
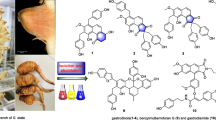
Abstract
Five new hybrid monoterpenoid indole alkaloids bearing an unusual 2,2-dimethyl-4-oxopiperidin-6-yl moiety, namely rauvotetraphyllines F–H (1, 3, 4), 17-epi-rauvotetraphylline F (2) and 21-epi-rauvotetraphylline H (5), were isolated from the aerial parts of Rauvolfia tetraphylla. Their structures were established by extensive spectroscopic analysis. The new alkaloids were evaluated for their cytotoxicity in vitro against five human cancer cell lines.
Graphical Abstract

Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
Rauvolfia genus of the Apocynaceae family, comprising about 60 species, is mainly distributed in America, Africa, Asia, and Oceania [1]. Plants of this genus are a rich source of monoterpenoid indole alkaloids, which have attracted great interests from biological and therapeutic aspects [2–5]. As part of a BioBioPha [http://www.chemlib.cn] objective to assemble a large-scale natural product library valuable in the discovery of new drug leads from nature, previous chemical study on the ethanolic extract of Rauvolfia tetraphylla had resulted in the isolation of five new indole alkaloids, rauvotetraphyllines A–E [6]. Further investigation of the remaining components led to the isolation of another five new alkaloids bearing an unusual 2,2-dimethyl-4-oxopiperidin-6-yl moiety, rauvotetraphyllines F–H (1, 3, 4), 17-epi-rauvotetraphylline F (2) and 21-epi-rauvotetraphylline H (5). The present paper describes the isolation, structure elucidation, and cytotoxic evaluation of the new compounds.
2 Results and Discussion
Compound 1, obtained as amorphous powder, possessed a molecular formula of C31H41N3O7, as evidenced by HR-ESI-MS (pos.) at m/z 568.3025 (calcd for C31H42N3O7, 568.3022), in combination with NMR spectra (Tables 1 and 3), requiring 13 degrees of unsaturation. In the UV spectrum, two characteristic maxima at 225 and 281 were detected, suggesting the existence of an unsubstituted indole chromophore [7]. The IR spectrum showed the presence of OH/NH (3404 cm−1) functionalities. The 1D-NMR spectra (Tables 1 and 3) revealed the presence of an unsubstituted indole moiety [δ H 7.38 (1H, d, J = 7.8 Hz), 7.26 (1H, d, J = 8.0 Hz), 7.02 (1H, dd, J = 8.0, 7.3 Hz), and 6.95 (1H, dd, J = 7.8, 7.3, Hz); δ C 139.7 (s), 138.2 (s), 128.9 (s), 121.9 (d), 119.7 (d), 118.5 (d), 111.9 (d), and 104.0 (s)], an ethylidene group [δ H 1.75 (3H, d, J = 6.9 Hz) and 5.93 (1H, q, J = 6.9 Hz); δ C 14.3 (q) and 125.2 (d)], and a glucose unit [δ H 4.64 (1H, d, J = 7.9 Hz), 3.29 (1H, m), 3.37 (1H, m), 3.28 (2H, m), 3.60 (1H, dd, J = 11.9, 4.9 Hz), and 3.74 (1H, dd, J = 11.9, 1.6 Hz); δ C 103.2 (d), 78.0 (d), 77.9 (d), 75.4 (d), 71.4 (d), and 62.7 (t)]. Comparison of its 13C NMR data with those of rauvotetraphylline B [6] revealed a remarkable resemblance except for a prominent difference as follows: the carbon signals assigned to 4,6-dimethylpyrid-2-yl unit in rauvotetraphylline B were not present, and there was a set of newly arisen resonances [δ C 56.5 (d), 47.4 (t), 213.2 (s), 55.7 (t), 55.5 (s), 25.6 (q), and 31.9 (q)] determined as a 2,2-dimethyl-4-oxopiperidin-6-yl moiety by HMBC correlations (Fig. 1) from H-17 to C-23, H-22 to C-23 and C-24, and H-24 to C-22, C-23, C-25, C-26, and C-27. The piperidinyl moiety was linked to C-16 through C-16–C-17 bond by HMBC correlations from H-16 to C-17 and C-22 and 1H-1H COSY correlation of H-16/H-17 (Fig. 1).

