
Abstract
Abrocitinib, an oral, once-daily, Janus kinase (JAK) 1-selective inhibitor, is approved for the treatment of adults and adolescents with moderate-to-severe atopic dermatitis (AD). Abrocitinib has shown rapid and sustained efficacy in phase 3 trials and a consistent, manageable safety profile in long-term studies. Rapid itch relief and skin clearance are more likely to be achieved with a 200-mg daily dose of abrocitinib than with dupilumab. All oral JAK inhibitors are associated with adverse events of special interest and laboratory changes, and initial risk assessment and follow-up monitoring are important. Appropriate selection of patients and adequate monitoring are key for the safe use of JAK inhibitors. Here, we review the practical use of abrocitinib and discuss characteristics of patients who are candidates for abrocitinib therapy. In general, abrocitinib may be used in all appropriate patients with moderate-to-severe AD in need of systemic therapy, provided there are no contraindications, e.g., in patients with active serious systemic infections and those with severe hepatic impairment, as well as pregnant or breastfeeding women. For patients aged ≥ 65 years, current long-time or past long-time smokers, and those with risk factors for venous thromboembolism, major adverse cardiovascular events, or malignancies, a meticulous benefit–risk assessment is recommended, and it is advised to start with the 100-mg dose, when abrocitinib is the selected treatment option.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Abrocitinib, an oral, once-daily Janus kinase (JAK) 1-selective inhibitor, is approved for the treatment of adults and adolescents with moderate-to-severe atopic dermatitis (AD). | |
Abrocitinib has shown a favorable benefit–risk profile in long-term studies with both the 100 and 200 mg/day doses. | |
In patients who are over 65 years of age, are current long-time or past long-time smokers, or have risk factors for cardiovascular events, venous thrombosis, infections, or malignancy, a thorough benefit–risk assessment is recommended and the lowest effective dose of abrocitinib is preferred. | |
Prior to starting treatment, clinicians should advise patients initiating abrocitinib about vaccination recommendations, the most frequent tolerability issues, family planning, risk of adverse events, and the need for blood monitoring. |
Introduction
Atopic Dermatitis Treatment Landscape
Atopic dermatitis (AD) can have a profound effect on patients’ quality of life (QoL) [1]. AD is a heterogeneous disease, and not all patients can sufficiently manage their AD with available topical or systemic therapies [2, 3]. Dupilumab was the first new systemic therapy to receive approval for the treatment of moderate-to-severe AD [4]. However, more than 60% of patients in dupilumab phase 3 randomized controlled trials did not achieve pre-established efficacy endpoints [5] and up to approximately 30% of dupilumab-treated patients in real-world studies did not achieve adequate disease control, as assessed using the patient-reported Atopic Disease Control Tool, after approximately 1 year of treatment [6,7,8], indicating the need for additional, more effective treatments.
JAK Inhibitor Safety Overview
To date, three Janus kinase (JAK) inhibitors—abrocitinib, baricitinib, and upadacitinib—have been approved for the treatment of moderate-to-severe AD in several countries worldwide, including the European Union; abrocitinib and upadacitinib, but not baricitinib, are also approved in the USA and Canada. Treatment with JAK inhibitors is associated with achievement of high-threshold efficacy endpoints (e.g., ≥ 90% improvement from baseline in the Eczema Area and Severity Index) [9,10,11], with an overall favorable tolerability and safety profile [12]. Head-to-head trials have shown advantages of abrocitinib and upadacitinib over dupilumab in regards to speed of onset and depth of responses [13,14,15]. However, the JAK class has been associated with rare serious adverse events (AEs), such as infections, major adverse cardiovascular events (MACE), venous thromboembolism (VTE), and malignancies, especially in patients aged ≥ 65 years, current long-time or past long-time smokers, and those at a higher baseline risk of these conditions (e.g., with history of atherosclerotic cardiovascular disease [ASCVD]) [12, 16, 17]. Lower incidence of these AEs was seen in patients with AD receiving abrocitinib indirectly compared with tofacitinib, another JAK inhibitor, used in patients with rheumatoid arthritis (RA) who had at least one additional cardiovascular (CV) risk factor; this is consistent with the younger age and lower prevalence of CV risk factors in patients with AD [18]. Abrocitinib may have an improved safety profile over tofacitinib since it provides more selective JAK1 inhibition and it would likely be used by a younger and healthier population without concomitant use of immunosuppressant therapies, as reported in the tofacitinib trial [16, 18, 19]. Indeed, integrated safety analysis of over 3000 patients with moderate-to-severe AD treated with abrocitinib found safety event rates to be lower than those observed in the tofacitinib studies in patients with RA [18].
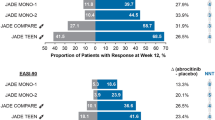
The efficacy and safety of abrocitinib, an oral, once-daily, JAK1-selective inhibitor, has been studied extensively in the JADE clinical development program, which involved more than 4000 adults and adolescents with moderate-to-severe AD [14, 20,21,22,23,24,25]. In the JADE clinical trials, abrocitinib dosages of 200 or 100 mg/day were efficacious as monotherapy [20,21,22] or in combination with medicated topical therapies [23,24,25] in improving skin lesions, pruritus, sleep disturbance, and QoL. Abrocitinib provided itch relief, as early as 24 h after the first dose, followed by rapid improvements in skin symptoms [14, 26]. Abrocitinib was infrequently discontinued as a result of AEs, which were mostly mild, transient, and manageable [19, 27, 28]. Long-term abrocitinib efficacy has been demonstrated at 48 and 96 weeks of treatment, with no unexpected safety findings observed [28,29,30].
