
Abstract
Four new 9,19-cycloartane triterpenoids, cimilactone E (1), cimilactone F (2), 2′-O-(E)-butenoyl-23-epi-26-deoxyactein (3), and 2′,12β-O-diacetylcimiracemonol-3-O-β-d-xylopyranoside (4), together with four known constituents (5–8) were isolated from the roots of Cimicifuga foetida. The new structures were elucidated by extensive spectroscopic analysis. In addition, compounds 7 and 8 showed significant Wnt signaling pathway inhibitory activity, with IC50 values of 3.33 and 13.34 μM, respectively, using the luciferase reporter gene assay.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
Wnt signaling pathway plays an important role in numerous biological processes, including axis formation, cell fate specification, cell polarity determination, and cell migration [1]. Pathologically, Wnt signaling pathway is frequently aberrant in wide spectrum of malignancies, such as colon cancer, liver cancer, leukemia, melanoma, pancreatic cancer, and breast cancer [2]. Thus, screening inhibitors of Wnt signaling pathway has been considered as effective therapeutic strategies to combat cancer [3].
Traditionally, the roots of Cimicifuga foetida have been used as a cooling and detoxifying remedy and officially listed in the Chinese Pharmacopoeia [4]. The theory of traditional Chinese medicine defines a tumor as a type of toxin. Base on this theory, we hypothesized that chemical constituents of Cimicifuga may have the effects against cancer, which led us to uncover a series of 9,19-cycloartane triterpenes. Many of them showed cytotoxic activity against tumor cell lines, in which five compounds isolated from C. yunnanensis induced apoptosis of MCF-7 cells via p53-dependent mitochondrial pathway, recently [5–14]. However, there have been no reports about the Wnt signaling pathway inhibitory activity of 9,19-cycloartane triterpenes by far. Therefore, to further screen inhibitors against Wnt signaling pathway from Cimicifuga spp., we carried out a study on the roots of Cimicifuga foetida from Yulong County of Yunnan province. Consequently, four new compounds, cimilactone E (1), cimicilactone F (2), 2(E)-O-butenoyl-23-epi-26-deoxyactein (3), 2′-O-acetylcimirace-moside H (4), and four known compounds cimicilactone A (5) [15], 12β-hydroxy-7(8)-en-cimigenol (6) [16], cimicifoetiside B (7) [17], and 2′-O-acetyl cimiracemoside M (8) [18] (see Fig. 1) were isolated from C. foetida. Furthermore, all compounds isolated were evaluated for their inhibition of Wnt signaling pathway. Among them, compounds 7 and 8 showed significant inhibitory activities (see Fig. 2), with IC50 values of 3.33 and 13.34 μM, respectively. The present paper described the isolation, structure elucidation, and biological activities of aforementioned compounds.

Structures of compounds 1–8

Inhibition of luciferase activity
2 Results and Discussion
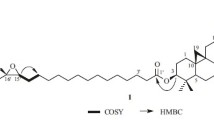