Key HMBC and 1H–1H COSY correlations of 1
The relative configuration of 1 was established by NMR analysis based on computer-generated 3D drawing with minimized energy by MM2 calculation (Fig. 2). ROESY correlations of H-16↔H-6β/H-14β and H-5↔H-21 suggested that 1 had the same stereochemistry as rauvotetraphylline B. The E-geometry of the ethylidene was indicated from ROESY correlations of H-15↔Me-18 and H-19↔H-21. The anti relationship of H-16 and H-17 was suggested by the large coupling constant (J 16,17 = 9.0 Hz), which could also be explained by that the molecule favors the conformation in which larger substituents are in the anti position. This was further supported by ROESY correlations of H-17↔Me-18. The R* configuration of C-17 was implied by ROESY correlations of H-6β↔H-22β and Me-18↔Me-26 (Fig. 2). Another noteworthy observation is that the chemical shift of H-15 (δ H 3.26) in 1 was relatively deshielded compared to that of H-15 (δ H 2.79) in its 17-epimer 2 (vide infra). This is attributed to paramagnetic deshielding caused by the proximity of the NH nitrogen atom to H-15 (Fig. 2). Thus, the structure of 1 was established as shown and named rauvotetraphylline F.

Key ROESY correlations of 1 and 2
Compound 2, isolated as amorphous powder, had the same molecular formula as 1 based on HR-ESI-MS (pos.), showing a quasi-molecular ion peak at m/z 568.3031 (calcd for C31H42N3O7, 568.3022). The 1H and 13C NMR spectra of 2 (Tables 1 and 3) were very similar in all respects to those of 2 except for the chemical shifts of H-5, H-6β, and H-15 in the 1H NMR spectrum. This discrepancy proved that compound 2 is a C-17 epimer of 1 while applying the same analysis carried out for 1. The paramagnetic deshielding experienced by H-15 in 1 was now experienced by H-5 and H-6β instead in 2 (Fig. 2), implying the S* configuration of C-17. This was further verified by strong ROESY correlations between H-22α and H-15/Me-18 and no correlation between Me-26 and Me-18. Therefore, the structure of 1 was elucidated as shown and named 17-epi-rauvotetraphylline F.
Compound 3 was obtained as amorphous powder. Its HR-ESI-MS revealed an [M + H]+ peak at m/z 390.2538 (calcd for C25H32N3O, 390.2545), suggesting the molecular formula C25H31N3O. The NMR data (Tables 1 and 3) were closely related to those of 1 except for the signals of a methylene group in 3 instead of an oxygenated methine group in 1, and the absence of a series of glucose resonances. The configuration of C-17 was designated as R* based on Me-18 showing ROESY correlation to Me-26, but no correlation to H-22. Consequently, the structure of 3 was determined and named rauvotetraphylline G.
Compound 4 was isolated as amorphous powder. Its molecular formula was determined as C27H33N3O3 by positive HR-ESI-MS at m/z 448.2613 (calcd for C27H34N3O3, 448.2600). The 13C NMR data (Table 3) were very similar to those of perakine [8]. The prominent difference between them was the aldehyde group in perakine changing into a 2,2-dimethyl-4-oxopiperidin-6-yl moiety [δ C 54.8 (d), 47.2 (t), 210.4 (s), 55.4 (t), 54.5 (s), 25.2 (q), 32.2 (q)] on the basis of HMBC correlations (Fig. 3) from H-21 to C-15 and C-20, H-22 to C-20, C-21, C-23, and C-24, and Me-26 to C-24, C-25, and C-27. The ROESY correlations (Fig. 4) of H-19↔H-3/H-14α, H-14β↔H-17, Me-18↔H-20, and H-20↔H-5/H-16 indicated that 4 possessed the same stereochemical characteristics as perakine. The R* configuration of C-21 was indicated by ROESY correlations of H-21↔H-14α/H-19, H-22α↔H-19, and H-22β↔Me-18, which was further supported by comparison of the 1H NMR spectra of 4 and its C-21 epimer 5 (vide infra) (Table 2). The proximity of the NH nitrogen atom to H-15 in 4 caused a marked downfield shift of H-15 (Δ = 0.30 ppm) (Fig. 4). Hence, the structure of 4 was assigned as shown and named rauvotetraphylline H.

