
Abstract
Ginger is an important spice crop with medicinal values and gingerols are the most abundant pungent polyphenols present in ginger, responsible for most of its pharmacological properties. The present study focuses on the molecular mechanism of gingerol biosynthesis in ginger using transcriptome analysis. Suppression Subtractive Hybridization (SSH) was done in leaf and rhizome tissues using high gingerol-producing ginger somaclone B3 as the tester and parent cultivar Maran as the driver and generated high-quality leaf and rhizome Expressed Sequence Tags (ESTs). The Blast2GO annotations of the ESTs revealed the involvement of leaf ESTs in secondary metabolite production, identifying the peroxisomal KAT2 gene (Leaf EST 9) for the high gingerol production in ginger. Rhizome ESTs mostly coded for DNA metabolic processes and differential genes for high gingerol production were not observed in rhizomes. In the qRT-PCR analysis, somaclone B3 had shown high chalcone synthase (CHS: rate-limiting gene in gingerol biosynthetic pathway) activity (0.54 fold) in the leaves of rhizome sprouts. The presence of a high gingerol gene in leaf ESTs and high expression of CHS in leaves presumed that the site of synthesis of gingerols in ginger is the leaves. A modified pathway for gingerol/polyketide backbone formation has been constructed explaining the involvement of KAT gene isoforms KAT2 and KAT5 in gingerol/flavonoid biosynthesis, specifically the KAT2 gene which is otherwise thought to be involved mainly in β-oxidation. The results of the present investigations have the potential of utilizing KAT/thiolase superfamily enzymes for protein/metabolic pathway engineering in ginger for large-scale production of gingerols.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Ginger is a valuable spice crop used in pharmaceutical industries for its aromatic and medicinal properties. Gingerols are the most potent and pharmacologically active compounds in ginger. Of the gingerols, [6]-gingerol is the most abundant bioactive compound with many medicinal properties viz. analgesic (Young et al. 2005), gastroprotective, anti-cancer (Zhu et al. 2017), cardiotonic, anti-inflammatory (Xu et al. 2018), anti-oxidant (Almatroodi et al. 2021), anti-pyretic, anti-hepatotoxic, anti-angiogenic (Kim et al. 2005), anti-hyperglycemic (Samad et al. 2017) and anti-platelet aggregation activities (Liao et al. 2012). Molecular docking studies have revealed the effectiveness of [6]-gingerol against cancer (Kumar et al. 2017) and against COVID-19 (Rathinavel et al. 2020 and Haridas et al. 2021), making [6]-gingerol a future drug of choice. Somaclonal variation is utilized to generate genetic variants in ginger due to breeding incompatibilities and lack of seed set in the crop. High variation in gingerol content was observed in ginger somaclones developed at Kerala Agricultural University (KAU) and somaclone B3 with high gingerol content was selected from the investigations.
Ginger has a large genome of 3099.75 Mbp (Genbank ID: GCA_018446385.1), and the DNA 1C value for ginger is given as 1.61 picograms (Smarda et al. 2014; Leitch et al. 2019) which is yet to be exploited fully. In ginger, the polyketide synthase (PKS) family of enzymes (e.g. Type III PKS) belonging to the phenylpropanoid pathway are responsible for the biosynthesis of a wide array of active secondary metabolites and medicinal value (Dibyendu 2015) including the gingerol backbone structure (Dennif et al. 1980 and Schroder 1997). The chalcone synthase (CHS) is involved in the biosynthesis of metabolically active gingerols (Denniff et al. 1980; Schroder 1997; Ramirez-Ahumada et al. 2006; and Ghosh and Mandi 2015). The present investigation utilizes Suppression Subtractive Hybridization to identify the genes involved in high gingerol production as SSH offers the advantage of comparing mRNA populations to isolate the differential cDNA. Suppression Subtractive Hybridization (SSH) has been successfully used for identifying high-quality differential ESTs responsible for the production of secondary metabolites/flavonoids (Park et al. 2004), flavor-enhancing genes encoding secondary metabolites induced by biotic stress (Gohain et al. 2012), and flavonoid genes acting as antimicrobial/antifungal agents (Nisha et al. 2018), in tea. Also, SSH-derived ESTs were utilized for studying gene up-regulation and flavonoid accumulation in lettuce (Park et al. 2007) and strawberry (Baldi et al. 2018). The ESTs for metabolites/flavonoid production were reported, in blueberry (Zifkin et al. 2012) using cDNA libraries, and for litchi (Lai et al. 2015), tea (Li et al. 2015), and banana (Muthusamy et al. 2016) using mRNA-sequencing. Besides, in silico analysis for ginger leaf, rhizome, and root ESTs in ginger provided valuable information on genes involved in the gingerol biosynthetic pathway (James et al. 2015). Also, EST-SSR markers were developed in ginger (Zingiber officinale Rosc.) with cross-transferability to other Zingiberaceae species (Awasthi et al. 2017). The present investigations thus aimed to study the molecular mechanism of gingerol biosynthesis in ginger using transcriptome analysis. The study included the successful generation of SSH-derived leaf and rhizome ESTs using genotypes with varied content of gingerols and analysis of EST sequences to find out the differential genes responsible for high gingerol production in ginger.
Materials and methods
Planting material
Selected ginger somaclone B3 (High gingerol), Athira (Medium gingerol), 132 M (Low gingerol) of KAU, and parent cultivar Maran (low gingerol) genotypes were received from the germplasm maintained at the Centre for Plant Biotechnology and Molecular Biology (CPBMB), KAU, Vellanikkara, Kerala, under the Department of Biotechnology-Biotechnology Industry Research Assistance Council (DBT-BIRAC) ginger research project of CPBMB.
Total RNA extraction and mRNA purification for SSH
Total RNA was extracted from young leaf tissues of ginger sprouts using TRIzol Reagent (Invitrogen), as directed in the manufacturer’s protocol (modified method developed by Chomcynski and Sacchi 1987). Since TRIzol is not specific for plant tissues, a few other modifications were incorporated into this method viz. the horizontal incubation of the tube with the homogenate (Pereira et al. 2017), the addition of chloroform into the homogenate before first centrifugation, additional chloroform step (Toni et al. 2018), one-hour incubation of extract in the freezer after addition of isopropanol and use of 3 M sodium acetate for precipitation (Deepa et al. 2014). For total RNA extraction from rhizomes, a protocol using acidic phenol (pH–4.2) was followed (Sreeja et al. 2018). Around 0.1 g of ground leaf tissue and 2 g of fresh rhizome tissue (one month before the harvesting stage) from the somaclone B3 and cultivar Maran was used for each isolation. The RNA samples were run on 1% agarose (formaldehyde) gel and quantified before performing SSH. The total RNA samples were pooled individually to purify out mRNA using the PolyATtract mRNA Isolation Kit (Promega).