Compound 1 was obtained as a white powder. The IR spectrum showed absorptions for hydroxyl groups at 3438 cm−1 and carbonyl groups at 1735 cm−1, respectively. Its molecular formula (C35H52O10) with ten degrees of unsaturation was deduced from HREIMS (m/z 632.3533 [M]+; calcd. 632.3560). In 1H NMR spectrum (Table 1), the characteristic cyclopropane methylene signals at δ H 0.15 and 0.50 (each 1H, d, J = 4.3 Hz), one secondary methyl signal at δ H 0.98 (d, J = 6.4 Hz), four tertiary methyl groups at δ H 0.84–1.26 (each 3H, s), an anomeric proton at δ H 4.83 (1H, d, J = 8.0 Hz) and two acetyl methyl groups at δ H 2.16 and 2.18 (each 3H, s) were observed. The 13C NMR and DEPT spectra of 1 exhibited 35 signals, of which 26 were attributed to the aglycon, five to a pentose residue, and four to two acetyl groups. The 13C NMR and DEPT spectrum of the aglycon of 1 also showed an ester carbonyl carbon at δ C 174.2 and two acetoxy carbonyl groups at δ C 171.1 and 170.5. The aforementioned data suggested that 1 was a 9,19-cycloartane tetranortriterpene glycoside with three carbonyl groups. The NMR spectroscopic data of 1 (Table 1) closely resembled that of cimilactone A (5), except for the presence of an additional acetoxy group. In the HMBC spectrum (Fig. 3), a correlation was observed between the proton at δ H 4.87 (1H, d, J = 8.0 Hz, H-1′) and the methine carbon at δ C 88.8 (C-3), suggesting that the sugar moiety was located at C-3. In 1H-1H COSY spectrum, the correlations of a downfield resonance at δ H 5.58 (1H, dd, J = 8.0 and 9.0 Hz, H-2′) with H-3′ (δ H 4.20, m) and H-1′ (δ H 4.83, d, J = 8.0 Hz), together with the HMBC correlations from the proton resonance (H-2′) to a carbonyl group (δ C 170.5, s) located the acetoxy group at C-2′. The sugar obtained after acid hydrolysis was identified as d-xylose by comparing its TLC and specific rotation with a standard. The relative configurations of H-3, H-5, H-12, H-16, and H-17 were established as in the α-orientation on the basis of the ROESY correlations of H-1′/H-3; H-3/H-5; H-12/H-17; H-16/H-17 and H-17/Me-18 (Fig. 3). Therefore, the structure of 1 was determined to be (3β,12β,16β)-12,2′–diacetoxy-3-hydroxy-24,25,26,27-tetranor-cycloartan-23,16-olide 3-O-β-d-xylopyranside (1), named cimilactone E.

Key 1H-1H COSY, HMBC and ROESY correlations of compound 1
Compound 2 was obtained as a white powder. The IR spectrum showed absorptions for hydroxyl (3442 cm−1), carbonyl group (1732 cm−1), and double bond (1632 cm−1), respectively. The HREIMS spectrum gave an [M]+ ion peak at m/z 658.3713 (calcd 658.3717), consistent with the molecular formula C37H54O10, requiring eleven rings or sites of unsaturation. The NMR data of 2 was similar to cimilactone E (1), except for the resonances of the sugar moiety. The 13C NMR spectrum revealed carbons assignable to a 2-butenoyl moiety at δ C 165.8 (s), 123.6 (d), 144.8 (d). Besides, the correlation from the proton resonance (δ H 5.68, dd, J = 7.9 and 9.0 Hz, H-2′) to the carbonyl group (δ C 166.2, s, C-1″) in the HMBC spectrum located the 2-butenoyl group at C-2′, which was further confirmed by the 1H-1H COSY correlations of the proton at δ H 5.68 (H-2′) with H-1′ and H-3′. In addition, the coupling constant (J = 15.6 Hz) of the two olefinic protons at δ H 6.12 (H-2″) and δ H 7.14 (H-3″) confirmed the E-geometry of a double bond in the 2-butenoyl moiety. The sugar obtained after acid hydrolysis was confirmed as d-xylose by comparing its TLC and specific rotation with a standard. The relative configurations of H-3, H-5, H-12, H-16, and H-17 were proposed as α-orientation, respectively, by the same way as that of 1. Therefore, the structure of 2 was identified as (3β,12β,16β)-12-acetoxy-3-hydroxy-24,25,26,27-tetranor-cycloartan-23,16-olide-3-O-{2′-O-(E)-2″-butenoyl}-β-d-xylopyran-side (2), named cimilactone F.
