
Abstract
Six new 9,19-cycloartane triterpene derivatives, as well as 3 known analogues (7–9), were isolated from the roots of Cimicifuga foetida L. Their structures were established on the basis of extensive spectroscopic analyses (IR, UV, ORD, HRESIMS, 1D and 2D NMR).
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
Cimicifuga foetida L. or actaea foetida also named “Shengma”, a well known medicinal plant widely distributed in China, has been used for alleviation fever, pain, and inflammation since ancient times in China [1–3]. Currently, it is prescribed as one of the source plants for the treatment of headache, sore throat, toothache, and uterine prolapse in the Chinese Pharmacopoeia 2010 [4]. Phytochemical investigation have shown that 9,19-cycloartane triterpenoids and their glycosides are the main constituents of Cimicifuga. Meanwhile, because of their structural diversity and significant antitumor activity, this kind of components attracted so much attention [5–14].
In our continuous search for the bioactive triterpenoids, six new cycloartane-type triterpenoids, namely 4′-O-acetyl-cimigenol-3-O-β-d-xylopyranoside (1), 2′,12-O-diacetyl-25-anhydrocimicigenol-3-O-β-d-xylopyranoside (2), 12β-hydroxy-1,19:9,11-didehydro-9,10-seco-cimigenol-3-O-β-d-xylopy-ranoside (3), (23S, 24R)-12β-hydroxy-7,8-dihydro-12-deacetyl-acetaeaepoxide-3-one. (4), 16,17-dide- hydro-2′,24-O-diacetylhydroshengmanol-3-O-β-d-xylopyranoside (5), and (23S,24S,25S)-16,23:23,- 26-diepoxy-24,25-dihydroxy-9,19-cycloart-1,2-en-3,12-dione (6), together with three known analogues, asiaticoside A (7), 24(S)-O-acetylhydroshengmanol-3-O-β-d-xylopyranoside (8), and cimisterol A (9) were isolated from the roots of C. foetida. All the new compounds were evaluated for their cytotoxicities against five selected human cancer cell lines (HL-60, SMMC-7721, A-549, MCF-7 and SW480).
2 Results and Discussion
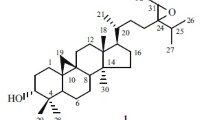
Compound 1 had the molecular formula of C37H58O10, which was determined by its HR-EIMS at m/z 662.4414 [M]+. The IR spectrum showed absorption for hydroxyl group at 3425 cm−1. The 1H NMR spectrum (Table 1) showed characteristic cyclopropane methylene signals at δ H 0.29 and 0.52 (each 1H, d, J = 3.8 Hz), a secondary methyl at δ H 0.86 (d, J = 6.5 Hz), six tertiary methyls at δ H 1.01 to 1.52 (each 3H, s), an acetyl group at δ H 1.98, and an anomeric proton at δ H 4.89 (d, J = 7.6 Hz). The 13C NMR and DEPT spectroscopic data (Table 2) of 1 displayed a characteristic cimigenol-type triterpenoid carbon resonances, corresponding to the methylene carbon of the cyclopropane ring at δ C 31.2 (C-19), four oxymethine carbons at δ C 89.0 (C-3), 80.6 (C-15), 72.2 (C-23), and 90.5 (C-24), and two oxygened quaternary carbons at δ C 112.3 and 71.3. Apart from above data, a glycosidic moiety signals [δ C 107.7 (d), 76.1 (d), 75.3 (d), 73.5 (d) and 63.5 (t)] were also obseverd in its 13C NMR spectrum. These data showed similarities as those of cimigenol-3-O-[2′-O-acetyl]-β-d-xylopyranoside [12]. However, a detailed comparison of their 1D NMR spectra revealed that they had different sugar unit. The upfield shift of H-2′ (δ H 5.56 → δ H 4.08) and the downfield shift of H-4′ (δ H 4.30 → δ H 5.44) in their 1H NMR spectra, along with the HMBC correlation (Fig. 1) of H-4′ (δ H 5.44) with the carbonyl group of the acetoxyl group (δ C 171.0) and of the anomeric proton with C-3, indicated that a 4′-O-acetyl-xylopyranosyl at C-3 in 1 replaced the 2′-O-acetyl-xylopyranosyl in cimigenol-3-O-[2′-O-acetyl]-β-d-xylopyranoside. The sugar unit of 1 was further confirmed by comparing its TLC and specific rotation with a standard after acid hydrolysis. Thus, the planar structure of 1 was determined.