This article provides a pragmatic approach to abrocitinib use in patients with AD using expert opinion to optimize patient selection, dosing, and monitoring. It is based on previously conducted studies and does not contain any new studies with human participants or animals performed by any of the authors. Recommendations are based on clinical trial data, safety evaluations of the JAK inhibitor class by regulatory authorities, and the authors’ experience [31,32,33].
Clinical Considerations Before Starting Abrocitinib
Abrocitinib may be used in most adults and adolescents with moderate-to-severe AD (Fig. 1) after individually assessing the benefit–risk balance for each patient [31,32,33]. In the USA, abrocitinib is indicated for use in adults and adolescents with moderate-to-severe AD with refractory disease that is not adequately controlled with other systemic therapies, including biologics or when use of those therapies is inadvisable [32]. The recommended dose is 100 mg in both adults and adolescents, and the 200 mg dose is recommended for patients not responding to 100 mg. In the European Union, abrocitinib is indicated for adults and adolescents with moderate-to-severe AD who are candidates for systemic therapy [31]. The recommended starting dose for both adults and adolescents is 100 or 200 mg, depending on individual patient characteristics, and the lowest effective dose should be considered for maintenance. In adolescents with body weight 25 to < 59 kg, abrocitinib should be started at 100 mg and increased to 200 mg if the patient does not respond adequately to the 100 mg dose; adolescents with body weight > 59 kg can be started at either 100 or 200 mg [31]. Abrocitinib is contraindicated in patients who are pregnant or breastfeeding [31, 32]. Patients who have active or untreated infections such as tuberculosis or hepatitis B or C should not take abrocitinib until these conditions are addressed appropriately [31, 32]. Patients who previously had hepatitis B or C, human immunodeficiency virus (HIV), or those who may be at high risk of undiagnosed disease should be assessed prior to initiating treatment for active infection.

Patient selection and considerations prior to initiation of abrocitinib. ALC absolute lymphocyte count, ANC absolute neutrophil count, CBC complete blood count, CVD, cardiovascular disease; CYP cytochrome P450, JAK Janus kinase, MACE major adverse cardiovascular events, PE pulmonary embolism; TB tuberculosis, VTE venous thromboembolism. aAbrocitinib is contraindicated, initiation is not recommended, or dose should be reduced for these groups of patients; verify the local product label to see the specific recommendations for your country or region
Abrocitinib can be used with or without topical corticosteroids but is not recommended to be used in combination withother JAK inhibitors, biologic immunomodulators, or other immunosuppressants such as cyclosporine and methotrexate [31, 32]. The combination of abrocitinib with these systemic therapies has not been studied and their concomitant use with abrocitinib in clinical practice is not recommended because of potential risk of additive immunosuppression [31]. Concomitant use of abrocitinib with phototherapy has also not been studied and is not recommended because of potential increased risk of malignancies, such as melanoma or nonmelanoma skin cancer. Abrocitinib is primarily eliminated by metabolism involving cytochrome P450 (CYP) enzymes, mainly CYP2C19 and CYP2C9, with minor involvement of CYP3A4 and CYP2B6 enzymes [34,35,36]. Co-administration of abrocitinib and moderate to strong CYP2C19/2C9 inducers, including rifampin (antituberculosis drug), phenytoin (antiepileptic drug), efavirenz (antiviral drug used for HIV infection), and apalutamide and enzalutamide (anticancer drugs used for prostate cancer), reduces the exposure to abrocitinib and is not recommended [31, 32, 34]. Furthermore, co-administration with strong CYP2C19 inhibitors, including fluvoxamine and fluoxetine (antidepressants), ticlopidine (antiplatelet drug), and fluconazole (antifungal drug), increases the exposure to abrocitinib and therefore requires dosage adjustments per label recommendations [31, 32, 34]. Organic anion transporter 3 (OAT3) inhibitors (e.g., probenecid, used for the treatment of gout and gouty arthritis, and teriflunomide, used for the treatment of multiple sclerosis) were found to increase abrocitinib exposure; however, these effects were not considered clinically relevant. Thus, no abrocitinib dose adjustment is necessary in patients taking OAT3 inhibitors [34].
Given the potential risk of MACE, VTE, and malignancies associated with JAK inhibitor use [16, 17, 33, 37], clinicians should consider the baseline risk before initiating abrocitinib. Higher rates of these AEs were seen in patients with RA who had increased risk for cardiovascular disease and received the JAK inhibitor, tofacitinib, indirectly compared with patients with AD who received abrocitinib treatment in randomized clinical trials and in an ongoing long-term extension study, highlighting the importance of evaluating the baseline risks for each unique patient [18]. Special considerations should be exercised when treating individuals aged ≥ 65 years because of high risk of serious AEs in this age group, current long-time or past long-time smokers, and those at increased risk of MACE (e.g., previous history of ASCVD, including myocardial infarction or stroke) or VTE (e.g., personal or family history of pulmonary embolism [PE] or deep vein thrombosis [DVT]) or with malignancy risk factors (e.g., current malignancy or history of malignancy) [16, 31,32,33]. Abrocitinib should be also used with caution in patients with other VTE risk factors, including obesity, prothrombotic disorder, recent trauma or major surgery, or prolonged immobilization [31, 38, 39]. Studies have also suggested that estrogen-containing oral contraceptive pills and hormone replacement therapy pose a risk for VTE, independent of JAK inhibitor use [40, 41]. However, no VTE cases were reported in abrocitinib-treated female patients who received hormonal contraceptives; a single case of VTE (nonfatal bilateral PE) was reported in a patient who received hormone replacement therapy [42]. The benefits and risks of JAK inhibitor use in patients with known malignancy should be evaluated on a case-by-case basis [16, 17, 31,32,33, 37].