Compound 3, was obtained as white powder, which gave an [M]+ ion at m/z 728.4120 [M]+ (calcd 728.4120) in HREIMS, consistent with the molecular formula of C41H60O11. The IR spectra showed absorption bands for hydroxyl groups at 3442 cm−1, carbonyl groups at 1730 cm−1 and double bond at 1632 cm−1, respectively. Typical proton signals of a cyclopropane methylene group (δ H 0.14 and 0.44, each 1H, d, J = 4.3 Hz); five tertiary methyl groups (δ H 0.83–1.47, each 3H, s); two secondary methyl group [(δ H 1.02 (3H, d, J = 6.0 Hz) and 1.67 (3H, d, J = 6.6 Hz)]; one acetyl methyl group (δ H 2.15, 3H, s); one 2(E)-butenoyl group [δ H 6.11 (1H, dd, J = 1.6 and 15.5 Hz), 7.12 (m) and 16.7 (3H, d, J = 6.6 Hz)], and an anomeric proton (δ H 4.88, 1H, d, J = 7.8 Hz) were observed in the 1H NMR spectrum (Table 1). The 13C NMR and DEPT spectrum (Table 1) of 3 exhibited 41 signals, of which 30 were attributed to the aglycon, five to a pentose residue, two to an acetyl group, and four to an 2-butenoyl group. Additionally, the aglycon of 3 also showed a hemiketal moiety at δ C 74.9 (d), 106.3 (s) and 68.5 (t), and an epoxyethane signals at δ C 62.6 (d) and 62.9 (s), which indicated that the aglycon of 3 was similar with 23-epi-deoxyacteol. A comparison of the spectroscopic data of 3 with those of 23-epi-26-deoxyactein showed that 3 closely resembles of it except for the presence of another tetra-carbon unit (2-butenoyl group) [19]. Additionally, the E-geometry of a double bond in the 2-butenoyl was confirmed in the same way with that of 2. In 1H-1H COSY spectrum, the correlations of a resonance at δ H 7.12 (m, H-3″) with H-2″ (δ H 6.11, dd, J = 1.6 and 15.6 Hz) and H-4″ (δ H 1.67, d, J = 6.6 Hz), together with the HMBC correlation from the proton resonance (δ H 5.67, dd, J = 7.8 and 10.2 Hz, H-2′) to a carbonyl group (δ C 166.2, s) located the 2(E)-butenoyl group at C-2′. Thus, 3 was characterized as 2′-O-2(E)-butenoyl-23-epi-26-deoxyactein (3).
Compound 4 was obtained as a white powder. The IR spectrum showed absorptions for hydroxyl (3439 cm−1), carbonyl groups (1730 cm−1), respectively. Its molecular formula (C39H60O12) with ten degrees of unsaturation was deduced from the analyses of 13C NMR and HREIMS data (m/z 720.4078 [M]+; calcd. 720.4085). In 1H NMR spectrum (Table 1), the signals due to a cyclopropane methylene group (δ H 0.15 and 0.46, each 1H, d, J = 3.6 Hz); six tertiary methyl groups (δ H 0.85–1.79, each 3H, s); one secondary methyl group (δ H 1.35, 3H, d, J = 6.0 Hz); two acetyl methyl group (δ H 2.17 and 2.10, each 3H, s); and an anomeric proton (δ H 4.82, 1H, d, J = 7.8 Hz) were observed. The 13C NMR and DEPT spectrum (Table 1) of 4 exhibited 39 signals, of which 30 were attributed to the aglycon, five to a pentose residue, and four to two acetyl groups. All above showed 4 was similar to cimiracemoside H, except for the presence of an additional acetoxy group. In 1H-1H COSY spectrum, the correlations of a resonance at δ H 4.19 (m, H-3′) with H-2′ (δ H 5.58, t, J = 9.0 Hz) and H-4′ (δ H 4.22, m), together with the HMBC correlation from a carbonyl group (δ C 170.5, 3H, s) to the proton resonance (δ H 5.58, H-2′) located the acetoxy group at C-2′. The ROESY correlations of H-17/H-22 and Me-21/H-24 proved H-22 in the α-orientation and H-24 as R configuration. Therefore, 4 was characterized as (3β,12β,16β,20S,22R,23S,24R)-16:23; 22:25-diepoxy-12,2′-diacetoxy-3,23,24-trihydroxy-9, 19-cyclocanostane-3-O-β-d-xylopyranoside (4), named 2′-O-acetyl cimiracemoside H.