Major correlations in 2D NMR spectra of compound 1
In the ROESY spectrum (Fig. 1), correlations of H-3/H-5 and H-15/H3-18 suggested that H-3 and H-15 were α- and β-oriented, respectively. Moreover, the configurations of C-23 and C-24 were assigned as R and S, respectively, by comparing the coupling constants of H-23 (9.0 Hz) and H-24 (0 Hz) of 1 with those of known compounds [15]. Therefore, compound 1 was established to be 4′-O-acetylcimigenol-3-O-β-d-xylopyranoside.
Compound 2 was obtained as a white powder and gave a molecular formula of C39H58O11 by its HR-EIMS (m/z 702.3974 [M]+). The 1H and 13C NMR spectra (Tables 1, 2) of 2 were very similar to those of 25-anhydrocimicigenol-3-O-[2′-O-acetyl]-β-d-xylopyranoside [16], with the exception of an additional acetyl group, which was assigned to C-12 on the basis of the HMBC correlation of H-12 (δ H 5.27) with the acetyl carbonyl carbon at δ C 170.9 and the 1H-1H COSY cross peak of H-12 (δ H 1.13) with H-11 (δ H 2.94). Significant ROESY correlation of H-12 with H-17 indicated a β-orientation of the substituent acetyl group at C-12. Therefore, the structure of 2 was determined as 2′,12-O-diacetyl-25-anhydrocimicigenol-3-O-β-d-xylopyranoside.
Compound 3 was isolated as a white powder, showing [M + Na]+ ion at m/z 657.3602 in the HR-ESIMS consistent with the empirical molecular formula C35H54O10 (calcd 657.3615), requiring 9 sites of unsaturation. The IR and UV spectra exhibited absorption bands for hydroxyl group (3431 cm−1) and conjugated double bond (λmax 249 nm; 1631 cm−1), respectively. The 1H NMR spectrum exhibited a sec-methyl signal at δ H 1.42 (3H, d, J = 6.4 Hz), six tert-methyls at δ H 0.95–1.54 (each 3H, s), two olefinic methine signals at δ H 5.92 (1H, br s) and 6.11 (1H, br s), and an anomeric methine signal at δ H 4.85 (1H, d, J = 7.6 Hz). The 13C NMR showed 35 carbon resonances (Table 2), of which 30 were attributed to a triterpene skeleton and five to a pentose. A DEPT NMR experiment permitted differentiation of the 30 carbon signals into seven methyls, five methylenes, eleven methines (including five oxygenated and two olefinic signals), and seven quaternary carbons (including two oxygenated and two olefinic signals). The diagnostic signals of two oxygen-bearing methine carbons at δ C 90.6 (C-24) and 70.4 (C-23), and a ketal carbon at δ C at 112.7 suggested that 3 was a cimigenol-type triterpene compound. Further inspection of the 1D NMR and HSQC spectra of 3, the characteristic cyclopropane methylene resonances H2-19 and two quaternary carbons (C-9 and C-10) were not observed at the characteristic high magnetic field. Besides, comparison the NMR spectra of 3 with those of 12β-hydroxycimigenol-3-O-β-d-xylopyranoside [17], the signals due to C-9, C-10, C-11, and C-19 showed a downfield shift from δ C 20.5, 26.1, 40.6, and 30.8 to 140.3, 138.7, 135.9, and 129.6, respectively, in 3. Such evidences indicated that 3 was a 9,10-seco-9,19-cyclolanostane glycoside with two double bonds. And the location of the double bonds (C10=C19 and C9=C11) could be further deduced. This was further supported by IR, UV and 2D NMR spectra (Fig. 2). Furthermore, the configurations of C-23 and C-24 were assigned as R and S, respectively, by the same way as 1. Ultimately, the structure of 3 was determined as 12β-hydroxy-10,19:9,11-didehydro-9,10-seco-cimigenol-3-O-β-d-xylopyranoside.