If abrocitinib is considered the appropriate treatment, a complete blood count (including platelets, absolute lymphocyte count [ALC], absolute neutrophil count [ANC], and hemoglobin), lipid parameter measurements, measurements of transaminases, renal function testing, and screening for hepatitis B or C and HIV in accordance with clinical guidelines and local regulations prior to immunosuppressive treatment should be performed before initiating treatment [31, 32]. Patients should also be evaluated and tested for tuberculosis before initiating abrocitinib; yearly screening should be considered for patients in highly endemic areas for tuberculosis. The use of screening tests, including interferon-γ release assays (e.g., QuantiFERON), tuberculin skin tests, enzyme-linked immunosorbent assay (ELISA) tests, and/or chest X-rays, before treating adult and adolescent patients with JAK inhibitors, varies depending on local regulations and clinical guidelines. Chest X-ray may be used in conjunction with the QuantiFERON test, or only if a patient tests positive using the QuantiFERON test according to local regulations. In the abrocitinib clinical trials, including adult and adolescent patients, a dose-dependent decrease in platelet count was noted, with a nadir at week 4 [19, 25]. Median platelet counts subsequently increased and reached a plateau at week 12, with final 12-week values remaining below baseline but within the normal range [19]. Thrombocytopenia (< 75 × 103 platelets/mm3) was rare but more common in patients aged ≥ 65 years and in those receiving abrocitinib 200 mg/day [19, 43]. No change over time was seen in ANC, ALC, or hemoglobin; however, rare cases of lymphopenia have been observed [19, 31, 32].
Blood lipid levels should be assessed before beginning abrocitinib and at 4 weeks following treatment initiation and managed, if needed, per dyslipidemia management guidelines [31, 32, 44, 45]. In clinical trials, abrocitinib was associated with a dose-related increase in low-density lipoprotein cholesterol (LDL-C) from baseline to week 16, with no notable change in the LDL-C/high-density lipoprotein cholesterol (HDL-C) ratio over 16 weeks [19].
Abrocitinib dose selection should consider patient characteristics. Patients aged ≥ 65 years are at a higher risk of AEs and should start with the 100-mg/day dosage [31,32,33]. Patients with mild (Child–Pugh class A, 5–6 points) or moderate (Child–Pugh class B, 7–9 points) hepatic impairment [46] can be treated with abrocitinib without dose adjustment; however, abrocitinib must not be used in patients with severe hepatic impairment (Child–Pugh class C, 10–15 points) [31, 32]. In abrocitinib clinical trials, both moderate (estimated glomerular filtration rate [eGFR] ≥ 30 to < 60 mL/min) and severe renal impairment (eGFR < 30 mL/min but not requiring dialysis) led to higher exposure to abrocitinib active metabolites, requiring dosage adjustment as stated in the label [31, 32, 47]. A lower abrocitinib dose (50 mg) is available for dose adjustment in special populations (i.e., patients with renal impairment or concomitant use of medications that interact with abrocitinib).
Advising the Patient Initiating Abrocitinib
Shared decision-making between the healthcare practitioner and the patient should take into account each patient’s risk factors and medical history before treatment initiation with abrocitinib. Risk factors for herpes zoster (HZ) during abrocitinib treatment included the higher dosage (200 mg/day), age ≥ 65 years, ALC < 1.0 × 103/mm3, and prior history of HZ [28]. If feasible, all patients should complete age-appropriate vaccines, including non-live HZ vaccination (e.g., Zoster Vaccine Recombinant, Adjuvanted) around 2–4 weeks before abrocitinib initiation, especially those who are at a higher risk of HZ (≥ 50 years old, immunosuppressed, or immunodeficient) [31, 32, 48, 49] (Fig. 2). In patients who have already initiated abrocitinib, non-live vaccines can be given at any point during therapy without stopping treatment. Patients should avoid vaccination with live vaccines immediately prior to, during, and immediately after abrocitinib treatment [31, 32]. We recommend stopping live vaccines up to at least 1 week prior to initiating abrocitinib, and a waiting time of 2–4 weeks after stopping treatment, depending on the vaccine product label.

Patient monitoring during abrocitinib treatment. CBC complete blood count, ER emergency room, MACE major adverse cardiovascular events, PE pulmonary embolism, UV ultraviolet, VTE venous thromboembolism
Potential tolerability concerns include nausea, headache, and acne [31]. Nausea typically occurred within the first week of treatment, was generally mild or moderate, and resolved spontaneously within a median time of 15 days [19]. Taking abrocitinib with food or reducing abrocitinib dose may help alleviate nausea [31, 50]. Headache (not severe or serious) usually occurred within the first week of treatment and resolved in a median time of 4 days [19]. Acne was not severe or serious, was less frequent with abrocitinib 100 mg, and should be managed according to guidelines (e.g., with topical clindamycin-benzoyl peroxide, retinoid, dapsone, azelaic acid, or oral antibiotics) [19, 51].
