
Abstract
Introduction
Deucravacitinib, an oral, selective, allosteric tyrosine kinase 2 inhibitor, blocks cytokine signaling involved in psoriasis pathogenesis. This ethnic-bridging study evaluated deucravacitinib pharmacokinetics, tolerability, and safety in healthy Chinese subjects.
Methods
This phase I, double-blind, single-/multiple-dose study randomized healthy Chinese subjects 4:1 to a single dose of deucravacitinib 6 mg or placebo (group 1) or deucravacitinib 12 mg or placebo (group 2) on day 1; groups 1 and 2 received deucravacitinib 6 mg and 12 mg once daily, respectively, or placebo on days 5–19. Blood samples were collected on days 1–5 (0 predose–96 h postdose), day 5 (0–24 h postdose), days 9 and 12 (0 h), and day 19 (0–24 h postdose). Deucravacitinib and metabolite (BMT-153261, BMT-158170) concentrations were determined using liquid chromatography/mass spectrometry; pharmacokinetic parameters were calculated using noncompartmental analysis. Urine was collected on days 1–4 (4 h predose–96 h postdose). Safety was monitored throughout.
Results
Forty healthy Chinese subjects (groups 1 and 2: deucravacitinib, n = 32; placebo, n = 8) were enrolled. Deucravacitinib was rapidly absorbed after single-/multiple-dose administration, with median time to maximal plasma concentration of 1.5–2.3 h. Systemic exposure after single or multiple doses increased approximately twofold with twofold dose increase. Modest deucravacitinib accumulation was observed after multiple-dose administration (1.3- to 1.4-fold increase in area under the curve [AUC] under one dosing interval). Metabolite-to-parent ratios for maximal plasma concentration and AUC remained consistent in each dose group. Mean urinary percent recovery and renal clearance were similar between dose groups. Most adverse events (AEs) were mild/moderate, with no serious treatment-related AEs, deaths, or discontinuations due to AEs.
Conclusion
Deucravacitinib was safe and well tolerated in healthy Chinese subjects. Deucravacitinib exhibited rapid absorption, dose-related increases in exposure, comparable half-life, and no evidence of time-dependent pharmacokinetics, suggesting minimal effect of Chinese ethnicity on deucravacitinib pharmacokinetics.
Clinical Trial Registration
NCT03956953.
Plain Language Summary
Deucravacitinib, a new oral medication, blocks an enzyme called tyrosine kinase 2 (TYK2), which is activated in plaque psoriasis. This reduces thick, scaly patches of skin, itching, and other symptoms. How a drug is absorbed and its effects can vary between patients of different races and ethnicities. We studied the safety of deucravacitinib in healthy Chinese volunteers. We also studied how bigger or smaller doses of deucravacitinib change how much of it is absorbed into the blood. We found that most side effects of deucravacitinib were mild or moderate compared to volunteers taking placebo, a look-alike pill that contains no drug. The most common side effects were skin rashes and headaches. No serious side effects were related to deucravacitinib. Deucravacitinib was quickly absorbed into the blood. The time it took for deucravacitinib to reach its maximum amount in the blood was similar regardless of how large of a dose was initially taken. Increasing the amount of deucravacitinib taken also increased the total amount of deucravacitinib absorbed, both in terms of the total amount absorbed and the maximum amount in blood at one time. These results in healthy Chinese volunteers were similar to the results of other studies in a general population of many races and ethnicities. Deucravacitinib works the same in Chinese patients as in patients of other ethnicities. Chinese patients will not need to adjust their dose when taking deucravacitinib.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Why carry out this study? |
Deucravacitinib is a novel tyrosine kinase 2 inhibitor approved for the treatment of plaque psoriasis. |
There may be racial or ethnic effects on drug pharmacokinetics and metabolism. |
This study evaluated the pharmacokinetics and safety of deucravacitinib in healthy Chinese subjects to determine if there were racial and ethnic disparities. |
What was learned from the study? |
Deucravacitinib was safe and well tolerated, and exhibited similar pharmacokinetics and dose effects in healthy Chinese subjects as reported for the general population in previous studies. |
Chinese ethnicity has minimal effect on deucravacitinib pharmacokinetics. |
Introduction
Tyrosine kinase 2 (TYK2) is an intracellular kinase that mediates signaling of cytokines (e.g., interleukin-23, type I interferons) involved in the pathogenesis of various immune-mediated, inflammatory diseases such as psoriasis, psoriatic arthritis, inflammatory bowel disease, and systemic lupus erythematosus [1]. Deucravacitinib, an oral, selective, allosteric TYK2 inhibitor, is approved in the USA, European Union, and other countries for the treatment of adults with moderate-to-severe plaque psoriasis who are candidates for systemic therapy [2, 3]. Deucravacitinib is highly selective for TYK2 because it binds to the regulatory domain of the kinase, whereas Janus kinase (JAK) 1, 2, and 3 inhibitors bind to the more conserved catalytic domain of the kinase [1]. As a result, deucravacitinib avoids the off-target effects associated with JAK 1, 2, and 3 inhibitors [4].