Key HMBC ( ) and 1H-1H COSY (
) and 1H-1H COSY ( ) correlations of compounds 3, 4 and 6
) correlations of compounds 3, 4 and 6
The molecular of compound 4 was assigned as C30H46O6 by HR-EIMS at m/z 502.3294 [M]+. The 1D NMR data of 4 (Tables 1, 2) showed that 4 was a highly oxygenated 9,19-cycloartane triterpene and resembled that of the aglycone of 7,8-dihydroactaeaepoxide-3-O-β-d-xylopyranoside [18]. However, the signals for the oxymethine at C-3 and the acetoxyl group at C-12 were absent. Instead a carbonyl group signal at δ C 215.3 and an upfield oxymethine at δ C 72.6 were observed, which indicated that the oxymethine (C-3) and the acetoxyl group (C-12) were replaced by a carbonyl group and a hydroxyl group, respectively. The evidence was established from HMBC correlations (Fig. 2) of H3-29 (δ H 1.23) and H3-30 (δ H 0.94) with C-3 (δ C 215.3), of H3-18 (δ H 1.51) with C-12 (δ C 72.6), together with the 1H-1H COSY correlations (Fig. 2) of H-1 (δ H 1.43, 1.71)/H-2 (δ H 2.30, 2.63), H-5 (δ H 1.57)/H-6 (δ H 0.99, 1.37)/H-7 (δ H 0.96, 1.30)/H-8 (δ H 1.66), H-11 (δ H 1.49, 2.64)/H-12 (δ H 4.10), H-15 (δ H 1.84, 1.96)/H-16(δ H 5.09)/H-17 (δ H 1.53). Thus, the planar structure of 4 was elucidated. Additionally, the substituent hydroxyl group at C-12 was assigned as β orientation by the ROESY correlation of H3-28 with H-12. Further inspection of the ROESY spectrum, the correlations of H-16 with H-22 and H-24, of H-22 with H-12, H-21, and H-28, and of H-24 with H-16, suggested that an α orientation of H-16, H-22, H-12, and H-24, respectively. So the configuration of C-23 was elucidated as S since C-23 and C-24 formed an oxirane. Ultimately, compound 4 was elucidated as (23S, 24R)-12β-hydroxy-7,8-dihydro-12-deacetylacetaeaepoxide-3-one.
Compound 5 was determined to have the molecular formula of C39H60O11 by its HR-EIMS (m/z 704.4138 [M]+). The 1H NMR spectrum (Table 1) displayed characteristic cyclopropane methylene signals at δ H 0.17 and 0.56 (each 1H, d, J = 3.8 Hz), a sec-methyl at δ H 1.00 (3H, d, J = 6.7 Hz), six tert-methyls at δ H 0.99–1.63 (each 3H, s), an acetyl methyl group at δ H 2.09 (3H, s), and an anomeric proton signal at δ H 4.86 (1H, d, J = 8.0 Hz), respectively, suggersting 5 to be a 9,19-cycloartane triterpene glycoside with a substituent acetyl group. The NMR data (Tables 1, 2) of 5 resembled those of 25-O-methyl-24-O-acetylhydroshengmanol-3-O-β-d-xylopyranoside [19], except for one more acetyl group for the sugar unit, one less substituent methoxy group at C-25, and the presence of two downfield signals at δ C 151.3 and 121.7 while the absence of an oxygen-bearing quaternary carbon and a methine resonance due to C-16 and C-17, respectively. On the basis of these observations, it was reasonable to deduce that 5 was a 16,17-dehydrated derivative with an additional acetyl group and a skimpy methoxy group of 25-O-methyl-24-O-acetylhydroshengmanol-3-O-β-d-xylopyranoside. As for 2, an acetyl group was determined to be at C-2′ for 5, which was further confirmed by the HMBC correlation between the H-2′ signal at δ H 5.20 and the carbonyl group signal at δ C 170.4. In the NMR spectra, the signals of the methoxy appeared in 25-O-methyl-24-O-acetylhydro-shengmanol-3-O-β-d-xylopyranoside couldn’t be observed in 5, while the chemical shift of C-25 shifted upfield from δ C 76.0 to 72.3. This suggested the methoxy group located at C-25 wasn’t appeared in 5. Additionally, the location of the double bond was further confirmed by the HMBC correlations of H-18 (δ H 1.27) and H-21 (δ H 1.00) with δ C 121.7, of H-15 (δ H 4.75) with δ C 151.3 and 121.7, and of H-23 (δ H 4.60) with δ C 151.3, respectively. The configuration of C-23 could be determined as β by ROESY correlation of H-23 with H-20. And the configuration of C-24 was assigned as S by comparison the coupling constant of H-24 (J = 2.5 Hz) with those of dahurinyl deacetate (J = 9 Hz, 24R) and isodahurinyl deacetate (J = 2.0 Hz, 24S) [20]. Therefore, 5 was elucidated as 16,17-didehydro-2′,24-O-diacetyl- hydroshengmanol-3-O-β-d-xylopyranoside.