Abrocitinib has not been studied in pregnant women; birth control is recommended in women of childbearing potential who are taking abrocitinib [31]. Women who are planning a pregnancy should discontinue abrocitinib for 4 weeks before attempting to conceive. Abrocitinib has no effects on male fertility and is not genotoxic [31, 32].
An increased infection risk during abrocitinib treatment has been reported in clinical trials; the most common serious infections were herpes simplex (< 5%), HZ (< 2%), and pneumonia (< 1%) [19, 28]. Herpes simplex medical history was a risk factor for herpes simplex infection [19]. Orally administered valaciclovir, aciclovir, or famciclovir may be used in patients who develop herpes simplex infection [52], and prophylactic use may be considered in individuals with a history of frequent herpes simplex outbreaks. In clinical trials, cases of HZ were typically mild or moderate [19]. Abrocitinib treatment may be temporarily stopped in case of HZ infection [31, 32], and patients may be treated with aciclovir, famciclovir, or valaciclovir per treatment guidelines [52, 53].
Healthcare professionals should discuss with their patients the risks of MACE, VTE, and malignancies associated with JAK inhibitors [33]. Age-appropriate malignancy screening (e.g., mammogram, prostate exam) should be performed in all patients with AD. Protection from the sun and monitoring for skin cancer are recommended in patients receiving abrocitinib [31, 32, 50].
Follow-up
Patients should be evaluated for clinical response to abrocitinib after the first 4 weeks of treatment, and laboratory monitoring for hematologic and lipid abnormalities should be conducted at this time and then every 8–12 weeks (Fig. 2) [19, 31, 32, 44, 54]. Interruption or discontinuation of abrocitinib may be needed if low levels of platelets (< 50 × 103/mm3), ANC (< 1 × 103/mm3), ALC (< 0.5 × 103/mm3), or hemoglobin (< 8 g/dL) are detected [31, 32]. Lipid levels should be assessed at the 4-week follow-up visit; in the case of hyperlipidemia, use of lipid-lowering therapy (e.g., statins) may be required [44]. Dose adjustments because of lipid abnormalities are not needed, nor is regular monitoring of renal and liver functions. Although a dose-related increase in creatine phosphokinase (CPK) was observed in clinical trials, no meaningful differences in the proportions of patients with CPK levels exceeding five- or tenfold the upper limit of normal were reported between abrocitinib doses and placebo [19]. Rhabdomyolysis has been reported rarely with JAK inhibitors [19, 55, 56]. Monitoring CPK levels in abrocitinib-treated patients who increase their physical activity or experience muscle pain is reasonable, as rhabdomyolysis can lead to kidney damage [57, 58].
Conclusions
Abrocitinib may be used in most patients with moderate-to-severe AD as indicated per label. In addition, cautious evaluation is needed in patients aged ≥ 65 years, current long-time or past long-time smokers, and those with risk factors for VTE, MACE, or malignancies [28, 33].
Abrocitinib availability at three doses (50, 100, and 200 mg) allows for treatment adjustment based on patients’ characteristics. Patients with tolerability concerns with abrocitinib 200 mg/day may have their dosage reduced to 100 mg/day with a reasonable likelihood of maintaining their response [24]. Clinical data showed that 60% of patients who switched to abrocitinib 100 mg and 22% of those who switched to placebo after achieving good response with abrocitinib 200 mg treatment for 3 months maintained control over their AD for almost a year thereafter. Additionally, for most of patients who experience flaring, a 3-month rescue treatment with abrocitinib 200 mg and topical medication successfully recaptured their previous response [24].
Clinicians can improve the benefit–risk ratio of JAK inhibitors by appropriate patient selection and evaluation as well as patient education.
References
Weidinger S, Beck LA, Bieber T, Kabashima K, Irvine AD. Atopic dermatitis. Nat Rev Dis Prim. 2018;4(1):1. https://doi.org/10.1038/s41572-018-0001-z.
Wollenberg A, Barbarot S, Bieber T, et al. Consensus-based European guidelines for treatment of atopic eczema (atopic dermatitis) in adults and children: part I. J Eur Acad Dermatol Venereol. 2018;32(5):657–82. https://doi.org/10.1111/jdv.14891.
Nogueira M, Torres T. Janus kinase inhibitors for the treatment of atopic dermatitis: focus on abrocitinib, baricitinib, and upadacitinib. Dermatol Pract Concept. 2021;11(4):e2021145. https://doi.org/10.5826/dpc.1104a145.
Guttman-Yassky E, Bissonnette R, Ungar B, et al. Dupilumab progressively improves systemic and cutaneous abnormalities in patients with atopic dermatitis. J Allergy Clin Immunol. 2019;143(1):155–72. https://doi.org/10.1016/j.jaci.2018.08.022.
Ariëns LFM, Bakker DS, van der Schaft J, Garritsen FM, Thijs JL, de Bruin-Weller MS. Dupilumab in atopic dermatitis: rationale, latest evidence and place in therapy. Ther Adv Chronic Dis. 2018;9(9):159–70. https://doi.org/10.1177/2040622318773686.
Strober B, Mallya UG, Yang M, et al. Treatment outcomes associated with dupilumab use in patients with atopic dermatitis: 1-year results from the RELIEVE-AD study. JAMA Dermatol. 2022;158(2):142–50. https://doi.org/10.1001/jamadermatol.2021.4778.
Oosterhaven JAF, Spekhorst LS, Zhang J, et al. Eczema control and treatment satisfaction in atopic dermatitis patients treated with dupilumab—a cross-sectional study from the BioDay registry. J Dermatolog Treat. 2022;33(4):1986–9. https://doi.org/10.1080/09546634.2021.1937485.