As noted in the introduction, the roots of C. foetida have been used as cooling and detoxification agents by Chinese people since ancient time. Previous reports have shown that many pure 9,19 cycloartane triterpenoids isolated from this species exhibited cytotoxic activity against 11 tumor cell lines (including HepG2, MDA-MB-A231, HL-60, SMMC-7721, A549, SK-BR-3, PANC-1, K562, U933, HEG-2, and SGC-7091) in vitro, respectively [5–7, 17, 20, 21]. But there is no report on inhibition of Wnt signaling pathway. The pure components isolated in the present paper were screened against Wnt signaling pathway using the luciferase reporter gene assay. The known compounds 7 and 8 showed notable activity with the IC50 values of 3.33 and 13.34 μM, respectively. To the best of our knowledge, this is the first time to report the inhibitory activity against Wnt signaling pathway of 9,19-cycloartane triterpenes. These data suggested that some chemical constituents from C. foetida might be valuable to against tumorigenesis through inhibition to Wnt signaling pathway.
3 Experiments Section
3.1 General Experimental Procedures
UV spectra were recorded in MeOH on a shimadizu UV-210A spectrometer. IR spectra were recorded on Shimadzu IR-450 spectrometer with KBr disc. Optical rotations were measured a Horiba SEAP-300 polarimeter. 1H NMR and 13C NMR spectra were recorded using a Bruker AM-600 spectrometer with TMS as internal standard, operating at 600 and 150 MHz, respectively. All compounds were measured in solvents pyridine-d 5 . ESIMS and HRESIMS were carried out on a Waters Autospec Premier-P776 spectrometer. TLC analysis was performed on silica gel GF254 plate (Qingdao Marine Chemical, Inc.). Lichroprep RP-18 (40–63 μm, Merck) and silica gel (200–300 mesh) was used for column chromatography. Semipreparative HPLC was carried out on an Agilent 1260 liquid chromatograph with a ZORBAX SB C-18 column (9.4 × 250 mm,5 μm,) and a ZORBAX XDB C-18 column (9.4 × 250 mm, 5 μm).
3.2 Plant Materials
The roots of C. foetida (82 kg) were collected from Yulong County of Yunnan province of China in September 2010 and authenticated by Prof. Shen-Ji Pei of Kunming Institute of Botany, where a voucher specimen (KUN No. 20100906) is deposited.
3.3 Extraction and Isolation
The air-dried roots of C. foetida (82 kg) were crushed with a blender and refluxed with 95 % MeOH for three times (5 h, each). The residue yield by removal of the solvent was dissolved in water to form a suspension. The aqueous suspension was successively partitioned with EtOAc and n-BuOH. The EtOAc (5.6 kg) fraction was absorbed on 12 kg silica gel and chromatographed on a prepacked (120 kg) silica gel column, eluting stepwise with CHCl3-MeOH (CHCl3, 100:1, 50:1, 20:1, 5:1) to give five fractions (I–V). Fr.IV (350 g) was subjected to silica gel chromatograph eluted with CHCl3-acetone (10: 1) to give five sub-fractions (Fr.IV. 1–5). Fr.IV.4 (0.5 g) was chromatographed repeatedly over HPLC (SB C-18 column, CH3CN-H2O, 7:3) to obtain compound 7 (5 mg) successively. Fr.IV.5 (5 g) was applied repeatedly to CC over RP-18 gel (60, 70, 80 and 90 % MeOH-H2O) to give fractions IV.5.1–4. Fr.IV.5.2 (60 mg) afforded compound 6 (5 mg), after repeated elution with a CHCl3-acetone (5:1, 3:2) system over silica gel CC and a CH3CN-H2O (65 %) over HPLC (SB C-18 column). Fr.IV.5.3 (450 mg) was separated by column chromatograph eluted with CHCl3-acetone (3:1), and by HPLC (SB C-18 column) with CH3CN/H2O (45 %) to obtain compounds 1 (10 mg), 4 (2.2 mg) and 5 (3 mg), respectively. Fr.IV.5. 4 (380 mg) was also separated by column chromatograph eluted with CHCl3-acetone (3:1), then HPLC (XDB C-18 column) with CH3CN-H2O (65 %) to yield compounds 2 (1.6 mg), 3 (1.8 mg) and 8 (5 mg).