Compound 6 was isolated as a white powder. Its molecular formula (C30H42O6) was deduced from HR-EIMS (m/z 498.2991 [M]+), corresponding to nine degrees of unsaturation. The 1H and 13C NMR spectroscopic data (Tables 1, 2) of 6 showed similarities with those of yunnanterpene A [21], except for the differences of rings A and C, and the chemical shifts of C-22, C-23, and C-24. Two methylene signals due to C-1 at δ C 33.3 and C-2 at δ C 37.4 appeared in the ring A of yunnanterpene A were absent from the 13C-DEPT spectrum of 6, respectively. Instead, two olefinic carbon signals at δ C 153.5 and 127.2 were observed. Besides, the signal due to C-3 showed an upfield shift from δ C 215.0 to 203.4. These evidences suggested the double bond was located at C-1 and C-2, which was further confirmed by the UV, IR (λmax 262 nm; 1669 cm−1), and the HMBC correlations (Fig. 2) of the olefinic protons at δ H 6.65 and 6.12 with the carbonyl carbon signals at δ C 204.3 (C-3), The other changes of the ring C was that the methine signal at δ C 72.1 (C-12) appeared in yunnanterpene A was absent instead of a quarternary carbonyl carbon signal at δ C 210.8 (C-12) in 6. Meanwhile, the 13C NMR signal due to C-11 showed a downfield shift from δ C 40.6 in yunnanterpene A to 46.1 in 6, and the signal due to C-13 exhibited an unfield from δ C 50.5 to 46.6, respectively. These observations indicated that the hydroxyl group was attached to C-12 was replaced by the carbonyl group, which was further confirmed by the HMBC correlations (Fig. 2) from H-11 (δ H 2.33 and 2.92) and H3-18 (δ H 1.38) to C-12 (δ C 210.8), respectively. All of the above observations were consistent with the HSQC, HMBC, and 1H-1H COSY correlations (Fig. 2). Besides, the downfield shift of C-22 and C-24 from δ C 37.5 and 83.7 to 41.3 and 86.3 and the upfield of C-23 from δ C 110.9 to 106.9 suggested the configurations of two compounds may be different. Furthermore, the diagnostic ROESY configurations of H-16 with H-16, H3-28 and H3-27, of H-24 with H-26α, of H3-27 with H-16 and H-26α, indicated that the α-orientation of H-16, H3-27, and H-24. And the configuration of C-23 was identified as S by comparision the chemical shifts of C-16 (δ C 73.0) and C-20 (δ C 25.7) with the 26-deoxyactein compound [C-16 (δ C 73.0) and C-20 (δ C 26.0)] [16] and 23-epi-26-deoxyactein [C-16 (δ C 74.5) and C-20 (δ C 23.0)] [22]. Hence, compound 6 was determined as (23S,24S,25S)-16,23:23,26-diepoxy-24,25-dihydroxy-9,19-cycloart-1,2-en-3,12-dione.
Three known compounds asiaticoside A (7) [23], 24(S)-O-acetylhydroshengmanol-3-O-β-d-xylopyranoside (8) [19], and cimisterol A (9) [13] were also isolated from this species. Their structures were identified by its 1D NMR spectra as well as comparison with reported data.
Compounds 1–6 isolated in the present study were evaluated for their cytotoxicities against five human cancer cell lines using MTT method, with cisplatin and taxol as the positive control. Unfortunately, none of them showed significant activity [24].