Kimball AB, Delevry D, Yang M, et al. Long-term effectiveness of dupilumab in patients with atopic dermatitis: results up to 3 years from the RELIEVE-AD study. Dermatol Ther (Heidelb). 2023;13(9):2107–20. https://doi.org/10.1007/s13555-023-00965-5.
Ständer S, Bhatia N, Gooderham MJ, et al. High threshold efficacy responses in moderate-to-severe atopic dermatitis are associated with additional quality of life benefits: pooled analyses of abrocitinib monotherapy studies in adults and adolescents. J Eur Acad Dermatol Venereol. 2022;36(8):1308–17. https://doi.org/10.1111/jdv.18170.
Guttman-Yassky E, Teixeira HD, Simpson EL, et al. Once-daily upadacitinib versus placebo in adolescents and adults with moderate-to-severe atopic dermatitis (Measure Up 1 and Measure Up 2): results from two replicate double-blind, randomised controlled phase 3 trials. Lancet. 2021;397(10290):2151–68. https://doi.org/10.1016/s0140-6736(21)00588-2.
Simpson EL, Lacour JP, Spelman L, et al. Baricitinib in patients with moderate-to-severe atopic dermatitis and inadequate response to topical corticosteroids: results from two randomized monotherapy phase III trials. Br J Dermatol. 2020;183(2):242–55. https://doi.org/10.1111/bjd.18898.
Le M, Berman-Rosa M, Ghazawi FM, et al. Systematic review on the efficacy and safety of oral Janus kinase inhibitors for the treatment of atopic dermatitis. Front Med (Lausanne). 2021;8: 682547. https://doi.org/10.3389/fmed.2021.682547.
Blauvelt A, Ladizinski B, Prajapati VH, et al. Efficacy and safety of switching from dupilumab to upadacitinib versus continuous upadacitinib in moderate-to-severe atopic dermatitis: results from an open-label extension of the phase 3, randomized, controlled trial (Heads Up). J Am Acad Dermatol. 2023;89(3):478–85. https://doi.org/10.1016/j.jaad.2023.05.033.
Reich K, Thyssen JP, Blauvelt A, et al. Efficacy and safety of abrocitinib versus dupilumab in adults with moderate-to-severe atopic dermatitis: a randomised, double-blind, multicentre phase 3 trial. Lancet. 2022;400(10348):273–82. https://doi.org/10.1016/s0140-6736(22)01199-0.
Blauvelt A, Teixeira HD, Simpson EL, et al. Efficacy and safety of upadacitinib vs dupilumab in adults with moderate-to-severe atopic dermatitis: a randomized clinical trial. JAMA Dermatol. 2021;157(9):1047–55. https://doi.org/10.1001/jamadermatol.2021.3023.
Ytterberg SR, Bhatt DL, Mikuls TR, et al. Cardiovascular and cancer risk with tofacitinib in rheumatoid arthritis. N Engl J Med. 2022;386(4):316–26. https://doi.org/10.1056/NEJMoa2109927.
Burmester GR, Cohen SB, Winthrop KL, et al. Safety profile of upadacitinib over 15 000 patient-years across rheumatoid arthritis, psoriatic arthritis, ankylosing spondylitis and atopic dermatitis. RMD Open. 2023;9(1):e002735. https://doi.org/10.1136/rmdopen-2022-002735.
Wollenberg A, Thyssen JP, Bieber T, Chan G, Kerkmann U. A detailed look at the European Medicines Agency’s recommendations for use of Janus kinase inhibitors in patients with atopic dermatitis. J Eur Acad Dermatol Venereol. 2023;37(10):2041–6. https://doi.org/10.1111/jdv.19255.
Simpson EL, Silverberg JI, Nosbaum A, et al. Integrated safety analysis of abrocitinib for the treatment of moderate-to-severe atopic dermatitis from the phase II and phase III clinical trial program. Am J Clin Dermatol. 2021;22(5):693–707. https://doi.org/10.1007/s40257-021-00618-3.
Gooderham MJ, Forman SB, Bissonnette R, et al. Efficacy and safety of oral Janus kinase 1 inhibitor abrocitinib for patients with atopic dermatitis: a phase 2 randomized clinical trial. JAMA Dermatol. 2019;155(12):1371–9. https://doi.org/10.1001/jamadermatol.2019.2855.
Simpson EL, Sinclair R, Forman S, et al. Efficacy and safety of abrocitinib in adults and adolescents with moderate-to-severe atopic dermatitis (JADE MONO-1): a multicentre, double-blind, randomised, placebo-controlled, phase 3 trial. Lancet. 2020;396(10246):255–66. https://doi.org/10.1016/s0140-6736(20)30732-7.
Silverberg JI, Simpson EL, Thyssen JP, et al. Efficacy and safety of abrocitinib in patients with moderate-to-severe atopic dermatitis: a randomized clinical trial. JAMA Dermatol. 2020;156(8):863–73. https://doi.org/10.1001/jamadermatol.2020.1406.
Bieber T, Simpson EL, Silverberg JI, et al. Abrocitinib versus placebo or dupilumab for atopic dermatitis. N Engl J Med. 2021;384(12):1101–12. https://doi.org/10.1056/NEJMoa2019380.
