
Abstract
Risankizumab is a humanized immunoglobulin (Ig) G1 monoclonal antibody developed and approved for the treatment of moderate-to-severe plaque psoriasis at a dose of 150 mg administered subcutaneously at weeks 0 and 4, and every 12 weeks thereafter. Ongoing trials are investigating the use of risankizumab in other inflammatory autoimmune diseases. Risankizumab exhibits linear pharmacokinetics when administered intravenously (0.01 mg/kg–1200 mg) or subcutaneously (0.25 mg/kg–300 mg), with a long terminal half-life of approximately 28 days. Following subcutaneous administration, peak plasma concentration was reached approximately 3–14 days after dosing, with an estimated bioavailability of 89%. Population pharmacokinetic analyses identified bodyweight, high titers of antidrug antibodies occurring in < 2% of evaluated subjects, baseline serum albumin, baseline high-sensitivity C-reactive protein, and baseline serum creatinine to be statistically correlated with risankizumab clearance, but none of them had a clinically meaningful impact on risankizumab efficacy in psoriasis patients following the clinical dosing regimen. Exposure–response analyses in psoriasis patients demonstrated that the maximum efficacy was achieved with the clinically approved regimen and there was no apparent correlation between risankizumab exposure and safety. A dedicated drug interaction cocktail study in patients with psoriasis demonstrated a lack of therapeutic protein–drug interaction potentials for risankizumab and various cytochrome P450 substrates. In this article, we review the clinical pharmacology data available to date for risankizumab, which supported the clinical development program and ultimately regulatory approvals for risankizumab in the treatment of patients with moderate-to-severe plaque psoriasis.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Risankizumab exhibits typical immunoglobulin (Ig) G1 monoclonal antibody pharmacokinetic characteristics with bi-exponential disposition, long elimination half-life (approximately 28 days), and linear pharmacokinetics when administered intravenously (0.01 mg/kg–1200 mg) or subcutaneously (0.25 mg/kg–300 mg). |
Bodyweight, high titers of antidrug antibodies, baseline serum albumin, baseline high-sensitivity C-reactive protein, and baseline serum creatinine were statistically correlated with risankizumab clearance in population pharmacokinetic analyses; however, exposure–response analyses demonstrated that these covariates had no clinically meaningful impact on risankizumab efficacy in psoriasis patients with the clinical dosing regimen of 150 mg administered at weeks 0 and 4, and every 12 weeks thereafter. |
The risankizumab clinical dosing regimen maximized efficacy as assessed by the Psoriasis Area and Severity Index (PASI) 90, PASI 100, and static Physicians Global Assessment 0/1 responses, with no apparent correlation between exposure and safety in patients with plaque psoriasis. |
A therapeutic protein drug interaction study and population pharmacokinetic analyses confirmed the expected lack of drug interaction potential for risankizumab as a perpetrator or a victim. |
1 Introduction
Interleukin (IL)-23 is a naturally occurring cytokine that is involved in inflammatory and immune responses. IL-23 drives the development, differentiation, and function of T helper (Th) 17 cells, which produce IL-17-A and -F, as well as other proinflammatory cytokines, and plays a key role in driving some inflammatory autoimmune diseases, including psoriasis [1].
Psoriasis is a chronic debilitating immunologic disease characterized by marked inflammation and thickening of the epidermis that results in thick, scaly plaques involving the skin, which can negatively impact the psychosocial well-being of patients. Furthermore, patients with psoriasis are at higher risk of developing comorbidities, including psoriatic arthritis, metabolic syndrome, cardiovascular disorders, or depression [2]. Psoriasis may be classified according to morphologic and clinical presentation: plaque psoriasis, guttate psoriasis, erythrodermic psoriasis (EP), generalized pustular psoriasis (GPP) and localized pustular psoriasis, and inverse or intertriginous psoriasis. Psoriasis is estimated to affect 2% of the population in the developed world [3], with plaque psoriasis being the most common form, affecting approximately 80–90% of patients, of whom 20% experience moderate-to-severe disease [4]. Both GPP and EP are rare forms of psoriasis that can be difficult to treat and can be fatal; approximately 10% of patients with GPP have a preceding history of psoriasis [5], and EP prevalence among psoriatic patients is estimated to be from 1 to 2.25% [6].