3 Experiments Section
3.1 General Experimental Procedures
Optical rotations were measured in MeOH with a Horiba SEAP-300 polarimeter. 1H and 13C NMR spectra were recorded in pyridine-d 5 on Bruker Avance III-600 MHz spectrometers (Bruker, Zürich, Switzerland), using TMS as internal standard for chemical shifts. Chemical shifts (δ) were expressed in ppm with reference to the TMS resonance. ESIMS, HRTOF-ESIMS and EIMS, HR-EIMS data were obtained using a VG Autospec-3000 and API QSTAR TOF spectrometer, respectively. Infrared spectra were recorded on a Shimadzu IR-450 instrument with KBr pellets. CD was detected with a Chirascan circular dichroism spectrograph (Applied Photophysis, England). Thin-layer chromatography was performed on precoated TLC plates (Silica gel GF254, Qingdao Marine Chemical, Inc.) and spots were visualized by heating after spraying with 10 % H2SO4 in EtOH. Semipreparative HPLC was performed on an Agilent 1100 liquid chromatograph with a YMC-Pack Pro C18 RS 10 mm × 250 mm column. Silica gel (mesh 200–300, Qingdao Marine Chemical, Inc.), Lichroprep RP-18 (40–63 µm, Merck), Amberlite IR-35 (10 mL) column and Sephadex LH-20 (Pharmacia) were used for column chromatography.
3.2 Plant Materials
The roots of Cimicifuga foetida (82 kg) were collected from Yulong County, Yunnan Province, in September 2010 and identified by Professor Pei shengji, Kunming Institute of Botany, Chinese Academy of Sciences. A voucher specimen (KUN No. 20100906) has been deposited at the State Key Laboratory of Phytochemistry and Plant Resources in West China, Kunming Institute of Botany, Chinese Academy of Sciences, PR China.
3.3 Extraction and Isolation
The air-dried roots of C. foetida (82 kg) were crushed with a blender and refluxed with 95 % EtOH at 70 °C for three times (5 h, each). The residue was yielded by removal of the solvent was dissolved in water to form a suspension. The aqueous suspension was successively partitioned with EtOAc and n-BuOH. The EtOAc (5.6 kg) fraction was absorbed on 12 kg silica gel and chromatographed on a prepacked (120 kg) silica gel column, eluting stepwise with CHCl3–MeOH (1:0, 100:1, 50:1, 20:1, 5:1) to give five fractions (A–E) Fraction C (230 g) was subjected to column chromatograph (CC) on silica gel (1.5 kg) and eluted with PE-Me2CO (5:1, 2:1, 0:1) to obtain C-1 (60 g), C-2 (40 g), and C-3 (105 g) as in the previous report [25]. Fraction C-2 (40 g) was purified using an ODS silica gel column with MeOH-H2O (60:40, 80:20, 100:0), followed by purification using preparative HPLC eluted with CH3CN-H2O (65:35), furnished compound 4 (2.3 mg). Similarly, using CH3CN-H2O (70:30) as eluent with a flow rate of 3 ml/min, compound 6 (9.6 mg) was purified from C-3 (105 g). Fraction D (200 g) was separated on silica gel eluted with CHCl3–Me2CO (gradient polarity from 15:1 to 5:1) to give ten subfractions (D-1-D-10). Fraction D-1 (10 g) was separated by CC (ODS silica gel) with MeOH–H2O (gradient polarity from 60:40 to 90:10) and purified by HPLC eluting with CH3CN–H2O (60:40, flow rate of 3 ml/min) to obtain 3 (6.0 mg) and 9 (2.5 mg) with retention times of 9.30 and 18.50 min, respectively. Sub-fraction D-6 (15 g) was subjected to silica gel (CH3Cl3–MeOH, gradient from 25:1 to 15:1) and then purified by an ODS silica gel column (MeOH–H2O, 70:30 to 100:0) and HPLC spectrum eluting with (CH3CN–H2O, 50:50, flow rate of 3 ml/min) to obtain 1 (6.2 mg) and 5 (5.1 mg) with retention times of 10.80 and 13.40 min, respectively. Sub-fraction D-8 (20 g) was chromatographed on a silica gel (CH3Cl3–MeOH, 30:1, 20:1, 15:1) and ODS (MeOH–H2O, 70:30 to 100:1), followed purified on HPLC CH3CN–H2O (67:33, flow rate of 3 ml/min) to yield 2 (5.3 mg), 7 (5.5 mg), and 8 (6.0 mg) with the retention times of 19.20, 26.70, and 12.40 min, respectively.