Blauvelt A, Silverberg JI, Lynde CW, et al. Abrocitinib induction, randomized withdrawal, and retreatment in patients with moderate-to-severe atopic dermatitis: results from the JAK1 Atopic Dermatitis Efficacy and Safety (JADE) REGIMEN phase 3 trial. J Am Acad Dermatol. 2022;86(1):104–12. https://doi.org/10.1016/j.jaad.2021.05.075.
Eichenfield LF, Flohr C, Sidbury R, et al. Efficacy and safety of abrocitinib in combination with topical therapy in adolescents with moderate-to-severe atopic dermatitis: the JADE TEEN randomized clinical trial. JAMA Dermatol. 2021;157(10):1165–73. https://doi.org/10.1001/jamadermatol.2021.2830.
Kim BS, Silverberg JI, Ständer S, et al. Rapid improvement of itch associated with atopic dermatitis with abrocitinib is partially independent of overall disease improvement: results from pooled phase 2b and 3 monotherapy studies. Dermatitis. 2021;32(1s):S39–44. https://doi.org/10.1097/der.0000000000000770.
Cork MJ, Deleuran M, Geng B, et al. Abrocitinib treatment in patients with moderate-to-severe atopic dermatitis: safety of abrocitinib stratified by age. In: Presented at the European Academy of Dermatology and Venereology (EADV) – 30th Congress; September 29–October 2, Virtual. 2021.
Simpson EL, Silverberg JI, Nosbaum A, et al. Integrated safety analysis of abrocitinib in 3802 patients with moderate-to-severe atopic dermatitis with over 5000 patient-years of exposure. J Am Acad Dermatol. 2023;89(3). supplement AB58.
Reich K, Silverberg JI, Papp KA, et al. Abrocitinib efficacy and safety in patients with moderate-to-severe atopic dermatitis: results from phase 3 studies, including the long-term extension JADE EXTEND study. J Eur Acad Dermatol Venereol. 2023;37(10):2056–66. https://doi.org/10.1111/jdv.19280.
Paller AS, Cork MJ, Flohr C, et al. P81 Long-term efficacy of abrocitinib in adolescents and adults with moderate-to-severe atopic dermatitis: a post hoc analysis of JADE EXTEND. Br J Dermatol. 2023;188(Supplement_4):ljad113.09. https://doi.org/10.1093/bjd/ljad113.109.
European Medicines Agency. Cibinqo® (abrocitinib). Summary of Product Characteristics (SmPC). Pfizer Europe MA EEIG: Belgium. 2024.
Cibinqo (abrocitinib) tablets, for oral use. Prescribing information. Pfizer Inc. 2023.
European Medicines Agency. EMA confirms measures to minimise risk of serious side effects with Janus kinase inhibitors for chronic inflammatory disorders [press release]. London, UK; 2023.
Wang X, Dowty ME, Wouters A, et al. Assessment of the effects of inhibition or induction of CYP2C19 and CYP2C9 enzymes, or inhibition of OAT3, on the pharmacokinetics of abrocitinib and its metabolites in healthy individuals. Eur J Drug Metab Pharmacokinet. 2022;47(3):419–29. https://doi.org/10.1007/s13318-021-00745-6.
Wang EQ, Le V, O’Gorman M, et al. Effects of hepatic impairment on the pharmacokinetics of abrocitinib and its metabolites. J Clin Pharmacol. 2021;61(10):1311–23. https://doi.org/10.1002/jcph.1858.
Bauman JN, Doran AC, King-Ahmad A, et al. The pharmacokinetics, metabolism, and clearance mechanisms of abrocitinib, a selective Janus kinase inhibitor, in humans. Drug Metab Dispos. 2022;50(8):1106–18. https://doi.org/10.1124/dmd.122.000829.
Bieber T, Katoh N, Simpson EL, et al. Safety of baricitinib for the treatment of atopic dermatitis over a median of 1.6 years and up to 3.9 years of treatment: an updated integrated analysis of eight clinical trials. J Dermatolog Treat. 2023;34(1):2161812. https://doi.org/10.1080/09546634.2022.2161812.
Stone J, Hangge P, Albadawi H, et al. Deep vein thrombosis: pathogenesis, diagnosis, and medical management. Cardiovasc Diagn Ther. 2017;7(Suppl 3):S276–S284. https://doi.org/10.21037/cdt.2017.09.01.
Olaf M, Cooney R. Deep venous thrombosis. Emerg Med Clin North Am. 2017;35(4):743–70. https://doi.org/10.1016/j.emc.2017.06.003.
Gialeraki A, Valsami S, Pittaras T, Panayiotakopoulos G, Politou M. Oral contraceptives and HRT risk of thrombosis. Clin Appl Thromb Hemost. 2018;24(2):217–25. https://doi.org/10.1177/1076029616683802.
Mori S, Ogata F, Tsunoda R. Risk of venous thromboembolism associated with Janus kinase inhibitors for rheumatoid arthritis: case presentation and literature review. Clin Rheumatol. 2021;40(11):4457–71. https://doi.org/10.1007/s10067-021-05911-4.
Thaci D, De Bruin‑weller M, Cork M, et al. Baseline use of oral contraceptives or hormone replacement therapy in patients with moderate-to-severe atopic dermatitis treated with abrocitinib in the phase 2 and phase 3 JADE clinical trial program and reported venous thromboembolic outcomes. In: Presented at the Annual European Academy of Dermatology and Venereology (EADV) Congress; October 11–14, Berlin, Germany. 2023.