The pharmacokinetics (PK) and safety of deucravacitinib in healthy subjects were previously reported in a first-in-human study (NCT02534636) [5]. Deucravacitinib was rapidly absorbed, attaining a time to maximum plasma concentration (tmax) within 1.0 h after dosing, after which the plasma concentration declined in an exponential manner, with a plasma half-life (t1/2) of 8–15 h [5]. Deucravacitinib demonstrated dose-proportional PK, with modest systemic accumulation (1.4- to 1.9-fold) observed after multiple dosing [5]. Additional PK analyses revealed that the major pathways of deucravacitinib elimination included metabolism by cytochrome P450 (CYP) 1A2 (~ 25% of dose), uridine 5′-diphospho-glucuronosyltransferase 1A9 (~ 24% of dose), carboxylesterase 2 (~ 15% of dose), CYP2B6/CYP2D6 (~ 6% of dose), direct urinary elimination (~ 13% of dose), and fecal excretion (~ 26% of dose). Deucravacitinib was generally safe and well tolerated when administered as single or multiple doses for up to 12 days to healthy subjects in the first-in-human study [5].
The PK and safety of drugs can vary among populations, reflecting racial and ethnic disparities in how a specific drug is metabolized in any given population [6,7,8]. The current ethnic bridging study was designed to evaluate the PK, safety, and tolerability of single and multiple doses of deucravacitinib in healthy Chinese subjects. Deucravacitinib 6 mg and 12 mg once daily were evaluated in previous phase II [9, 10] and phase III [11, 12] trials. Based on the doses studied in these trials, the current ethnic bridging study also evaluated deucravacitinib 6 mg and 12 mg.
Methods
Study Design
This was a phase I, double-blind, randomized, placebo-controlled, single- and multiple-dose study (Fig. 1). The study was conducted at a single clinical facility in China (Beijing Anzhen Hospital, Capital Medical University, Beijing, China) in accordance with the Declaration of Helsinki and the International Conference on Harmonization E6 Guidelines on Good Clinical Practice. An institutional review board reviewed and approved the study protocol and related documents, and all subjects provided written informed consent before any study procedures were performed. This study is registered at www.clinicaltrials.gov as NCT03956953.

Study design. QD once daily
On day 1, eligible healthy subjects were randomized 4:1 to a single oral dose of deucravacitinib 6 mg or matching placebo (group 1) or a single oral dose of deucravacitinib 12 mg or placebo (group 2). On day 5 through day 19, subjects in group 1 received deucravacitinib 6 mg once daily or placebo, and subjects in group 2 received deucravacitinib 12 mg once daily or placebo. Subjects were admitted to the clinical facility on day − 1 and were required to fast (i.e., nothing to eat or drink except water) for approximately 10 h before and 4 h after study drug administration on days 1, 5, and 19.