Key HMBC correlations of 4

Key ROESY correlations of 4 and 5
Compound 5, obtained as amorphous powder, had the same molecular formula as 4, possessing a quasi-molecular ion peak at m/z 448.2602 (calcd for C27H34N3O3, 448.2600). The 1H and 13C NMR spectra of 5 (Tables 2 and 3) were almost identical to those of 4 except for the upfield shift of H-15 (Δ = −0.30 ppm) and downfield shifts of Me-18 (Δ = 0.15 ppm) and H-19 (Δ = 0.11 ppm) in the 1H NMR spectrum. This can be rationalized in terms of paramagnetic deshielding experienced by Me-18 and H-19 in 5 and H-15 in 4 (Fig. 4), revealing the S* configuration of C-21. This was further supported by significant ROESY correlation (Fig. 4) of H-22α↔H-15 and no correlation of H-22α↔H-19 or H-22β↔Me-18. Therefore, the structure of 5 was elucidated as shown and named 21-epi-rauvotetraphylline H.
The contribution of artifacts on structural diversity of alkaloids from Rauvolfia species is not ignorable as acidic or basic conditions are often used during isolation process, in spite that many artifacts from this genus are generally presented in literatures as naturally occurring compounds [9]. Considering that the presence of aldehyde group at C-16/C-20 is common for sarpagine/perakine type alkaloids [8, 10, 11], it’s plausible to deduce that, like triacetonamine [12], a common artifact of plant extractions, the 2,2-dimethyl-4-oxopiperidine moiety might also be an artifact produced by reaction of aldehyde group with acetone/ammonia since the latter were used as eluents during the isolation procedures. These artifacts represent a unique type of sarpagine/perakine series bearing an unusual piperidine unit brought about by using common eluents.
All of the isolated compounds were evaluated for their in vitro growth inhibitory effects against five human tumor cell lines (HL-60, SMMC-7721, A-549, MCF-7 and SW-480) with cisplatin and taxol serving as positive controls by the MTT method [13]. Regrettably, all tested compounds were inactive (IC50 values > 40 μM).
3 Experimental Section
3.1 General Experimental Procedures
Optical rotations were measured on a Jasco P-1020 automatic digital polarimeter. UV data were obtained from online HPLC analysis. IR spectra (KBr) were obtained on a Bruker Tensor-27 infrared spectrophotometer. NMR spectra were acquired with a Bruker DRX-500 or Bruker Avance III 600 instrument (Bruker BioSpin GmbH, Rheinstetten, Germany) with deuterated solvent signals used as internal standards. ESI-MS (including HR-ESI-MS) were measured on API QSTAR Pulsar i mass spectrometers. Silica gel (200–300 mesh, Qingdao Marine Chemical Inc., China) and Sephadex LH-20 (Amersham Biosciences, Sweden) were used for column chromatography. Medium pressure liquid chromatography (MPLC) was performed on a Büchi Sepacore System equipping with pump manager C-615, pump modules C-605, and fraction collector C-660 (Büchi Labortechnik AG, Switzerland), and columns packed with Chromatorex C-18 (40–75 μm, Fuji Silysia Chemical Ltd., Japan). Fractions were monitored by TLC (Qingdao Marine Chemical Inc., China) in combination with reversed-phase HPLC (Agilent 1200, Extend-C18 column, 5 μm, 4.6 × 150 mm).