Subtractive cDNA library construction
Suppression Subtractive Hybridization was performed using the PCR-select cDNA Subtraction Kit (Clontech) as instructed in the manufacturer’s protocol (Diatchenko et. al. 1996). A forward subtraction was carried out using a concentrated mRNA population of approximately 2 µg from the tester and driver respectively for generating corresponding ds cDNA. The cDNA library was constructed by eluting and ligating the cDNAs into a pTZ57R/T vector (Thermo Scientific) and subsequent transformation into competent DH5α E. coli cells. For EST library preparation, the ampicillin-resistant (white) colonies were selected and maintained as grids on LB-Ampicillin (10%) plate at 37 °C for overnight incubation. The colonies maintained as grids were further verified for the presence of inserts, using colony PCR with M13 universal primers. The stab cultures on 2% LB agar were prepared for each clone with proper labeling and stored at 4 °C for future use.
Sequencing, analysis, and functional annotation
Twenty-seven clones from leaf ESTs and 42 clones from rhizome ESTs were Sanger sequenced by outsourcing the samples to the DNA sequencing facility of Scigenom, Kochi, Kerala, using universal M13 forward and reverse primers. The region covering vector and adaptor sequences were detected and removed from the raw and trimmed sequences using the VecScreen (www.ncbi.nlm.nih.gov/tools/vecscreen) and Bioedit (Biological Sequence Alignment Editor) tool. The assembly of edited forward and reverse sequences was performed using CAP3 Sequence Assembly Program (http://doua.prabi.fr/software/cap3) and BLAST tools were used for homology search. The functional annotation for the gene of interest was carried out using the program KAAS-KEGG Automatic Annotation Server (http://www.genome.jp/tools/kaas/). The Blast2GO software program was implemented for high throughput functional annotation, represented through the Gene Ontology.
qRT-PCR for CHS gene expression analysis
Total RNA was extracted from rhizome sprouts (young leaves) using PureLink Plant RNA Reagent (Ambion, Life technologies), as directed in the manufacturer’s protocol. Around 100 mg of sprouts from the somaclone B3, variety Athira, somaclone 132 M, and parent cultivar Maran were used for each isolation. The RNA samples were run on 1% agarose (formaldehyde) gel and quantified before performing qRT-PCR. The first-strand cDNA was synthesized using the Revert Aid First Strand cDNA Synthesis Kit (Thermo Scientific), as per the manufacturer’s protocol. The forward and reverse primers for the test gene CHS (Ghosh and Mandi 2015) and the endogenous housekeeping gene Actin (Yamanouchi et. al. 2002) were as follows: CHS F (5’ACTTCTATTTCCGCGTCACC3’), CHS R (5’TCACCCTCGTCTTCTCACAG3’), Act F (5’TCCATCTTGGCATCTCTCAG3’) and Act R (5’GTACCCGCATCAGGCATCTG3’). The master mix was prepared using 10 µL SYBR Premix Ex Taq II (Tli RNaseH Plus) (2X), 1.0 µL each of forward and reverse primers (10 µM), 0.4 µL ROX Reference Dye (50X), and the final volume was made up to 18 µL using distilled water. The cDNA samples were loaded on 96 well plates followed by the addition of the master mix and proper pipetting. The PCR profile was followed as given by DSS Takara Bio India i.e., initial denaturation at 95 °C for 3 min followed by a PCR cycle with 95 °C for 10 s of denaturation and 62 °C for 30 s of annealing for 40 cycles. For each variety, two technical replicates were used. Relative quantification was done by relating the PCR signal in the target transcript (B3, Athira, 132 M) to that of the control transcript (Maran). The value for the control (Maran) was set at zero. The fold expression change was analyzed in CHS by keeping the actin gene as an internal control. The relative expression ratio was calculated by using the mathematical model Comparative ΔCt method by Livak and Schmittgen (2001).
Results and discussion
Suppression Subtractive Hybridization was adapted in the investigations to subtract out the common sequences present in high gingerol somaclone B3 and parent cultivar Maran and to identify the genes specifically involved in the upregulation of gingerol biosynthesis in B3. The CHS gene expression analysis of rhizome sprouts of selected somaclones and the parent cultivar was done to show the activity of CHS even at a very young stage and to establish the role of leaves in gingerol biosynthesis.
Total RNA isolation and identification of differential leaf and rhizome ESTs using SSH
In the modified protocol for total RNA isolation, the horizontal incubation of the tubes with powdered leaf sample and TRIzol at room temperature for 5 min (Pereira et al. 2017), was carried out for the proper dissociation of nucleoprotein complexes by increasing the surface area of contact. Additional chloroform wash was given to remove the left-over impurities which also included insoluble lipids (Toni et al. 2018). Again, an hour of incubation of extract in the freezer after the addition of isopropanol and the use of 3 M sodium acetate (Deepa et al. 2014) facilitated proper precipitation of the RNA sample since the positively charged sodium acetate neutralized the negative charges on the sugar-phosphate backbone of nucleic acid, making the molecule less soluble in water. The details of total RNA isolation from rhizomes have been discussed in our previous publication (Sreeja et al. 2018). For SSH, high-quality total RNA with good band integrity was obtained from leaves (Fig. 1a) and rhizomes (Sreeja et al. 2018). Three distinct RNA bands corresponding to 28S rRNA, 18S rRNA, and 5S rRNA + tRNA were present (Fig. 1a). The concentration of total RNA from leaves of B3 and Maran were 1.09 and 1.21 µg/µL respectively and a good concentration of purified mRNA was obtained for B3 (0.37 µg/µL) and Maran (1.54 µg/µL). The A260/A280 and A260/A230 values for total RNA and mRNA of leaves were more than 1.8 and 1.0 respectively.

a Agarose-formaldehyde gel electrophoresis showing total RNA isolated from ginger leaves of somaclone B3 and control cultivar Maran using modified TRIzol method. Lane1: Transcript RNA marker (0.2–10 kb), Lane 2: Total RNA from somaclone B3, and Lane 3: Total RNA from control cultivar Maran b Second PCR product after experimental subtraction of leaf tester sample. Lane 1 and Lane 6: ϕX174 DNA/Hae III digest marker, Lane 3 and Lane 4: Subtracted and unsubtracted leaf tester cDNA. c Second PCR product after experimental subtraction of rhizome tester sample. Lane 1 and Lane 5: ϕX174 DNA/Hae III digest marker, Lane 2 and Lane 3: Subtracted and unsubtracted rhizome tester cDNA
A single forward experimental subtraction was performed using mRNA populations from the B3 (tester) and Maran (driver). The electrophoresis results revealed the subtracted fragments ranging from 0.3–1.5 kb and unsubtracted fragments of 0.3–2.0 kb for the leaf sample (Fig. 1b). The subtracted and unsubtracted fragments for the rhizome sample ranged from 0.11–1.1 kb and 0.19- > 2.0 kb (Fig. 1c) respectively. The subtracted sample smears were further eluted from the gel for cloning, using the Thermo Scientific GeneJET Extraction Kit which yielded a PCR product of 25.47 ng/µL and 38.56 ng/µL for leaf and rhizome respectively. After transformation 26 white colonies for leaf and 42 white colonies for rhizome were obtained for performing colony PCR and the amplicons showed high molecular weight ranging from 400 bp to 1 kb [Online resource 1 and Online resource 2].