Participants
Healthy adult male and female subjects (born in mainland China with both parents ethnically Chinese) without any evidence of organ dysfunction or clinically significant abnormalities in medical history, physical examination findings, 12-lead electrocardiogram recordings, and clinical laboratory evaluations were eligible for study inclusion. Subjects were required to be between 18 and 45 years of age with a body mass index of 18.0–24.0 kg/m2 and total body weight ≥ 50 kg. Female subjects were required to have proof that they were not pregnant at the time of screening. Key exclusion criteria were a history of serious bacterial, fungal, or viral infection; any significant acute or chronic illness, including infection within 7 days of study admission; a history of or suspected immunodeficiency or other factor that would increase the risk of developing an infection; use or intended use of over-the-counter medication or Chinese herbal preparations within 14 days before dosing and during the study; and use of any prescription drugs or over-the-counter acid controllers within 4 weeks before study drug administration.
Pharmacokinetic Sampling and Bioanalytical Methodology
Blood specimens for PK analysis were collected at selected time points on days 1–5 (0 [predose] to 96 h postdose), day 5 (0–24 h postdose), days 9 and 12 (0 h), and day 19 (0–24 h postdose). Plasma samples processed from the collected blood specimens were analyzed for deucravacitinib and its metabolite (BMT-153261 and BMT-158170) concentrations using validated liquid chromatography/mass spectrometry assays. The lower limit of quantitation of deucravacitinib, BMT-153261, and BMT-158170 in plasma was 0.5 ng/mL. Urine collection was performed during the single-dose period on days 1–4 (4 h predose to 96 h postdose). A validated assay was used to analyze urine samples for deucravacitinib only; the lower limit of quantitation of deucravacitinib in urine was 0.2 ng/mL.
Pharmacokinetic Analyses
PK parameters for deucravacitinib and its metabolites were area under the concentration–time curve (AUC) from 0 to the time of the last quantifiable concentration (AUC0–t), AUC from 0 to infinity (AUC0–∞), AUC over one dosing interval (AUCtau), tmax, t1/2, maximum plasma concentration (Cmax), apparent oral total body clearance (CLT/F; deucravacitinib only), total amount of drug recovered in urine (URt; deucravacitinib only), total percentage of administered dose recovered unchanged in urine (pURt; deucravacitinib only), and renal clearance (CLR; deucravacitinib only). PK analyses included all subjects who received at least one dose of study drug and had sufficient concentration–time data available to calculate at least one key PK parameter (i.e., Cmax or AUC). PK parameters were estimated from plasma and urine concentration data by non-compartmental analysis using Phoenix WinNolin (version 8.1; Certara Inc., Princeton, NJ, USA).
Safety Analyses
Safety was evaluated throughout the study via adverse event monitoring using the Medical Dictionary for Regulatory Activities (MedDRA version 22.0). Physical examinations, vital sign measurements, 12-lead electrocardiogram recordings, and clinical laboratory evaluations were performed at selected time points during the study. Any subject who received at least one dose of study drug was included in the safety analyses.
Statistical Analyses
No formal statistical testing of study results was performed. The sample sizes specified in this study were not based on statistical power considerations but were typical for single- and multiple-dose PK studies where both active drug and placebo are administered within groups.
The plasma concentrations of deucravacitinib and each metabolite were summarized by dose and time point during the single- and multiple-dose periods; the urine concentrations of deucravacitinib only were summarized during the single-dose period. Additionally, semi-logarithmic and linear plots of the arithmetic mean and standard deviation by dose and time point were produced for the single- and multiple-dose periods. Descriptive statistics (n, mean, standard deviation, geometric mean, percent coefficient of variation, median, minimum, and maximum) were used to summarize plasma and urine concentrations as well as PK parameters. PK data management was performed using SAS version 9.4 (SAS Institute Inc, Cary, NC, USA).
Incidences of adverse events, serious adverse events, adverse events by severity, and adverse events related to study drug were summarized. Marked abnormalities in clinical laboratory parameters, electrocardiogram recordings, vital sign measurements, and physical examination findings occurring up to 30 days after study drug administration were also summarized.
Results
Baseline Subject Demographics and Clinical Characteristics
A total of 40 subjects were randomized and received study drug in group 1 (deucravacitinib, n = 16; placebo, n = 4) and group 2 (deucravacitinib, n = 16; placebo, n = 4). Thirty-nine subjects completed the study; one subject in the deucravacitinib 6 mg single-dose cohort discontinued as a result of pregnancy (additional details are provided in the “Safety” section). Baseline subject demographics and clinical characteristics were similar across groups (Table 1).