Simpson EL, Silverberg JI, Nosbaum A, et al. Safety of abrocitinib in 3582 patients with monderate-to-severe atopic dermatitis with over 900 patients exposed for almost 2 years. In: Presented at the 31st European Academy of Dermatology and Venereology (EADV) Hybrid Congress; September 7–10, Milan, Italy. 2022.
Grundy SM, Stone NJ, Bailey AL, et al. 2018 AHA/ACC/AACVPR/AAPA/ABC/ACPM/ADA/AGS/APHA/ASPC/NLA/PCNA guideline on the management of blood cholesterol: a report of the American College of Cardiology/American Heart Association task force on clinical practice guidelines. Circulation. 2019;139(25):e1082–143. https://doi.org/10.1161/cir.0000000000000625.
Mach F, Baigent C, Catapano AL, et al. 2019 ESC/EAS guidelines for the management of dyslipidaemias: lipid modification to reduce cardiovascular risk. Eur Heart J. 2020;41(1):111–88. https://doi.org/10.1093/eurheartj/ehz455.
Tsoris A, Marlar CA. Use of the Child Pugh score in liver disease. Statpearls. Treasure Island (FL): StatPearls; 2022.
Wang EQ, Le V, Winton JA, et al. Effects of renal impairment on the pharmacokinetics of abrocitinib and its metabolites. J Clin Pharmacol. 2022;62(4):505–19. https://doi.org/10.1002/jcph.1980.
US Food and Drug Administration. Shingrix. Prescribing Information. 2023. https://www.fda.gov/media/108597/download
Dooling KL, Guo A, Patel M, et al. Recommendations of the advisory committee on immunization practices for use of herpes zoster vaccines. MMWR Morb Mortal Wkly Rep. 2018;67(3):103–8. https://doi.org/10.15585/mmwr.mm6703a5.
Cibinqo 100 mg film-coated tablets (summary of product characteristics). Pfizer; 2021.
Zaenglein AL, Pathy AL, Schlosser BJ, et al. Guidelines of care for the management of acne vulgaris. J Am Acad Dermatol. 2016;74(5):945–73.e33. https://doi.org/10.1016/j.jaad.2015.12.037.
Richez C, Morel J, Cornec D, et al. Practical management of patients on Janus kinase inhibitor (JAKi) therapy: practical fact sheets drawn up by the Rheumatism and Inflammation Club (CRI), a group endorsed by the French Society for Rheumatology (SFR). Jt Bone Spine. 2019;86(Suppl 1):eS2–eS103. https://doi.org/10.1016/s1297-319x(19)30154-x.
Werner RN, Nikkels AF, Marinović B, et al. European consensus-based (S2k) guideline on the management of herpes zoster—guided by the European Dermatology Forum (EDF) in cooperation with the European Academy of Dermatology and Venereology (EADV), part 2: treatment. J Eur Acad Dermatol Venereol. 2017;31(1):20–9. https://doi.org/10.1111/jdv.13957.
Gauer RL, Braun MM. Thrombocytopenia. Am Fam Physician. 2012;85(6):612–22.
Cohen SB, van Vollenhoven RF, Winthrop KL, et al. Safety profile of upadacitinib in rheumatoid arthritis: integrated analysis from the select phase III clinical programme. Ann Rheum Dis. 2020;80(3):304–11. https://doi.org/10.1136/annrheumdis-2020-218510.
King B, Maari C, Lain E, et al. Extended safety analysis of baricitinib 2 mg in adult patients with atopic dermatitis: an integrated analysis from eight randomized clinical trials. Am J Clin Dermatol. 2021;22(3):395–405. https://doi.org/10.1007/s40257-021-00602-x.
Valiyil R, Christopher-Stine L. Drug-related myopathies of which the clinician should be aware. Curr Rheumatol Rep. 2010;12(3):213–20. https://doi.org/10.1007/s11926-010-0104-3.
Apple FS, Quist HE, Doyle PJ, Otto AP, Murakami MM. Plasma 99th percentile reference limits for cardiac troponin and creatine kinase MB mass for use with European Society of Cardiology/American College of Cardiology Consensus Recommendations. Clin Chem. 2003;49(8):1331–6. https://doi.org/10.1373/49.8.1331.
Acknowledgements
Medical Writing and Editorial Assistance
Editorial and medical writing support under the guidance of the authors was provided by Kristine De La Torre, PhD, at ApotheCom, San Francisco, CA, USA, and was funded by Pfizer Inc., New York, NY, USA, in accordance with Good Publication Practice (GPP 2022) guidelines (Ann Intern Med. 2022; 10.7326/M22-1460).
Authorship
All named authors meet the International Committee of Medical Journal Editors (ICMJE) criteria for authorship for this article, take responsibility for the integrity of the work, and have given their approval for this version to be published.
Funding
Sponsorship for this study and Rapid Service Fee were funded by Pfizer Inc.
Author information
Authors and Affiliations
Contributions
Melinda J. Gooderham, Marjolein de Bruin-Weller, Stephan Weidinger, Michael J. Cork, Lawrence F. Eichenfield, Eric L. Simpson, Athanasios Tsianakas, Urs Kerkmann, Claire Feeney and William Romero contributed to the study concept, interpreted the data, provided critical feedback on the manuscript, approved the final manuscript for submission, and are accountable for the accuracy and integrity of the manuscript.