Assembly and annotation of ESTs
After sequencing, quality ESTs were obtained from 19 leaf clones and 24 rhizome clones. Trimming, removal of vector and adaptor sequences, and assembly of forward and reverse sequences were done to obtain a readable sequence length ranging from 268 to 863 bp and 154 to 937 bp for the leaf and rhizome ESTs respectively. The sequences from 18 leaf and rhizome ESTs, showed sequence similarity with significant proteins/enzymes from the database (Tables 1 and 2). Five leaf ESTs were involved in the production of plant secondary metabolites (Table 3) and leaf EST 12 coded for an unknown protein. Seven novel rhizome ESTs (EST 2, EST 11, EST 15, EST 17, EST 18, EST 20, and EST 22) with unknown functions were obtained and differential genes involved in the secondary metabolic pathway were not reported from rhizome ESTs. The 19 forward and reverse leaf ESTs and 24 forward and reverse rhizome ESTs were submitted in the public database with GenBank Accession ID (JZ979518 to JZ979555) and (JZ979556 to JZ979603) as given in Online resource 3 and Online resource 4 respectively.
Blast2GO functional annotations of ESTs
Leaf EST classifications (Fig. 2, (a) and (b)) were done as per the Blast2GO results. The species distribution chart for the leaf EST sequences showed the maximum hits with Musa acuminata subsp. malaccensis which is from the order Zingiberales of Zingiber officinale. The major biological process taking place in leaves was the metabolic process and the major molecular function reported was the ATP binding followed by the transferase activity. The gene ontology of important biological activities and molecular functions in leaves are represented in Fig. 3, (a) and (b) respectively. Transferase (major enzyme class reported, Fig. 4) class of enzymes, 3-ketoacyl- CoA thiolase (EC 2.3.1.16) and acetyl-CoA-acetyltransferase (EC 2.3.1.9), have conserved domains in Leaf EST 9 and were involved in the formation of secondary metabolites. The Blast2GO data for rhizome ESTs showed the maximum hits with Daucus carota subsp. sativus. As per the GO count, the major biological process taking place in rhizomes was the cellular (DNA integration and DNA recombination) and metabolic (DNA metabolism) process (Fig. 5a), and the major molecular function reported in rhizomes were the zinc ion binding function (Fig. 5b). The biological activities and molecular functions for rhizome ESTs are presented in (Fig. 6, (a) and (b)) respectively.

Leaf ESTs coding for a biological processes and b molecular functions

Gene Ontology (GO) counts for a biological processes and b molecular functions in leaf ESTs

Enzyme code distribution for leaf ESTs of somaclone B3

Gene Ontology counts for a biological processes and b molecular functions in rhizome ESTs

Rhizome ESTs coding for a biological processes and b molecular functions
Chalcone synthase gene expression
High-quality total RNA from rhizome sprouts of (young leaves) B3, Athira, 132 M, and Maran having concentrations of 1.20, 1.60, 1.52, and 2.00 µg/µL respectively were used for qRT-PCR. The concentration of first-strand cDNA for B3, Athira, 132 M, and Maran were 1.50, 1.71, 1.70, and 1.60 µg/µL respectively. A system-generated graph showed the expression fold change for the CHS gene in B3 (0.54), Athira (0.20), and 132 M (−1.56) when Maran was set as a calibrator (Fig. 7). The expression data analysis for the CHS gene confirmed higher secondary metabolic activity for gingerol in somaclone B3 (0.54) even in the initial sprouting stage. The qPCR result supports our SSH experimental outcome assuming the leaf as the site of synthesis of gingerols/flavonoids and rhizomes as the site of accumulation, distribution, and storage of the metabolites. Ghasemzadeh et al 2016 also observed the highest CHS activity in three-month-old leaves of ginger as compared to older leaves. Some of the previous studies also support our presumption that the site of synthesis of gingerols is the leaves in ginger. The gingerol/flavonoid biosynthesis decreased in ginger leaves and increased in rhizomes from 3 to 9 months after planting (MAP) with the highest values in leaves from 3 to 6 MAP and the highest values in rhizomes from 6 to 9 MAP (Ghasemzadeh et al. 2016). The transport of secondary metabolites was thus observed from leaves to rhizomes for the final accumulation of the metabolites in rhizomes (Ghasemzadeh et al. 2010, 2016; Jiang et al. 2018; Li et al. 2019). Also, Tanweer et al. 2020, highlighted the importance of leaf gingerol for medicinal purposes, again confirming the synthesis of gingerols in leaves.

Expression fold change for CHS gene with Maran as calibrator. Somaclone B3 shows the highest CHS (rate limiting gene in gingerol biosynthesis) gene expression at the sprouting stage of rhizome
To validate the role of the 3-ketoacyl-CoA thiolase gene in gingerol biosynthesis, Rani et al. 2020, performed a qRT-PCR analysis in ginger sprouts, keeping KAT2 as a test gene and actin as a housekeeping gene and observed higher expression of KAT2 gene in high gingerol yielding varieties /clones (viz. B3) as compared to cultivar Maran.
Leaf EST 9 encoding KAT2: degradative thiolase involved in secondary metabolism
The KAT2 gene was identified as the differential gene responsible for the higher gingerol biosynthesis in ginger somaclone B3. KAT2 belongs to the Type I thiolase family of enzymes which are degradative thiolase (Igual et. al. 1992; Peretó et. al. 2005) and are normally peroxisomal. Type I thiolase (KAT2) is involved in fatty acid β-oxidation and carries out thiolysis of acetyl-CoA units from the thiol end. Previous studies in Arabidopsis could locate three loci encoding KAT viz. KAT1, KAT2, and KAT5 (Germain et. al. 2001) are closely related to the type I thiolase from humans, mice, and yeast as well as Neurospora crassa and Oryza sativa, located in peroxisomes. The Arabidopsis KAT2 locus showed high expression throughout the life cycle with a strong expression during germination and later stages (Wiszniewski et. al. 2012) with higher transcript levels than KAT1 and KAT5 (Germain et. al. 2001; Kamada et. al. 2003). The KAT2 gene has shown contribution toward jasmonic acid (biotic and abiotic stress response) (Castillo et. al. 2004; Afitlhile et. al. 2005; Chen et. al. 2020) biosynthesis, peroxisomal benzoic acid (building blocks for specialized metabolites) synthesis (Bussell et. al. 2014) and reproductive success in plants (Footitt et. al. 2007). Also, the mutations in the KAT2 gene terminated the mobilization of TAGs and the degradation of lipids during seedling growth, showing the influence of the KAT2 gene on lipid metabolism (Germain et. al. 2001).
In the present study Leaf EST 9 also showed a conserved domain for acetyl-CoA acetyltransferases (ACAT) which are Type II thiolase or biosynthetic thiolase (Igual et. al. 1992; Peretó et. al. 2005) and are commonly cytosolic. Type II thiolase performs acetoacetyl CoA synthesis in the mevalonate biosynthesis pathway through Claisen-condensation-reaction.