Pharmacokinetics of Deucravacitinib and Metabolites
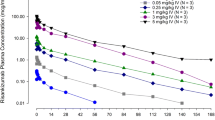
Deucravacitinib was rapidly absorbed after a single dose (Fig. 2; Table 2) or following multiple daily doses of 6 mg or 12 mg (Fig. 3; Table 3), with median tmax ranging between 1.5 and 2.3 h. Values for Cmax, AUC0–t, and AUC0–∞ after a single dose (Table 2) and steady-state values for Cmax and AUCtau after multiple doses (Table 3) increased by approximately twofold with a twofold increase in deucravacitinib dose. Modest accumulation of deucravacitinib, which was consistent between doses, was observed after multiple doses (1.3- to 1.4-fold increase in AUCtau) (Table 3). CLT/F values were dose independent after a single dose (Table 2) or following multiple daily doses (Table 3). The metabolite-to-parent ratio for Cmax and AUC remained consistent for BMT-153261 and BMT-158170 in the 6-mg and 12-mg dose groups after single- or multiple-dose administration of deucravacitinib (Tables 4, 5). Mean percentage of urinary recovery and mean CLR values of deucravacitinib were similar after a single dose of 6 mg or 12 mg (Table 2).

Mean plasma concentration versus time at day 1 after a single dose of deucravacitinib: a deucravacitinib, b BMT-158170, and c BMT-153261. LLOQ lower limit of quantitation

Mean plasma concentration of deucravacitinib versus time at day 5 and day 19 after multiple dosing: a deucravacitinib 6 mg once daily, b deucravacitinib 12 mg once daily. LLOQ lower limit of quantitation, QD once daily
Safety
Twenty subjects (50%) developed at least one treatment-emergent adverse event considered related to deucravacitinib; the most common treatment-related events were mild rash (n = 14 [35%]) and mild headache (n = 8 [20%]; Table 6). Two severe adverse events of elevated creatine phosphokinase levels were reported (one at each dose in the multiple-dose group), which were considered related to deucravacitinib. Both events resolved without medical treatment and without the need to delay or discontinue deucravacitinib administration. All other adverse events also resolved without medical treatment, and none resulted in study discontinuation. One serious adverse event (hospitalization secondary to an elective abortion), which was considered not related to deucravacitinib treatment, was reported during the study. No deaths were reported. No clinically significant abnormal electrocardiogram results or changes in vital signs were observed.
Discussion
This is the first study to evaluate the PK, safety, and tolerability of single and multiple doses of the TYK2 inhibitor deucravacitinib in healthy Chinese subjects. The phase I study demonstrated that deucravacitinib was rapidly absorbed after oral administration of single or multiple doses. Systemic exposure to deucravacitinib after a single dose and at steady-state exposures after multiple doses increased in a dose-proportional manner. Modest accumulation of deucravacitinib in both dose groups was observed after multiple dosing, and deucravacitinib was rapidly eliminated. Similar to deucravacitinib, systemic exposure to the metabolites of deucravacitinib was dose proportional and the metabolite-to-parent ratio remained consistent between dose groups after single- or multiple-dose administration. The mean percentage of urinary recovery and the CLR of deucravacitinib were similar between dose groups after single-dose administration. Deucravacitinib was safe and well tolerated in healthy Chinese subjects.
The PK profile of deucravacitinib in healthy Chinese subjects was comparable to that previously described in a phase I, first-in-human study of healthy non-Chinese subjects [5]. The t1/2 value after single dosing and the Cmax and AUCtau values after multiple dosing were generally comparable in both studies, suggesting limited ethnic variability. Moreover, the PK parameters of deucravacitinib in healthy Chinese subjects and in healthy non-Chinese subjects in the first-in-human study were linear and dose proportional at the dose ranges evaluated in these studies. Similar to non-Chinese subjects [5], deucravacitinib exhibited rapid absorption, dose-related increases in exposure, comparable terminal t1/2, and no evidence of time-dependent PK in Chinese subjects, suggesting minimal effect of Chinese ethnicity on deucravacitinib PK. The lack of substantial differences in the PK profile of deucravacitinib between healthy Chinese subjects and healthy non-Chinese subjects in the first-in-human study is consistent with the absorption, metabolism, and elimination mechanism of deucravacitinib. PK differences between Chinese and non-Chinese subjects for deucravacitinib, a small molecule metabolized via drug-metabolizing enzymes and transporters, could arise as a result of differences in enzyme or transporter activity between the various ethnicities. However, as multiple enzymes and pathways are involved in the metabolism of deucravacitinib, it is unlikely that genetic polymorphisms in any one enzyme would meaningfully alter deucravacitinib exposure.