Compound 1: white powder (MeOH); \( [\alpha ]_{\rm{D}}^{20} \): −41.67 (c 0.24, MeOH); UV (MeOH) λ max (log ε): 209 (2.32) nm; IR (KBr) v max: 3438 (OH), 2964, 1735 (C=O), 1631(C=C), 1442, 1375, 1242, 1032, 982 cm−1; positive HRESIMS m/z 632.3533 [M]+, (C35H52O10, calcd. 632.3560), 1H and 13C NMR data, see Table 1.
Compound 2: white powder (MeOH); \( [\alpha ]_{\rm{D}}^{20} \): −90.67 (c 0.06, MeOH); UV (MeOH) λ max (log ε): 206 (4.29) nm; IR (KBr) v max: 3442 (OH), 2963, 2936, 1732 (C=O), 1655(C=C), 1444, 1365, 1245, 1184, 1072, 984 cm−1; positive HRESIMS m/z 658.3713 [M]+, (C37H54O10, calcd. 658.3717), 1H and 13C NMR data, see Table 1.
Compound 3: white powder (MeOH); \( [\alpha ]_{\rm D}^{20} \): −49.18 (c 0.18, MeOH); UV (MeOH) λ max (log ε): 206 (4.14) nm; IR (KBr) v max: 3441 (OH), 2932, 1730 (C=O), 1651(C=C), 1443, 1365, 1250, 1184, 1072, 1032, 982 cm−1; positive HRESIMS m/z 728.4120 [M]+, (C41H60O11, calcd. 728.4136), 1H and 13C NMR data, see Table 1.
Compound 4: white powder (MeOH); \( [\alpha ]_{D}^{20} \): −19.67 (c 0.13, MeOH); UV (MeOH) λ max (log ε): 201 (3.58) nm; IR (KBr) v max: 3439 (OH), 2939, 1735 (C=O), 1632(C=C), 1462, 1377, 1244, 1159, 1047, 981, 602–576 cm−1; positive HREIMS m/z 720.4078 [M]+, (C39H60O12, calcd. 720.4085), 1H and 13C NMR data, see Table 1.
The known compounds, cimilactone A (5), 12β-hydroxy-7(8)-en-cimigenol (6), cmicifoetiside B (7) and 2′-O-acetyl cimiracemoside M (8) were identified by comparing their physical and spectroscopic data with reported data.
3.4 Acidic Hydrolysis of 1–4
A solution of each new compound (1 mg) in 0.5 N HCl (3 ml) was refluxed for 4 h. The reaction mixture was diluted in 10 mL water and extracted with chloroform. After separating the organic layer, the aqueous phase was neutralized with Ag2CO3 to obtain some white precipitate. The precipitate residue was dissolved in pyridine and analyzed by TLC in n-BuOH-acetone-H2O (4:3:1, v/v), which had the same R f value with D-(+)-xylose [22].
3.5 Luciferase Activity
The Wnt signaling inhibitory activity of the eight 9, 19-cycloartane triterpenes (1–8) using the luciferase reporter gene assay as previously described [2]. Briefly, HEK293W cells were seeded in 96 well plate, and the luciferase activities were measured after incubation with the triterpenes for 24 h, using the Dual-Lucy Assay Kit (Promega) according to the manufacturer’s instructions.