3.4 4′-O-Acetylcimigenol-3-O-β-d-xylopyranoside (1)
White powder; \([\alpha ]_{\text{D}}^{20}\) −3.59 (c 0.2, MeOH); IR (KBr) ν max 3425, 2964, 2936, 2870, 1742, 1626, 1458, 1413, 1379, 1308, 1252, 1170, 1047, 979 cm−1; 1H and 13C NMR data see Tables 1, 2; HREIMS m/z 662.4414 [M]+ (calcd for 662.4394).
3.5 2′,12-O-Diacetyl-25-anhydrocimicigenol-3-O-β-d-xylopyranoside (2)
White powder; \([\alpha ]_{\text{D}}^{20}\) −1.63 (c 1.8, MeOH); IR (KBr) ν max 3442, 2935, 2871, 1736, 1629, 1455, 1413, 1239, 1159, 1071, 1044, 982 cm−1; 1H and 13C NMR data see Tables 1, 2; HREIMS m/z 702.3974 [M]+ (calcd for 702.3979).
3.6 12β-Hydroxy-1,19:9,11-didehydro-9,10-seco-cimigenol-3-O-β-d-xylopyranoside (3)
White powder; \([\alpha ]_{\text{D}}^{20}\) −1.63 (c 0.01, MeOH); IR (KBr) ν max 3431, 2931, 2873, 1631, 1456, 1384, 1238, 1161, 1041, 975 cm−1; 1H (C5D5N, 600 MHz) and 13C NMR (C5D5N, 150 MHz) data see Tables 1, 2; ESIMS m/z 657 [M + Na]+; HRESIMS m/z 657.3602 (calcd for 657.3615).
3.7 (23S,24R)-12β-Hydroxy-7,8-dihydro-12-deacetyl- acetaeaepoxide-3-one. (4)
White powder; \([\alpha ]_{\text{D}}^{20}\) −18.17 (c 1.1, MeOH); IR (KBr) ν max 3441, 2966, 2932, 2875, 1705, 1628, 1465, 1383, 1248, 1061 cm−1; 1H and 13C NMR data see Tables 1, 2; HREIMS m/z 502.3294 [M]+ (calcd for 502.3294).
3.8 16,17-Didehydro-2′,24-O-diacetylhydroshengmanol-3-O-β-d-xylopyranoside (5)
White powder; \([\alpha ]_{\text{D}}^{20}\) −10.28 (c 0.01, MeOH); IR (KBr) ν max 3431, 2929, 2870, 2853, 1739, 1629, 1459, 1375, 1237, 1240, 1167, 1125, 1072, 1046, 980 cm−1; 1H and 13C NMR data see Tables 1, 2; HREIMS m/z 704.4138 [M]+ (calcd for 704.4136).
3.9 (23S,24S,25S)-16,23:23,26-Diepoxy-24,25-diihydroxy-9,19-cycloart-1,2-en-3,12-dione (6)
White powder; \([\alpha ]_{\text{D}}^{20}\) −24.21 (c 0.5, MeOH); IR (KBr) ν max 3440, 2931, 2871, 1712, 1669, 1457, 1380, 1278, 1167, 1019, 940 cm−1; 1H and 13C NMR data see Tables 1, 2; HRESIMS m/z 498.2991 [M]+ (calcd for 498.2981).
3.10 Acidic Hydrolysis of 1, 2 and 5
The new compounds 1, 2 and 5 (4 mg of each) were dissolved in MeOH (5 mL) and refluxed with 0.5 N HCl (3 mL) for 4 h. Each reaction mixture was diluted with H2O and extracted with CHCl3 (3 × 10 mL). The water layer was then neutralized by Ag2CO3, and the precipitate filtered to give a monosaccharide. Each monosaccharide of those compounds had an R f (EtOAc/CHCl3/MeOH/H2O, 3:2:2:1) and specific rotation \([\alpha ]_{\text{D}}^{20}\) +24.3 (c 0.10, H2O) corresponding to those of d-xylose (Sigma-Aldrich).