Although the sample size in this study (pharmacokinetic population, N = 32; safety population, N = 40) is generally consistent with those in other phase I trials that evaluated the pharmacokinetics and safety of other agents used to treat plaque psoriasis (N = 24–89) [13,14,15], the relatively small sample size, combined with the short study duration and population of healthy volunteers, may limit how study results may be generalized to patients with plaque psoriasis who will receive longer-term deucravacitinib treatment. A larger, phase III randomized controlled trial (NCT04167462) has been conducted in mainland China, Taiwan, and South Korea to further evaluate the efficacy and safety of deucravacitinib in a population from this region with moderate to severe plaque psoriasis.
This study demonstrated that deucravacitinib administered as single or multiple doses of 6 mg or 12 mg once daily was safe and well tolerated in healthy Chinese subjects. Most adverse events were mild or moderate in severity and there were no serious treatment-related adverse events, deaths, or discontinuations due to adverse events. Elevations in creatine phosphokinase were transient and resolved without medical treatment or deucravacitinib delay or discontinuation. The safety and tolerability profile of deucravacitinib in healthy Chinese subjects is consistent with that described in the phase I, first-in-human deucravacitinib trial in healthy non-Chinese subjects [5].
Conclusion
The TYK2 inhibitor deucravacitinib, administered as single or multiple doses, was safe and well tolerated in healthy Chinese subjects. Similar to non-Chinese subjects, deucravacitinib exhibited rapid absorption, dose-related increases in exposure, comparable terminal t1/2, and no evidence of time-dependent PK in Chinese subjects, suggesting minimal effect of Chinese ethnicity on deucravacitinib PK. Consequently, ethnic-specific dose adjustment is not considered necessary for deucravacitinib in the Chinese population.
Data Availability
The datasets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.
References
Burke JR, Cheng L, Gillooly KM, et al. Autoimmune pathways in mice and humans are blocked by pharmacological stabilization of the TYK2 pseudokinase domain. Sci Transl Med. 2019;11(502):eaaw1736.
Sotyktu [package insert]. Princeton, NJ, USA: Bristol Myers Squibb Company; Sept 2022.
Sotyktu [summary of product characteristics]. Dublin, Ireland: Bristol Myers Squibb Pharmaceutical Operations; Mar 2023.
Winthrop KL. The emerging safety profile of JAK inhibitors in rheumatic disease. Nat Rev Rheumatol. 2017;13(4):234–43.
Catlett IM, Aras U, Hansen L, et al. First-in-human study of deucravacitinib: a selective, potent, allosteric small-molecule inhibitor of tyrosine kinase 2. Clin Transl Sci. 2023;16:151–64.
Bjornsson TD, Wagner JA, Donahue SR, et al. A review and assessment of potential sources of ethnic differences in drug responsiveness. J Clin Pharmacol. 2003;43(9):943–67.
Eliasson E. Ethnicity and adverse drug reactions. BMJ. 2006;332(7551):1163–4.
Baehr A, Peña JC, Hu DJ. Racial and ethnic disparities in adverse drug events: a systematic review of the literature. J Racial Ethn Health Disparities. 2015;2(4):527–36.
Papp K, Gordon K, Thaci D, et al. Phase 2 trial of selective tyrosine kinase 2 inhibition in psoriasis. N Engl J Med. 2018;379(14):1313–21.
Mease PJ, Deodhar AA, van der Heijde D, et al. Efficacy and safety of selective TYK2 inhibitor, deucravacitinib, in a phase II trial in psoriatic arthritis. Ann Rheum Dis. 2022;81(6):815–22.