Despite showing a very less sequence identity of ~ 38%, KAT2 and ACAT display more shared structural characteristics and spatial positioning of the active site residues among themselves and with CHS (pivotal for the biosynthesis of flavonoids viz. gingerols in ginger). Hence indicates a common evolutionary origin (Mathieu et al. 1997; Modis and Wierenga 2000) and may be concluded as the prime reason for the involvement of these enzymes in secondary metabolite production viz. flavonoid and isoprenoid compounds respectively. Since land plants have gene lineages corresponding to KAT2 and KAT5 recommending conservation of different functions for these two genes, the data obtained for Arabidopsis with respect to the role of KAT in secondary metabolite production suggests a similar role for the KAT gene in Zingiber officinale.
KAT2: Contributes to the KAT5-generated cytosolic acetyl-CoA pool for gingerol/flavonoid biosynthesis
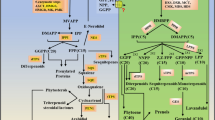
The role of the phenylpropanoid pathway in the construction of the basic backbone of flavonoids (provides p-coumaroyl-CoA from Phe) has been neatly explained (Saito et al. 2013), but the involvement of the polyketide pathway (provides malonyl-CoA for C2 chain elongation by CHS) (Tohge et al. 2018) is yet to be fully explained. KAT5 is one of the major genes acting in the polyketide pathway for the production of cytosolic acetyl-CoA which is further degraded into the building block malonyl-CoA. Malonyl-CoA is a substrate for the biosynthesis of core flavonoid naringenin by CHS. Many studies have shown the co-regulation of the cytosolic KAT5 with genes involved in flavonoid biosynthesis (Germain et al. 2001; Carrie et al. 2007; Yonekura-Sakakibara et al. 2008; Wiszniewski et al. 2012; de Souza et. al. 2020).
Based on the present investigations, efforts have been made to explain the possible contribution of the peroxisomal KAT2 gene to the KAT5-generated cytosolic acetyl-CoA pool, which is the major substrate for gingerol/flavonoid biosynthesis. The β-oxidation of the fats stored as triacylglycerols (TAGs) provide energy for the post-germinative process in plants (Chapman et al. 2012). The lipases break down TAGs into fatty acids in the peroxisomes and fatty acids get activated (esterified with CoA moiety) by long-chain acyl-CoA synthetases, which is crucial for the entry to the β-oxidation cycle (Fulda et al. 2002). The final step in β-oxidation is performed by KAT2 which releases acetyl-CoA and a shortened fatty acyl-CoA which can carry on with β-oxidation further (Hayashi et al. 1998; Germain et al. 2001). The acetyl-CoA thus produced can enter the glyoxylate cycle to form four-carbon metabolites to be converted to sucrose as a source of carbon and energy (Graham 2008; Theodoulou and Eastmond 2012). The acetyl-CoA cannot as such cross the peroxisomal membrane and is metabolized into citrate, by citrate synthase enzyme (CSY) which can readily traverse the peroxisomal membrane to enter the cytosol (Rottensteiner and Theodoulou 2006) for fatty acid biosynthesis as well as to the mitochondrial TCA cycle for respiration (Raymond et al. 1992). In Arabidopsis, CSY2 and CSY3 convert acetyl-CoA to citrate for export to mitochondria (Hu et al. 2012) which contributes to the mitochondrial pool of acetyl-CoA and further production of mitochondrial citrate. Other than entering the mitochondria, peroxisomal citrate is also cleaved by another important cytosolic enzyme ATP-citrate lyase (ACL) which converts citrate and CoA to oxaloacetate and acetyl-CoA, contributing to the cytosolic acetyl-CoA (Fatland et. al. 2002). An alternate route for producing acetyl-CoA is the formation of fatty acyl-CoAs from free fatty acids, ATP, and CoA in chloroplasts (de Souza et al. 2020). As depicted in Fig. 8, acyl-CoAs from chloroplasts could break down to acetyl-CoA in the cytosol and one of the intermediates, 3-ketoacyl-CoA is acted upon by KAT5 to produce cytosolic acetyl-CoA. The cytosolic acetyl-CoA thus formed is the major substrate for flavonoid biosynthesis and undergoes carboxylation by acetyl-CoA carboxylase (ACC) to form malonyl-CoA which acts as the extender unit in the reaction catalyzed by Chalcone synthase for the biosynthesis of gingerols. The possible contribution of KAT2 in cytosolic acetyl-CoA production for flavonoid/gingerol biosynthesis is illustrated in Fig. 8 (modified from de Souza et al. 2020) which indicates how acetyl-CoA generated from peroxisomes could get involved in gingerol biosynthesis. The detailed pathway for gingerol biosynthesis is given as Online resource 5 and is also explained by Ramirez-Ahumada et al. 2006.

Flow chart showing possible pathway for gingerol biosynthesis and the role of KAT2 and KAT5 enzymes in generation of extender unit, acetyl-CoA for gingerol production. The enzymes are highlighted in yellow. ACS, acetyl-CoA synthetase; ACAD, acyl-CoA dehydrogenase; ECH, enoyl-CoA hydratase; HCD, hydroxyacyl-CoA dehydrogenase; PAL, Phe ammonia lyase; C4H, cinnamate 4-hydroxylase; 4CL, 4-coumarate:CoA ligase (as modified from de Souza et al. 2020). The predicted route for KAT2 generated peroxisomal acetyl-CoA and its regeneration as cytosolic acetyl-CoA is highlighted in bold red arrows. Cytosolic acetyl-CoA is carboxylated by ACC to malonyl-CoA which is the extender unit for flavonoids/gingerol
Earlier studies in yeast suggested metabolic engineering for the overproduction of mitochondrial citrate (Tang et. al. 2013) leading to increased cytosolic acetyl-CoA and malonyl-CoA for higher flavonoid production (Tang et. al. 2013). Similarly, the present investigation explains the role of the peroxisomal KAT2 gene towards citrate generation in the cytosol, leading to increased cytosolic acetyl-CoA and malonyl-CoA in ginger for higher gingerol production (Fig. 8). Peroxisomal citrate transported to mitochondria may also add carbon flux towards mitochondrial citrate generation leading to increased cytosolic acetyl-CoA pool.
Future prospects and conclusion
The future prospects for KAT enzymes or the major isoforms like KAT2/KAT5 can be, post-translational modification, active site modification (Tan et. al. 2020), and protein engineering (Liu et. al. 2020) for drug development which could unravel the true potential of this enzyme for synthesizing important secondary metabolites. It’s known that polyketide biosynthesis is performed by PKS via decarboxylative Claisen condensation reactions with possibly some additional modifications catalyzed by modifying domains of PKSs. Such modifications are responsible for the great variety of secondary metabolites being produced by PKSs. The modified pathways can be further engineered using microbial systems as “factories” for the mass production of secondary metabolites which is another new emerging field of metabolite engineering (Pyne et. al. 2019; Huccetogullari et. al. 2019; Cravens et. al. 2019; Birchfield and McIntosh 2020; Mishra et. al. 2021).
