Basic Research Programs of Yunnan Province (No. 2016FA030) and the National Natural Science Foundation of China (31370377). The authors are grateful to Dr. Sheng-Zhou Huang for identification of plant samples.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflict of interest.
Electronic supplementary material
Below is the link to the electronic supplementary material.
13659_2018_166_MOESM1_ESM.pdf
Supplementary material 1 (PDF 1986 kb) 1D and 2D NMR spectra, and HRESIMS of compounds 1–3 are available as Supplementary Information
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Deng, Y., Bao, MF., Shi, BB. et al. Three New Indole Alkaloids from Tabernaemontana divaricata. Nat. Prod. Bioprospect. 8, 183–188 (2018). https://doi.org/10.1007/s13659-018-0166-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13659-018-0166-x