Armstrong AW, Gooderham M, Warren RB, et al. Deucravacitinib versus placebo and apremilast in moderate to severe plaque psoriasis: efficacy and safety results from the 52-week, randomized, double-blinded, placebo-controlled phase 3 POETYK PSO-1 trial. J Am Acad Dermatol. 2023;88(1):29–39.
Strober B, Thaçi D, Sofen H, et al. Deucravacitinib versus placebo and apremilast in moderate to severe plaque psoriasis: efficacy and safety results from the 52-week, randomized, double-blinded, Program fOr Evaluation of TYK2 inhibitor psoriasis second phase 3 trial. J Am Acad Dermatol. 2023;88(1):40–51.
Khalilieh S, Hodsman P, Xu C, Tzontcheva A, Glasgow S, Montgomery D. Pharmacokinetics of tildrakizumab (MK-3222), an anti-IL-23 monoclonal antibody, after intravenous or subcutaneous administration in healthy subjects. Basic Clin Pharmacol Toxicol. 2018;123(3):294–300.
Khatri A, Eckert D, Oberoi R, et al. Pharmacokinetics of risankizumab in Asian healthy subjects and patients with moderate to severe plaque psoriasis, generalized pustular psoriasis, and erythrodermic psoriasis. J Clin Pharmacol. 2019;59(12):1656–68.
Zhuang Y, Calderon C, Marciniak SJ Jr, et al. First-in-human study to assess guselkumab (anti-IL-23 mAb) pharmacokinetics/safety in healthy subjects and patients with moderate-to-severe psoriasis. Eur J Clin Pharmacol. 2016;72(11):1303–10.
Acknowledgements
We thank the participants in this study. Principal investigators were Shan Jing and Yang Lin; sub-investigators were Liu Tianyang and Luo Fangfang.
Medical Writing/Editorial Assistance
Writing and editorial assistance was provided by Jieming Fang, MD, of Peloton Advantage, LLC, an OPEN Health company, funded by Bristol Myers Squibb, in accordance with Good Publication Practice (GPP3) guidelines.
Authorship
All named authors meet the International Committee of Medical Journal Editors (ICMJE) criteria for authorship for this article, take responsibility for the integrity of the work as a whole, and have given their approval for this version to be published.
Funding
This study was sponsored by Bristol Myers Squibb. Bristol Myers Squibb was involved in the design of the study, data collection, and interpretation, writing of this manuscript, and the decision to submit for publication. Bristol Myers Squibb also funded the journal’s Rapid Service Fee.
Author information
Authors and Affiliations
Contributions
Shan Jing, Yang Lin, Randy Dockens, David Marchisin, Bing He, Ihab G. Girgis, Bindu Murthy, and Urvi Aras contributed to data analysis and data interpretation. Anjaneya Chimalakonda contributed to the conception and design of the study, data acquisition, data analysis, and data interpretation. All authors contributed to the drafting of the article and commented on previous versions of the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Conflict of Interest
Shan Jing and Yang Lin declare that they have no competing interests. Randy Dockens, David Marchisin, Bing He, Bindu Murthy, and Urvi Aras declare that they are employees of and shareholders in Bristol Myers Squibb. Ihab G. Girgis and Anjaneya Chimalakonda declare that they were employees and shareholders in Bristol Myers Squibb at the time of study conduct; Ihab G. Girgis is now an employee of CSL Behring and Anjaneya Chimalakonda is now an employee of Boehringer Ingelheim.
Ethical Approval
The study was conducted at a single clinical facility in China (Beijing Anzhen Hospital, Capital Medical University, Beijing, China) in accordance with the Declaration of Helsinki and the International Conference on Harmonization E6 Guidelines on Good Clinical Practice. An institutional review board reviewed and approved the study protocol and related documents, and all subjects provided written informed consent before any study procedures were performed.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Jing, S., Lin, Y., Dockens, R. et al. Pharmacokinetics and Safety of the Tyrosine Kinase 2 Inhibitor Deucravacitinib in Healthy Chinese Subjects. Dermatol Ther (Heidelb) 13, 3153–3164 (2023). https://doi.org/10.1007/s13555-023-01050-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13555-023-01050-7