An effort has been extended in the current study to unravel the molecular basis of gingerol biosynthesis in ginger, by generating leaf EST encoding the peroxisomal KAT2 gene for 3-ketoacyl-CoA thiolase 2, responsible for the possible upregulation of gingerol. The study explains the possible contribution of KAT2-generated peroxisomal acetyl-CoA in the cytosolic acetyl-CoA pool which subsequently gives rise to malonyl-CoA, the extender unit for gingerol biosynthesis. Although the peroxisomal KAT2 gene is otherwise known for β-oxidation-related functions, the similar contributions of KAT2/KAT5 isoforms towards cytosolic acetyl-CoA generation is an interesting outcome of the present study. The expression level of the rate-limiting gene, chalcone synthase, involved in the gingerol biosynthetic pathway in rhizome leaf sprouts, confirmed gingerol biosynthesis even at the sprouting stage. In this study, no differential genes for secondary metabolites were reported in ginger rhizomes, and it is presumed that the leaf is the site of synthesis of secondary metabolites in ginger and the rhizome is the site of storage of metabolites. This work supports recent studies done on the thiolase family of enzymes as potential candidates for active site modification/binding pocket engineering that paves way for future protein engineering and drug development studies in the pharmaceutics/medical sector, in an energy-efficient and PKS-independent way. Integrating proteomics and metabolomics towards engineering enzymes with better stability, substrate specificity, and catalytic activity will lead to robust KAT enzymes producing a range of desired value-added products.
Abbreviations
- KAT:
-
3-Ketoacyl-CoA thiolase
- SSH:
-
Suppression subtraction hybridization
- EST:
-
Expressed sequence tags
- GO:
-
Gene ontology
- PKS:
-
Polyketide synthase
- KAU:
-
Kerala Agricultural University
- CHS:
-
Chalcone synthase
- CPBMB:
-
Centre for Plant Biotechnology and Molecular Biology
- DBT-BIRAC:
-
Department of Biotechnology-Biotechnology Industry Research Assistance Council
- MAP:
-
Months after planting
- ACAT:
-
Acetyl-CoA acetyltransferase
- CSY:
-
Citrate synthase
- ICAR:
-
Indian Council of Agricultural Research
- ACL:
-
ATP-citrate lyase
- ACC:
-
Acetyl-CoA carboxylase
References
Afitlhile MM, Fukushige H, Nishimura M, Hildebrand DF (2005) A defect in glyoxysomal fatty acid β-oxidation reduces jasmonic acid accumulation in Arabidopsis. Plant Physiol Biochem 43(6):603–609. https://doi.org/10.1016/j.plaphy.2005.03.016
Almatroodi SA, Alnuqaydan AM, Babiker AY, Almogbel MA, Khan AA, Rahmani AH (2021) 6-gingerol, a bioactive compound of ginger attenuates renal damage in streptozotocin-induced diabetic rats by regulating the oxidative stress and inflammation. Pharm 13:317. https://doi.org/10.3390/pharmaceutics13030317
Assembly [Internet]. Bethesda (MD): National Library of Medicine (US), National Center for Biotechnology Information; [2021] – Accession No. GCF_018446385.1, Zingiber officinale, chromosome; [cited 2021/05/21]. Available from: https://www.ncbi.nlm.nih.gov/assembly/GCF_018446385.1
Awasthi P, Singh A, Sheikh G, Mahajan V, Gupta AP, Gupta S, Bedi YS, Gandhi SG (2017) Mining and characterization of EST-SSR markers for Zingiber officinale Roscoe with transferability to other species of Zingiberaceae. Physiol Mol Biol Plants 23(4):925–931. https://doi.org/10.1007/s12298-017-0472-5
Baldi P, Orsucci S, Moser M, Brilli M, Giongo L, Si-Ammour A (2018) Gene expression and metabolite accumulation during strawberry (Fragaria ananassa) fruit development and ripening. Planta 248(5):1143–1157. https://doi.org/10.1007/s00425-018-2962-2
Birchfield AS, McIntosh CA (2020) Metabolic engineering and synthetic biology of plant natural products–A minireview. Curr Plant Biol 24:100163. https://doi.org/10.1016/j.cpb.2020.100163
Bussell JD, Reichelt M, Wiszniewski AAG, Gershenzon J, Smith SM (2014) Peroisomal ATP-binding cassette transporter COMATOSE and the multifuctional protein ABNORMAL INFLORESCENCE MERISTEM are required for the production of benzoylated metabolites in Arabidopsis seeds. Plant Physiol. 164:48–54. https://doi.org/10.1104/pp.113.229807
Carrie C, Murcha MW, Miller AH, Smith SM, Whelan J (2007) Nine 3-ketoacyl-CoA thiolases (KATs) and acetoacetyl-CoA thiolases (ACATs) encoded by five genes in Arabidopsis thaliana are targeted either to peroxisomes but not to mitochondria. Plant Mol Biol 63:97–108. https://doi.org/10.1007/s11103-006-9075-1
Castillo MC, Martinez C, Buchala A, Metrau J-P, Leon J (2004) Gene-specific involvement of β-oxidation in wound-activated responses in Arabidopsis. Plant Physiol 135:85–94. https://doi.org/10.1104/pp.104.039925
Chapman KD, Dyer JM, Mullen RT (2012) Biogenesis and functions of lipid droplets in plants. J Lipid Res 53:215–226. https://doi.org/10.1194/jlr.r021436
Chen X, Snyder CL, Truksa M, Shah S, Weselake RJ (2011) sn-Glycerol-3-phosphate acyltransferases in plants. Plant Signal Behav 6(11):1695–1699. https://doi.org/10.4161/psb.6.11.17777
Chen Y, Yan Y, Wu T-T, Zhang G-L, Yin H, Chen W, Wang S, Chang F, Gou J-Y (2020) Cloning of wheat keto-acyl thiolase 2B reveals a role of jasmonic acid in grain weight determination. Nat Commun 11:6266. https://doi.org/10.1038/s41467-020-20133-z
Chomczynski P, Sacchi N (1987) Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal Biochem 162(1):156–159. https://doi.org/10.1006/abio.1987.9999
Cravens A, Payne J, Smolke CD (2019) Synthetic biology strategies for microbial biosynthesis of plant natural products. Nat Commun 10:2142. https://doi.org/10.1038/s41467-019-09848-w
Czerniawski P, Bednarek P (2018) Glutathione-S-transferases in the biosynthesis of sulfur-containing secondary metabolites in Brassicaceae plants. Front Plant Sci 9:1639. https://doi.org/10.3389/fpls.2018.01639