Biologics have emerged as a promising alternative treatment option to conventional systemic therapies, such as methotrexate and retinoids, which have potential cumulative toxicities for patients with psoriasis. IL-17 and IL-12/23 inhibitors, such as ustekinumab (a p40 IL-12/23 inhibitor) [7], guselkumab [8] and tildrakizumab (IL-23 inhibitors) [9], and brodalumab, ixekizumab, and secukinumab (IL-17 inhibitors) [10], have demonstrated efficacy in treating this chronic disease.
Risankizumab is a humanized immunoglobulin (Ig) G1 monoclonal antibody that selectively binds with high affinity (≤ 29 pM) to the p19 subunit of the human cytokine IL-23, and inhibits its interaction with the IL-23 receptor and the downstream IL-23-dependent cell signaling and proinflammatory effects. In contrast with ustekinumab, risankizumab does not bind to human IL-12, which shares the p40 subunit with IL-23 [11].
As of June 2019, risankizumab was approved in multiple countries and regions, including the United States, the European Union, Japan, Canada, Switzerland, and Brazil, for the treatment of patients with moderate-to-severe chronic plaque psoriasis. In addition, risankizumab was also approved in Japan for the treatment of psoriatic arthritis and GPP/EP.
This review provides an integrated summary of the clinical pharmacology assessments of risankizumab, including pharmacokinetics, immunogenicity, and exposure–response analyses for efficacy and safety that supported the full characterization of the risankizumab clinical profile in psoriasis patients.
2 Clinical Studies
An overview of all studies conducted to date that are pertinent to risankizumab clinical pharmacology is presented in Table 1 (three phase I studies in healthy subjects and psoriasis patients) and Table 2 (seven phase II and III studies in patients with plaque psoriasis or GPP/EP); these studies evaluated the pharmacokinetics, immunogenicity, safety, and tolerability, and, in phase II and III studies, the efficacy of risankizumab. Population pharmacokinetic and exposure–response analyses for efficacy and safety, as well as analyses for the impact of immunogenicity on pharmacokinetics, safety, and efficacy, were performed using combined data from these phase I, II, and III studies to support the overall characterization of risankizumab clinical pharmacology, as well as the clinical dosing recommendation [12,13,14,15]. High-level results and conclusions from these studies and analyses are summarized in the following sections.
3 Clinical Pharmacokinetics of Risankizumab
Risankizumab exhibits pharmacokinetic characteristics consistent with typical IgG1 monoclonal antibodies, with no apparent target-mediated disposition following intravenous or subcutaneous administration. The role of intrinsic and extrinsic factors in the pharmacokinetics of risankizumab are detailed below.
3.1 Single-Dose Pharmacokinetics
Single-dose pharmacokinetics of risankizumab were evaluated in two studies, Study 1 in Caucasian, Japanese, and Chinese healthy volunteers, and Study 2 in patients with psoriasis following either intravenous or subcutaneous administrations across doses ranging from 0.01 to 5 mg/kg intravenously, 200 to 1200 mg intravenously, 0.25 to 1 mg/kg subcutaneously, and 18 to 300 mg subcutaneously [13, 16]. Intensive plasma samples for pharmacokinetic assessment were collected in these studies.
In both single-dose studies, risankizumab pharmacokinetics were linear (dose-independent) in the evaluated dose ranges (Table 3). No apparent target-mediated disposition was observed. Risankizumab terminal elimination half-life (t½) ranged from 18 to 34 days in healthy subjects and in patients with psoriasis across dose groups. The linear pharmacokinetic characteristics of risankizumab were not unexpected given that IL-23 is a soluble target expressed at low systemic concentrations, and target-mediated disposition would not be anticipated for the range of doses tested. Risankizumab peak plasma concentrations were achieved between 3 and 14 days (median values) following subcutaneous administration (Fig. 1; Table 3).

Risankizumab plasma concentration (geometric means) versus time profiles following IV and SC administration in subjects with moderate-to-severe psoriasis (Study 2). IV intravenous, SC subcutaneous
There was no apparent difference in risankizumab pharmacokinetics between healthy subjects and patients with psoriasis [13, 16]. Risankizumab exposures were 22–31% higher in healthy Chinese and Japanese subjects compared with healthy Caucasian subjects; this small difference in exposure was a result of the fixed-dosing regimen and the lower body weight for Chinese and Japanese subjects compared with Caucasian subjects [13].