References
K.M. Cadigan, R. Nusse, Genes Dev. 11, 3286–3305 (1997)
X.Y. Li, Y.Y. Wang, C.G. Yuan, X.J. Hao, Y. Li, Nat. Prod. Bioprospect. 3, 24–28 (2013)
A. Gregorieff, H. Clevers, Genes Dev. 2005(19), 877–890 (2005)
Pharmacopoeia Commission of the People’s Republic of China, The Pharmacopoeia of Chinese People’s Republic (The Chemical Industry Publishing House, Beijing, 2010), pp. 68–69
Y. Nian, M.X. Zhang, Y. Li, Y.Y. Wang, J.C. Chen, L. Lu, L. Zhou, M.H. Qiu, Phytochemistry 72, 1473–1481 (2011)
Y. Nian, H.Y. Wang, J. Su, L. Zhou, M.H. Qiu, Fitoterapia 83, 293–297 (2012)
L. Lu, J.C. Chen, Y. Li, C. Qing, Y.Y. Wang, Y. Nian, M.H. Qiu, Chem. Pharm. Bull. 60, 571–577 (2012)
D.S. Li, Y. Nian, Y. Sun, M.H. Qiu, Helv. Chim. Acta 94, 632–638 (2011)
L.R. Sun, J. Yan, L. Lu, S.J. Pei, Z.R. Li, L. Zhou, X.M. Zhang, M.H. Qiu, Helv. Chim. Acta 90, 1313–1318 (2007)
L. Lu, J.C. Chen, H.J. Song, Y. Li, Y. Nian, M.H. Qiu, Chem. Pharm. Bull. 58, 729–733 (2010)
D.F. Zhu, Y. Nian, H.Y. Wang, Z.R. Zhang, Y.B. Song, R.T. Li, M.H. Qiu, J. Nat. Med. 12, 0294–0296 (2014)
H.Y. Wang, Y. Nian, C.Y. Ma, J.Q. Liu, Y.B. Song, L. Zhou, M.H. Qiu, Chin. J. Chem. 30, 1265–1268 (2012)
Y. Nian, H.Y. Wang, L. Zhou, J. Su, Y. Li, M.H. Qiu, Planta Med. 79, 60–69 (2013)
Y. Nian, H. Zhu, W.R. Tang, Y. Luo, J. Du, M.H. Qiu, J. Nat. Prod. 76, 896–902 (2013)
Y. Liu, D. Chen, J. Si, G. Tu, D. An, J. Nat. Prod. 65, 1486–1488 (2002)
Y. Nian, H.Y. Wang, L. Zhou, J. Su, Y. Li, M.H. Qiu, Planta Med. 78, 1–10 (2012)
L.R. Sun, C. Qing, Y.L. Zhang, S.Y. Jia, Z.R. Li, S.J. Pei, M.H. Qiu, L.G. Michael, X.Q. Samuel, Beilstein J. Organ. 3(3) (2007)
Y.S. Fan, W. Jia, A.H. Zhao, J. Tang, H.Q. Duan, Chin. Chem. Lett. 17, 1477–1480 (2006)
S.N. Chen, W. Li, D.S. Fabricant, B.D. Santarsiero, A. Mesecar, J.F. Fitzloff, H.H.S. Fong, N.R. Farnsworth, J. Nat. Prod. 65, 601–605 (2002)
Z. Tian, R.L. Pan, J.Y. Si, P.G. Xiao, Fitoterapia 77, 39–42 (2006)
J.Y. Chen, P.L. Li, X.L. Tang, X.L. Tang, S.J. Wang, Y.T. Jiang, L. Shen, B.M. Xu, Y.L. Shao, G.Q. Li, J. Nat. Prod. 77, 1997–2005 (2014)
Y. Shao, A. Harris, M. Wang, H. Zhang, A.C. Geoffrey, M. Bowman, E. Lemmo, J. Nat. Prod. 63, 905–910 (2000)
Acknowledgments
This project was supported by Program for National Natural Science Foundation of China (Nos. U1132604 and 81302670), The Major Deployment Program of the CAS (No. KSZD-EW-Z-004-01), and Foundation of State Key Laboratory of Phytochemistry and Plant Resources in West China (P2008-ZZ05). Authors also were particularly grateful to Research Group of Prof. Yan Li for bioassay tests.
Conflict of Interest
All authors declare no conflict of interest.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Di-Fan Zhu and Guo-Lei Zhu have contributed equally to this work.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
About this article

Cite this article
Zhu, DF., Zhu, GL., Kong, LM. et al. Cycloartane Glycosides from the Roots of Cimicifuga foetida with Wnt Signaling Pathway Inhibitory Activity. Nat. Prod. Bioprospect. 5, 61–67 (2015). https://doi.org/10.1007/s13659-015-0053-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13659-015-0053-7