de Souza LP, Garbowicz K, Brotman Y, Tohge T, Fernie AR (2020) The acetate pathway supports flavonoid and lipid biosynthesis in Arabidopsis. Plant Physiol 182(2):857–869. https://doi.org/10.1104/pp.19.00683
Deepa K, Sheeja TE, Santhi R, Sasikumar B, Cyriac A, Deepesh PV, Prasath D (2014) A simple and efficient protocol for isolation of high quality functional RNA from different tissues of turmeric (Curcuma longa L.). Physiol Mol Biol Plants 20(2):26–271. https://doi.org/10.1007/s12298-013-0218-y
Denniff P, Macloed I, Whiting DA (1980) Studies in the biosynthesis of [6]-gingerol, pungent principle of ginger (Zingiber officinale). J Chem Soc Perkin Trans 1:2637–2644. https://doi.org/10.1039/P19800002637
Diatchenko L, Lau YFC, Campbell AP, Chenchik A, Moqadam F, Huang B, Lukyanov S, Lukyanov K, Gurskaya N, Sverdlov ED, Siebert PD (1996) Suppression subtractive hybridization: a method for generating differentially regulated or tissue-specific cDNA probes and libraries. Proc Natl Acad Sci USA 93(12):6025–6030
Dibyendu DM (2015) A brief review on plant type III polyketide synthases, an important group of enzymes of secondary metabolism. Res J Recent Sci 4:2277–2502
Dixon DP, Skipsey M, Edwards R (2010) Roles for glutathione transferases in plant secondary metabolism. Phytochemistry 71(4):338–350. https://doi.org/10.1016/j.phytochem.2009.12.012
Fatland BL, Ke J, Anderson MD, Mentzen WI, Cui LW, Allred CC, Johnston JL, Nikolau BJ, Wurtele ES (2002) Molecular characterization of a heteromeric ATP-citrate lyase that generates cytosolic acetyl-CoA in Arabidopsis. Plant Physiol 130:740–756. https://doi.org/10.1104/pp.008110
Footitt S, Cornah JE, Pracharoenwattana I, Bryce JH, Smith SM (2007) The Arabidopsis 3-ketoacyl-CoA thiolase-2 (Kat2-1) mutant exhibits increased flowering but reduced reproductive success. J Exp Botony 58(11):2959–2968
Fulda M, Shockey J, Werber M, Wolter FP, Heinz E (2002) Two long-chain acyl-CoA synthetases from Arabidopsis thaliana involved in peroxisomal fatty acid β-oxidation. Plant J 32:93–103. https://doi.org/10.1046/j.1365-313x.2002.01405.x
Germain V, Rylott EL, Larson TR, Sherson SM, Bechtold N, Carde JP (2001) Requirement for 3-ketoacyl-CoA thiolase-2 in peroxisome development, fatty acid beta-oxidation and breakdown of triacylglycerol in lipid bodies of Arabidopsis seedlings. Plant J 28:1–12. https://doi.org/10.1046/j.1365-313x.2001.01095.x
Ghasemzadeh A, Jaafar HZE, Rahmat A (2010) Identification and concentration of some flavonoid components in Malaysian young ginger (Zingiber officinale Roscoe) varieties by a high performance liquid chromatography method. Mol 15:6231–6243. https://doi.org/10.3390/molecules15096231
Ghasemzadeh A, Jaafar HZE, Ashkani S, Rahmat A, Juraimi AS, Puteh A, Mohamed MTM (2016) Variation in secondary metabolite production as well as antioxidant and antibacterial activities of Zingiber zerumbet (L.) at different stages of growth. BMC Complement Alternat Med 16:104. https://doi.org/10.1186/s12906-016-1072-6
Ghosh S, Mandi S (2015) SNP in chalcone synthase is associated with variation of 6-gingerol content in contrasting landraces of Zingiber officinale Roscoe. Gene 566:184–188. https://doi.org/10.1016/j.gene.2015.04.042
Gohain B, Borchetia S, Bhorali P, Agarwal N, Bhuyan LP, Rahman A, Sakata K, Mizutani M, Shimizu B, Gurusubramaniam G, Ravindranath R, Kalita MC, Hazarika M, Das S (2012) Understanding Darjeeling tea flavor on a molecular basis. Plant Mol Biol 78:577–597. https://doi.org/10.1007/s11103-012-9887-0
Graham IA (2008) Seed storage oil mobilization. Annu Rev Plant Biol 59:115–142. https://doi.org/10.1146/annurev.arplant.59.032607.092938
Haapalainen AM, Merilainen G, Wierenga RK (2006) The thiolase superfamily: condensing enzymes with diverse reaction specificities. Trends Biochemical Sci 31(1):64–71. https://doi.org/10.1016/j.tibs.2005.11.011
Haridas M, Sasidhar V, Nath P, Sabu AA, Rammanohar P (2021) Compounds of Citrus medica and Zingiber officinale for COVID-19 inhibition: in silico evidence for cues from Ayurveda. Futur J Pharm Sci 7:13. https://doi.org/10.1186/s43094-020-00171-6
Hariharan T, Johnson PJ, Cattolico RA (1998) Purification and characterization of phosphoribulokinase from the marine chromophytic alga Heterosigma carterae. Plant Physiol 117(1):321–329. https://doi.org/10.1104/pp.117.1.321
Hayashi M, Toriyama K, Kondo M, Nishimura M (1998) 2,4-dichlorophenoxybutyric acid-resistant mutants of Arabidopsis have defects in glyoxysomal fatty acid β-oxidation. Plant Cell 10:183–195. https://doi.org/10.1105/tpc.10.2.183
Hu J, Baker A, Bartel B, Linka N, Mullen RT, Reumann S, Zolman BK (2012) Plant peroxisomes: biogenesis and function. Plant Cell 24(6):2279–2303. https://doi.org/10.1105/tpc.112.096586
Huccetogullari D, Luo ZW, Lee SY (2019) Metabolic engineering of microorganisms for production of aromatic compounds. Microb Cell Fact 18:41. https://doi.org/10.1186/s12934-019-1090-4
Igual JC, Gonzalez-Bosch C, Dopazo J, Perez-Ortin JE (1992) Phylogenetic analysis of the thiolase family. Implications for the evolutionary origin of peroxisomes. J Mol Evol 35:147–155. https://doi.org/10.1007/bf00183226
James P, Baby B, Charles S, Nair LS, Nazeem PA (2015) Computer aided gene mining for gingerol biosynthesis. Bioinformation 11(6):316–321. https://doi.org/10.6026/97320630011316
Jiang Y, Huang M, Wisniewski M, Li H, Zhang M, Tao X, Liu Y, Zou Y (2018) Transcriptome analysis provides insights into gingerol biosynthesis in ginger (Zingiber officinale). Plant Genome 11(3):180034. https://doi.org/10.3835/plantgenome2018.06.0034
Kamada T, Nito K, Hayashi H, Mano S, Hayashi M, Nishimura M (2003) Functional differentiation of peroxisomes revealed by expression profiles of peroxisomal genes in Arabidopsis thaliana. Plant Cell Physiol 44:1275–1289. https://doi.org/10.1093/pcp/pcg173