Corresponding author
Ethics declarations
Conflicts of Interest
Melinda J. Gooderham has received grants, personal fees, honoraria, and/or nonfinancial support from Pfizer Inc., AbbVie, Amgen, Akros Pharma, Arcutis, AnaptysBio, Aristea, Bausch Health, Bristol Myers Squibb, Boehringer Ingelheim, Celgene, Dermavant, Dermira, Eli Lilly and Company, Galderma, InMagene, JAMP Pharma, Janssen, Kyowa Kirin, LEO Pharma, MedImmune, Merck, Moonlake, Meiji, Novartis, Roche, Sanofi Genzyme, Regeneron Pharmaceuticals, Sun Pharma, Takeda, Tarsus, UCB, Union and Ventyx. Marjolein de Bruin-Weller is a consultant, advisory board member, and/or speaker for Pfizer Inc., AbbVie, Almirall, Arena, Aslan, Eli Lilly and Company, Galderma, Janssen, LEO Pharma, Regeneron Pharmaceuticals, and Sanofi Genzyme. Stephan Weidinger has received institutional research grants from Pfizer Inc., La Roche-Posay, LEO Pharma, and Sanofi Deutschland GmbH; has performed consultancies for Pfizer Inc., AbbVie, Almirall, Boehringer Ingelheim, Eli Lilly and Company, Galderma, Kymab, LEO Pharma, Novartis, Regeneron Pharmaceuticals, and Sanofi Genzyme; has lectured at educational events sponsored by Pfizer Inc., AbbVie, Almirall, Galderma, LEO Pharma, Novartis, Regeneron Pharmaceuticals, and Sanofi Genzyme; and is involved in performing clinical trials with many pharmaceutical industries that manufacture drugs used for the treatment of psoriasis and atopic dermatitis. Michael J. Cork has been a clinical trial investigator for Pfizer Inc., Atopix, Galapagos, Hyphens, Johnson & Johnson, Kymab, LEO Pharma, L’Oreal/La Roche-Posay, Novartis, Regeneron Pharmaceuticals, and Sanofi Genzyme; and an advisory board member, consultant, and/or invited lecturer for Pfizer Inc., Abbvie, Amlar, Astellas, Atopix, Boots, Dermavant, Galapagos, Galderma, Hyphens, Johnson & Johnson, Kymab, LEO Pharma, L’Oreal/La Roche-Posay, Menlo Therapeutics, Novartis, Oxagen, Procter & Gamble, Reckitt Benckiser, Regeneron Pharmaceuticals, and Sanofi Genzyme. Lawrence F. Eichenfield has served as a scientific adviser, consultant, and/or clinical study investigator for Pfizer Inc., AbbVie, Amgen, Apogee, Arcutis, Aslan, Attovia, Bristol Myers Squibb, Castle Biosciences, Dermavant, Eli Lilly and Company, Forte, Galderma, Incyte, Janssen, Johnson & Johnson, LEO Pharma, Novartis, Ortho Dermatologics, Regeneron Pharmaceuticals, Sanofi Genzyme, Target RWE and UCB. Eric L. Simpson has received grants from Pfizer Inc., Eli Lilly and Company, Kyowa Kirin, LEO Pharma, Merck, and Regeneron Pharmaceuticals, and personal fees from Pfizer Inc., Bausch Health (Valeant), Dermira, Eli Lilly and Company, Galderma, LEO Pharma, Menlo Therapeutics, Novartis, Regeneron Pharmaceuticals, and Sanofi Genzyme. Athanasios Tsianakas has acted as a clinical investigator and received honoraria for lectures and consulting from Pfizer Inc. Claire Feeney is an employee and shareholder of Pfizer Inc. Urs Kerkmann was an employee and shareholder of Pfizer Pharma GmbH at the time the study was conducted. William Romero was an employee and shareholder of Pfizer Inc. at the time the study was conducted. He is currently an employee of Eli Lilly and Company Limited.
Ethical Approval
This article is based on previously conducted studies and does not contain any new studies with human participants or animals performed by any of the authors.
Additional information
Prior Publication: This practical approach article summarizes recommendations for abrocitinib use based on published clinical trial data, safety evaluations of the JAK inhibitor class by regulatory authorities, and the authors’ experience. The published primary studies from the JADE clinical program describing the abrocitinib safety data summarized in this article are listed below:
Gooderham MJ et al. JAMA Dermatol. 2019;155(12):1371–1379. (ClinicalTrials.gov number, NCT02780167).
Simpson EL et al. Lancet. 2020;396(10246):255–266. (ClinicalTrials.gov number, NCT03349060).
Silverberg JI et al. JAMA Dermatol. 2020;156(8):863–873. (ClinicalTrials.gov number, NCT03575871).
Bieber T et al. N Engl J Med. 2021;384(12):1101–1112. (ClinicalTrials.gov number, NCT03720470).
Eichenfield LF et al. JAMA Dermatol. 2021;157(10):1165–1173. (ClinicalTrials.gov number, NCT03796676).
Blauvelt A et al. J Am Acad Dermatol. 2022;86(1):104–112. (ClinicalTrials.gov number, NCT03627767).
Reich K et al. Lancet. 2022;400(10348):273–282. (ClinicalTrials.gov number, NCT04345367).
Reich K et al. J Eur Acad Dermatol Venereol. 2023;37(10):2056–2066. (ClinicalTrials.gov number, NCT03422822).
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Gooderham, M.J., de Bruin-Weller, M., Weidinger, S. et al. Practical Management of the JAK1 Inhibitor Abrocitinib for Atopic Dermatitis in Clinical Practice: Special Safety Considerations. Dermatol Ther (Heidelb) 14, 2285–2296 (2024). https://doi.org/10.1007/s13555-024-01200-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13555-024-01200-5