3.2 Plant Material
The aerial parts of Rauvolfia tetraphylla were collected in Xiaomenglun of Yunnan Province, China, in June 2010 and identified by Mr. Yu Chen of Kunming Institute of Botany, Chinese Academy of Sciences. The voucher specimen (No. BBP0234020RT) was deposited at BioBioPha Co., Ltd.
3.3 Extraction and Isolation
The air-dried and powdered aerial parts of R. tetraphylla (7.5 kg) were extracted three times with EtOH-H2O (95:5, v/v; 3 × 20 L, each 5 days) at room temperature, and the solvent was removed under reduced pressure to give crude extract (ca. 400 g), which was then fractionated by silica gel column chromatography (CC) eluted with a gradient solvent system (containing 0.2 % ammonia) of petroleum ether-acetone and then MeOH to yield seven fractions A–G. Fraction D, eluted by acetone, was separated on silica gel CC (CHCl3–MeOH–ammonia, 100:1:0.5 → 0:100:0.5) to give three subfractions D1–D3. Fraction D1 was purified further by silica gel CC (CHCl3–MeOH–ammonia, 50:1:0.1) and then prep. TLC (CHCl3–MeOH–ammonia, 10:1:0.1) to afford 4 (9 mg) and 5 (6 mg). Fraction D2 was separated by silica gel CC (CHCl3–MeOH–ammonia, 40:1:0.1) and then prep. TLC (CHCl3–MeOH–ammonia, 9:1:0.1) to afford 3 (10 mg). Fraction G, eluted by MeOH, was separated further by silica gel CC (CHCl3–MeOH–ammonia, 10:1:0.1 → 0:10:0.1), repeated MPLC (40 → 45 % MeOH in H2O), and then Sephadex LH-20 (MeOH) to afford 1 (72 mg) and 2 (26 mg).
3.4 Rauvotetraphylline F (1)
White amorphous powder; [α] 15D +10.1 (c 0.19, CHCl3); UV (MeOH) λ max: 225, 281, 290 (sh) nm; IR (KBr) ν max 3404, 2961, 2921, 1700, 1470, 1453, 1384, 1338, 1320, 1302, 1076, 1031, 745 cm−1; 1H and 13C NMR data see Tables 1 and 3; ESI-MS (pos.): m/z 568 [M + H]+; HR-ESI-MS (pos.): m/z 568.3025 (calcd for C31H42N3O7, 568.3022).
3.5 17-epi-Rauvotetraphylline F (2)
White amorphous powder; [α] 16D +14.9 (c 0.20, CHCl3); UV (MeOH) λ max: 225, 281, 290 (sh) nm; IR (KBr) ν max 3396, 2962, 2923, 1699, 1626, 1471, 1451, 1384, 1337, 1300, 1075, 1030, 746 cm−1; 1H and 13C NMR data see Tables 1 and 3; ESI-MS (pos.): m/z 568 [M + H]+; HR-ESI-MS (pos.): m/z 568.3031 (calcd for C31H42N3O7, 568.3022).
3.6 Rauvotetraphylline G (3)
White amorphous powder; [α] 14D −22.4 (c 0.19, CHCl3); UV (MeOH) λ max: 225, 280, 290 (sh) nm; IR (KBr) ν max 3421, 3143, 3057, 2961, 2925, 2855, 1705, 1626, 1473, 1301, 1240, 1169, 741 cm−1; 1H and 13C NMR data see Tables 1 and 3; ESI-MS (pos.): m/z 390 [M + H]+; HR-ESI-MS (pos.): m/z 390.2538 (calcd for C25H32N3O, 390.2545).