Kim EC, Min JK, Kim TY, Lee SJ, Yang HO, Han S, Kim YM, Kwon YG (2005) [6]-gingerol, a pungent ingredient of ginger, inhibits angiogenesis in vitro and in vivo. Biochem Biophys Res Commun 335(2):300–308. https://doi.org/10.1016/j.bbrc.2005.07.076
Kumar M, Shylaja MR, Nazeem PA, Babu T (2017) 6-Gingerol is the most potent anti-cancerous compound in ginger (Zingiber officinale Rosc.). J Develop Drugs 6(1):1–6. https://doi.org/10.4172/2329-6631.1000167
Lai B, Hu B, Qin Y-H, Zhao J-T, Wang H-C, Hu G-B (2015) Transcriptomic analysis of Litchi chinensis pericarp during maturation with a focus on chlorophyll degradation and flavonoid biosynthesis. BMC Genomics 16:1–15. https://doi.org/10.1186/s12864-015-1433-4
Leitch IJ, Johnston E, Pellicer J, Hidalgo O and Bennett MD (2019) Angiosperm DNA C-values database (release 9.0, Apr 2019). https://cvalues.science.kew.org/
Li CF, Zhu Y, Yu Y, Zhao Q-Y, Wang S-J, Wang X-C, Yao M-Z, Luo D, Chen L, Yang Y-J (2015) Global transcriptome and gene regulation network for secondary metabolite biosynthetic of tea plant (Camellia sinensis). BMC Genomics 16(1):1–21. https://doi.org/10.1186/s12864-015-1773-0
Li Z, Chen Z-X, Tang J-M, Jiang Y-S, Liao Q-H, LiuY-Q and Liu G-H (2019) Metabolomic analysis of bioactive compounds in mature rhizomes and daughter rhizomes in ginger (Zingiber officinale). Preprint doi: https://doi.org/10.21203/rs.2.17463/v1
Liao Y, Leu Y, Chan Y, Kuo P, Wu T (2012) Anti-platelet aggregation and vasorelaxing effects of the constituents of the rhizomes of Zingiber officinale. Molecules 17:8928–8937. https://doi.org/10.3390/molecules17088928
Liu L, Zhou S, Deng U (2020) The 3-ketoacyl-CoA thiolase: an engineered enzyme for carbon chain elongation of chemical compounds. Appl Microbiol Biotechnol. https://doi.org/10.1007/s00253-020-10848-w
Livak KJ, Schmittgen TD (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 25(4):402–408. https://doi.org/10.1006/meth.2001.1262
Mathieu M, Modis Y, Zeelen JP, Engel CK, Abagyan AR, Rasmussen B, Lamzin VS, Kunau WH, Wierenga RK (1997) The 1.8Å crystal structure of the dimeric peroxisomal 3-ketoacyl-CoA thiolase of Saccharomyces cerevisiae: implications for substrate binding and reaction mechanism. J Mol Biol 273:714–728. https://doi.org/10.1006/jmbi.1997.1331
Mishra S, Sahu PK, Agarwal V, Singh N (2021) Exploiting endophytic microbes as micro-factories for plant secondary metabolite production. Appl Microbiol Biotechnol. https://doi.org/10.1007/s00253-021-11527-0
Modis Y, Wierenga RK (2000) Crystallographic analysis of the reaction pathway of Zoogloea ramigera biosynthetic thiolase. J Mol Biol 297:1171–1182. https://doi.org/10.1006/jmbi.2000.3638
Muthusamy M, Uma S, Backiyarani S, Saraswathi MS, Chandrasekar A (2016) Transcriptomic changes of drought-tolerant and sensitive banana cultivars exposed to drought stress. Frontiers Plant Sci 7:1609. https://doi.org/10.3389/fpls.2016.01609
Nisha SN, Prabu G, Mandal AKA (2018) Biochemical and molecular studies on the resistance mechanisms in tea [Camellia sinensis (L.) O. Kuntze]. Physiol Mol Biol Plants 24(5):867–880. https://doi.org/10.1007/s12298-018-0565-9
Okamura E, Tomita T, Sawa R, Nishiyama M, Kuzuyama T (2010) Unprecedented acetoacetyl-Coenzyme A synthesizing enzyme of the thiolase superfamily involved in the mevalonate pathway. Proc Natl Acad Sci USA 107:11265–11270. https://doi.org/10.1073/pnas.1000532107
Park J-S, Kim J-B, Hahn B-S, Kim K-H, Ha S-H, Kim J-B, Kim Y-H (2004) EST analysis of genes involved in secondary metabolism in Camellia sinensis (tea), using suppression subtractive hybridization. Plant Sci 166:953–961. https://doi.org/10.1016/j.plantsci.2003.12.010
Park J-S, Choung M-G, Kim J-B, Hahn B-S, Kim J-B, Bae S-C, Roh K-H, Kim Y-H, Cheon C-I, Sung M-K, Cho K-J (2007) Genes up-regulated during red coloration in UV-B irradiated lettuce leaves. Plant Cell Rep 26:507–516. https://doi.org/10.1007/s00299-006-0255-x
Pereira WJ, Bassinello PZ, Brondani C, Vianello RP (2017) An improved method for RNA extraction from common bean seeds and validation of reference genes for qPCR. Crop Breed Appl Biotechnol 17:150–158. https://doi.org/10.1590/1984-70332017v17n2a22
Pereto J, Lopez-Garcia P, Moreira D (2005) Phytogenetic analysis of eukaryotic thiolases suggests multiple proteobacterial origins. J Mol Evol 61:65–74. https://doi.org/10.1007/s00239-004-0280-8
Petrussa E, Braidot E, Zancani M, Peresson C, Bertolini A, Patui S, Vianello A (2013) Plant flavonoids-biosynthesis, transport and involvement in stress responses. Int J Mol Sci 14(7):14950–14973. https://doi.org/10.3390/ijms140714950
Pye VE, Christensen CE, Dyer JH, Arent S, Henriksen A (2010) Peroxisomal plant 3-ketoacyl-CoA thiolase structure and activity are regulated by a sensitive redox switch. J Biol Chem 285(31):24078–24088. https://doi.org/10.1074/jbc.M110.106013
Pyne ME, Narcross L, Martin VJJ (2019) Engineering plant secondary metabolism in microbial systems. Plant Physiol 179:844–861. https://doi.org/10.1104/pp.18.01291
Ramirez-Ahumada MD, Timmermann BN, Gang DR (2006) Biosynthesis of curcuminoids and gingerols in turmeric (Curcuma longa) and ginger (Zingiber officinale): identification of curcuminoid synthase and hydroxycinnamoyl-CoA thioesterases. Phytochem 67:2017–2029. https://doi.org/10.1016/j.phytochem.2006.06.028
Rani M, Shylaja MR, Mathew D (2020) Analysis of differentially expressed ESTs for gingerol production and validation of 3-ketoacyl-CoA thiolase gene expression in high gingerol yielding genotypes of ginger (Zingiber officinale Rosc.). Res J Agric Sci 11(2):461–466. https://doi.org/10.2139/ssrn.3666175
Rathinavel T, Palanisamy M, Palanisamy S, Subramanian A, Thangaswamy S (2020) Phytochemical 6-gingerol-A promising drug of choice for COVID-19. Int J Adv Sci Eng 6(4):1482–1489. https://doi.org/10.29294/IJASE.6.4.2020.1482-1489