References
J.A. Compton, A. Culham, J.G. Gibbings, S.L. Jury, Biochem. Syst. Ecol. 26, 185–197 (1998)
J.A. Compton, A. Culham, S.L. Jury, Taxon 47, 593–634 (1998)
J.X. Li, Z.Y. Yu, Curr. Med. Chem. 13, 2927–2951 (2006)
Pharmacopoeia Commission of the People’s Republic of China, The Pharmacopoeia of Chinese People’s Republic (The Chemical Industry Publishing House, Beijing, 2010), pp. 68–69
S. Kadota, J.X. Li, K. Tanaka, T. Namba, Tetrahedron 51, 1143–1166 (1995)
J.X. Li, S. Kadota, X.F. Pu, T. Namba, Tetrahedron 35, 4575–4576 (1994)
C.J. Li, Y.H. Li, P.G. Xiao, T.J. Mabry, W.H. Watson, M. Krawiec, Phytochemistry 42, 489–494 (1996)
N.Q. Zhu, Y. Jiang, M.F. Wang, C.T. Ho, J. Nat. Prod. 64, 627–629 (2001)
Z. Tian, R.L. Pan, J.Y. Si, P.G. Xiao, Fitoterapia 77, 39–42 (2006)
Y. Nian, Y.L. Zhang, J.C. Chen, L. Lu, M.H. Qiu, C. Qing, J. Nat. Prod. 73, 93–98 (2010)
L.R. Sun, J. Yan, L. Zhou, Z.R. Li, M.H. Qiu, Molecules 16, 5701–5708 (2011)
Y. Nian, X.M. Zhang, Y. Li, Y.Y. Wang, J.C. Chen, L. Lu, L. Zhou, M.H. Qiu, Phytochemistry 72, 1473–1481 (2011)
Y. Nian, H.Y. Wang, J. Su, L. Zhou, M.H. Qiu, Fitoterapia 83, 293–297 (2012)
D.F. Zhu, G.L. Zhu, L.M. Kong, N.M. Bao, L. Zhou, Y. Nian, M.H. Qiu, Nat. Prod. Bioprospect 5, 61–67 (2015)
Y. Nian, H.Y. Wang, J. Su, L. Zhou, G. Feng, Y. Li, M.H. Qiu, Tetrahedron 68, 6521–6527 (2012)
L. Zhou, J.S. Yang, G.Z. Tu, J.H. Zou, Chem. Pharm. Bull. 54, 823–826 (2006)
A. Kusano, M. Shibano, G. Kusano, Chem. Pharm. Bull. 43, 1167–1170 (1995)
Z. Ali, I.K. Shabana, R.S. Pawar, D. Ferreira, I.A. Khan, J. Nat. Prod. 70, 107–110 (2007)
A. Kusano, M. Shibano, G. Kusano, Chem. Pharm. Bull. 44, 167–172 (1996)
Y. Shao, A. Harris, M.H. Wang, H.J. Zhang, G.A. Cordell, M. Bowman, E. Lemmo, J. Nat. Prod. 63, 905–910 (2000)
Y. Nian, H. Zhu, W.R. Tang, Y. Luo, J. Du, M.H. Qiu, J. Nat. Prod. 76, 896–902 (2013)
S.N. Chen, W.K. Li, D.S. Fabricant, B.D. Santarsiero, A. Mesecar, J.F. Fitzloff, H.H.S. Fong, N.R. Farnsworth, J. Nat. Prod. 65, 601–605 (2002)
J.C. Gao, F. Huang, J.C. Zhang, G.Y. Zhu, M.S. Yang, P.G. Xiao, J. Nat. Prod. 69, 1500–1502 (2006)
L.J. Reed, H. Muench, Am. J. Hyg. 27, 493–497 (1938)
H.Y. Wang, Y. Nian, C.Y. Ma, J.Q. Liu, Y.B. Song, L. Zhou, M.H. Qiu, Chin. J. Chem. 30, 1265–1268 (2012)
Acknowledgments
This project was supported by Program for National Natural Science Foundation of China (Nos. U1132604 and 81302670), The Major Deployment Program of the CAS (No. KSZDEW-Z-004-03), and Foundation of State Key Laboratory of Phytochemistry and Plant Resources in West China (P2008-ZZ05).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
All authors declare no conflict of interest.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Zhu, GL., Zhu, DF., Wan, LS. et al. Six New 9,19-Cycloartane Triterpenoids from Cimicifuga foetida L.. Nat. Prod. Bioprospect. 6, 187–193 (2016). https://doi.org/10.1007/s13659-016-0097-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13659-016-0097-3