3.7 Rauvotetraphylline H (4)
White amorphous powder; [α] 14D +9.9 (c 0.20, CHCl3); UV (MeOH) λ max: 220, 262 nm; IR (KBr) ν max 3433, 2965, 2934, 1741, 1707, 1592, 1453, 1380, 1364, 1295, 1033, 773, 753 cm−1; 1H and 13C NMR data see Tables 2 and 3; ESI-MS (pos.): m/z 448 [M + H]+; HR-ESI-MS (pos.): m/z 448.2613 (calcd for C27H34N3O3, 448.2600).
3.8 17-epi-Rauvotetraphylline H (5)
White amorphous powder; [α] 15D +47.3 (c 0.20, CHCl3); UV (MeOH) λ max: 220, 263 nm; IR (KBr) ν max 3433, 2965, 2936, 1742, 1707, 1592, 1453, 1376, 1268, 1230, 1177, 1032, 774, 753 cm−1; 1H and 13C NMR data see Tables 2 and 3; ESI-MS (pos.): m/z 448 [M + H]+; HR-ESI-MS (pos.): m/z 448.2602 (calcd for C27H34N3O3, 448.2600).
3.9 Cytotoxicity Bioassay
The cytotoxicity assay was performed according to an MTT [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide] method [14], by use of the following five human cancer cell lines: HL-60, SMMC-7721, A-549, MCF-7, and SW-480. The IC50 values were calculated by Reed and Muench’s method [15].
References
Editorial Committee of Flora of China, Flora of China (Science Press, Beijing, 1995), p. 157
D.L. Bemis, J.L. Capodice, P. Gorroochurn, A.E. Katz, R. Buttyan, Int. J. Oncol. 29, 1065–1073 (2006)
C.W. Wright, J.D. Phillipson, S.O. Awe, G.C. Kirby, D.C. Warhurst, J. Quetin-Leclercq, L. Angenot, Phytother. Res. 10, 361–363 (1996)
N.J. Martin, S.F. Ferreiro, F. Barbault, M. Nicolas, G. Lecellier, C. Paetz, M. Gaysinski, E. Alonso, O.P. Thomas, L.M. Botana, P. Raharivelomanana, Phytochemistry 109, 84–95 (2015)
N. Neuss, Indole and Biogenetically Related Alkaloids, vol. Chapter 17 (Academic Press, New York, 1980)
Y. Gao, D.S. Zhou, L.M. Kong, P. Hai, Y. Li, F. Wang, J.K. Liu, Nat. Prod. Bioprospect. 2, 65–69 (2012)
R. Verpoorte, J. Nat. Prod. 49, 1–25 (1986)
L. Li, H.P. He, H. Zhou, X.J. Hao, Nat. Prod. Res. Dev. 19, 235–239 (2007)
M. Lounasmaa, P. Hanhinen, J. Nat. Prod. 63, 1456–1460 (2000)
R. Sheludko, I. Gerasimenko, H. Kolshorn, J. Stöckigt, J. Nat. Prod. 65, 1006–1010 (2002)
L. Katoa, R.M. Bragaa, I. Kochb, L.S. Kinoshitab, Phytochemistry 60, 315–320 (2002)
L.F. Bjeldanes, G.W. Chang, J. Agric. Food Chem. 23, 1010–1011 (1975)
J.L. Pousset, J. Poisson, L. Olivier, J. Le Men, M.M. Janot, C.R. Acad, Science 261, 5538 (1965)
T.J. Mosmann, Immunol. Methods 65, 55–63 (1983)
L.J. Reed, H. Muench, Am. J. Hyg. 27, 493–497 (1938)
Acknowledgments
This work was financially supported by “Large-scale Compound Library” Project of National Development and Reform Commission of China.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare no conflict of interest.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Gao, Y., Zhou, DS., Hai, P. et al. Hybrid Monoterpenoid Indole Alkaloids Obtained as Artifacts from Rauvolfia tetraphylla . Nat. Prod. Bioprospect. 5, 247–253 (2015). https://doi.org/10.1007/s13659-015-0074-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13659-015-0074-2