Raymond P, Spiteri A, Dieuaide M, Gerhardt B, Pradet A (1992) Peroxisomal β-oxidation of fatty acids and citrate formation by a particulate fraction from early germinating sunflower seeds. Plant Physiol Biochem 30:153–161
Rottensteiner H, Theodoulou FL (2006) The ins and outs of peroxisomes: co-oxidation of membrane transport and peroxisomal metabolism. Biochimica Biophysica Acta 1763:1527–1540. https://doi.org/10.1016/j.bbamcr.2006.08.012
Saito K, Yonekura-Sakakibara K, Nakabayashi R, Higashi Y, Yamazaki M, Tohge T, Fernie AR (2013) The flavonoid biosynthetic pathway in Arabidopsis: structural and genetic diversity. Plant Physiol Biochem 72:21–34. https://doi.org/10.1016/j.plaphy.2013.02.001
Samad MB, Mohsin MNAB, Razu BA, Hossain MT, Mahzabeen S, Unnoor N, Muna IA, Akhter F, Kabir AU, Hannan JMA (2017) [6]-gingerol, from Zingiber officinale, potentiates GLP-1 mediated glucose-stimulated insulin secretion pathway in pancreatic β-cells and increases RAB8/RAB10-regulated membrane presentation of GLUT4 transporters in skeletal muscle to improve hyperglycemia in Leprdb/db type 2 diabetic mice. BMC 17:395. https://doi.org/10.1186/s12906-017-1903-0
Schroder J (1997) A family of plant specific polyketide synthases: facts and predictions. Trends Plant Sci 2:373–378. https://doi.org/10.1016/S1360-1385(97)87121-X
Smarda P, Bures P, Horova L, Leitch IJ, Mucina L, Pacini E, Tichy L, Grulich V, Rotreklova O (2014) Ecological and evolutionary significance of genomic GC content diversity in monocots. Proc Natl Acad Sciences 111:E4096–E4102. https://doi.org/10.1073/pnas.1321152111
Sreeja S, Shylaja MR (2018) A modified protocol for isolation of high quality total RNA from ginger (Zingiber officinale Rosc.) rhizomes. J Tropical Agriculture 56(1):45–49
Subbaiah CC, Huber SC, Sachs MM, Rhoads DM (2007) Sucrose synthase: expanding protein function. Plant Signal Behav 2:28–29. https://doi.org/10.4161/psb.2.1.3646
Tan Z, Clomburg JM, Cheong S, Qian S, Gonzalez R (2020) A polyketoacyl-CoA thiolase-dependent pathway for the synthesis of polyketide backbones. Nat Catalysis 3:593–603. https://doi.org/10.1038/s41929-020-0471-8
Tang X, Feng H, Chen WN (2013) Metabolic engineering for enhanced fatty acids synthesis in Saccharomyces cerevisiae. Metab Eng. https://doi.org/10.1016/j.ymben.2013.01.003
Tanweer S, Mehmood T, Zainab S, Ahmad Z, Shehzad A (2020) Comparison and HPLC quantification of antioxidant profiling of ginger rhizome, leaves and flower extracts. Clin Phytoscience 6:12. https://doi.org/10.1186/s40816-020-00158-z
Theodoulou FL, Eastmond PJ (2012) Seed storage oil catabolism: a story of give and take. Curr Opin Plant Biol 15:322–328. https://doi.org/10.1016/j.pbi.2012.03.017
Tohge T, de Souza P, Fernie AR (2018) On the natural diversity of phenylacylated-flavonoid and their in planta function under conditions of stress. Phytochem Reviews 17:279–290. https://doi.org/10.1007/s11101-017-9531-3
Toni LS, Garcia AM, Jeffrey DA, Jiang X, Stauffer BL, Miyamoto SD, Sucharov CC (2018) Optimization of Phenol-Chloroform RNA Extraction. Methodsx 5:599–608. https://doi.org/10.1016/j.mex.2018.05.011
Wiszniewski AAG, Smith SM, Bussell JD (2012) Conservation of two lineages of peroxisomal (Type I) 3-ketoacyl-CoA thiolases in land plants, specialization of the genes in Brassicaceae, and characterization of their expression in Arabidopsis thaliana. J Exp Botony 63(17):6093–6103. https://doi.org/10.1093/jxb/ers260
Xu T, Qin G, Jiang W, Zhao Y, Xu Y, Lv X (2018) 6-gingerol protects heart by suppressing myocardial ischemia/reperfusion induced inflammation via the PI3K/Akt-dependent mechanism in rats. Evidence-based complem Altern Med. https://doi.org/10.1155/2018/6209679
Yamanouchi U, Yano M, Lin MA, Yamada K (2002) A rice spotted leaf gene, Spl7, encoes a heat stress transcription factor protein. Proc Natl Acad Sci USA 99(11):7530–7535. https://doi.org/10.1073/pnas.112209199
Yonekura-Sakakibara K, Tohge T, Matsuda F, Nakabayashi R, Takayama H, Niida R, Watanabe-Takahashi A, Inoue E, Saito K (2008) Comprehensive flavonol profiling and transcriptome coexpression analysis leading to decoding gene-metabolite correlations in Arabidopsis. Plant Cell 20:2160–2176. https://doi.org/10.1105/tpc.108.058040
Young HY, Luo YL, Cheng HY, Hsieh WC, Liao JC, Peng WH (2005) Analgesic and anti-inflammatory activities of [6]-gingerol. J Ethnopharmacol 96(1–2):207–210. https://doi.org/10.1016/j.jep.2004.09.009
Zhu Y, Wang F, Zhao Y, Wang P, Sang S (2017) Gastroprotective [6]-gingerol aspirinate as a novel chemopreventive prodrug of aspirin for colon cancer. Sci Rep 7:40119. https://doi.org/10.1038/srep40119
Zifkin M, Jin A, Ozga JA, Zaharia LI, Schernthaner JP, Gesell A, Abrams SR, Kennedy JA, Constabel CP (2012) Gene expression and metabolite profiling of developing highbush blueberry fruit indicates transcriptional regulation of flavonoid metabolism and activation of abscisic acid metabolism. Plant Physiol 158(1):200–224. https://doi.org/10.1104/pp.111.180950
Funding
We would like to thank KAU (Grant Number R7/61877/13) and the Indian Council of Agricultural Research (ICAR) [(grant number 7(5)-2012-EPD)] for providing financial support for the research work. Also, our sincere thanks to the DBT-BIRAC (grant number BT/CRS0068/CRS-03/13) ginger research project for providing the ginger germplasm and equipment facilities for our study.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interests
The authors have no competing interests to declare that are relevant to the content of this article.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Sreeja, S., Shylaja, M.R., Nazeem, P.A. et al. Peroxisomal KAT2 (3-ketoacyl-CoA thiolase 2) gene has a key role in gingerol biosynthesis in ginger (Zingiber officinale Rosc.). J. Plant Biochem. Biotechnol. 32, 451–466 (2023). https://doi.org/10.1007/s13562-022-00825-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13562-022-00825-x