3.2 Multiple-Dose Pharmacokinetics
Multiple-dose pharmacokinetics of risankizumab were evaluated in one global phase II study and four global phase III studies in patients with moderate-to-severe plaque psoriasis, as well as two Japanese phase II/III studies in patients with moderate-to-severe plaque psoriasis (with or without psoriatic arthritis), GPP, or EP [17,18,19,20,21,22]. Serial intensive pharmacokinetic samples were collected in the global phase II study, while in global phase III studies or Japanese phase II/III studies, fewer pharmacokinetic samples were collected.
An approximately dose proportional increase in risankizumab exposure was observed following repeated administration of 90 and 180 mg subcutaneous doses in the global phase II study [17, 23], and between 75 and 150 mg subcutaneous doses in the Japanese phase II/III studies [13].
Following multiple dosing of risankizumab 150 mg subcutaneously at weeks 0 and 4, and once every 12 weeks thereafter, in global phase III studies, steady-state risankizumab plasma concentrations were approximately achieved by week 16. Risankizumab geometric mean trough plasma concentrations (Ctrough) at weeks 16 and 52 were 1.84 µg/mL and 1.52 µg/mL, respectively, based on pooled data across all four global phase III studies. There was no indication of time-dependent kinetics. A summary of risankizumab Ctrough across all phase III studies after administration of risankizumab 150 mg subcutaneously at weeks 0 and 4, and every 12 weeks thereafter, in patients with psoriasis is shown in Table 4.
Risankizumab geometric mean Ctrough values in Japanese patients with psoriasis, at weeks 16 and 52, were 2.06 µg/mL and 1.64 µg/mL, respectively, based on pooled data of Japanese patients across all four global phase III and Japanese phase II/III studies [13], approximating the exposures in non-Japanese subjects. In addition, risankizumab exposures in Japanese patients with moderate-to-severe plaque psoriasis (with or without psoriatic arthritis) were comparable with those with GPP or EP [13].
3.3 Population Pharmacokinetics
The population pharmacokinetics of risankizumab were characterized using data from a phase I study in healthy Caucasian, Japanese, and Chinese subjects (Study 1), and a phase I study (Study 2), a phase II study (Study 3), and four phase III studies (Study 4, UltIMMa-1; Study 5, UltIMMa-2; Study 6, IMMhance; and Study 7, IMMvent) in patients with moderate-to-severe plaque psoriasis, referred to as the global population pharmacokinetic model [12]. These analyses included risankizumab pharmacokinetic data following single- and multiple-dose administration across doses ranging from 0.01 to 5 mg/kg intravenously, 200 to 1200 mg intravenously, 0.25 to 1 mg/kg subcutaneously, and 18 to 300 mg subcutaneously.
A total of 1899 adult male and female subjects who received at least one dose of risankizumab were included in the global population pharmacokinetic analyses [12]. A two-compartment model with first-order absorption and elimination described the time course of risankizumab plasma concentrations well in both healthy subjects and patients with moderate-to-severe plaque psoriasis.
For a typical 90 kg individual (approximately the median bodyweight for patients with psoriasis recruited in the clinical studies), risankizumab plasma clearance (CL), volume of distribution of the central (Vc) and peripheral (Vp) compartments, steady-state volume of distribution (Vss), and t½ were estimated to be 0.31 L/day, 6.52 L, 4.67 L, 11.2 L and 28 days, respectively. The interindividual variability for risankizumab CL, Vc, and absorption rate constant (Ka) were 24%, 34%, and 63%, respectively.
From the results of the covariate analyses, moderate-to-severe psoriasis disease, baseline Psoriasis Area and Severity Index (PASI) score, presence of neutralizing antibodies (NAbs) to risankizumab, age, sex, race, liver function markers, country, and common concomitant medications were not statistically significant covariates for risankizumab pharmacokinetic parameters. Although baseline serum albumin, baseline high-sensitivity C-reactive protein (hs-CRP), and baseline serum creatinine were found to be statistically significant covariates for risankizumab CL, they had negligible impact on risankizumab exposure. Risankizumab exposures following the clinical regimen of 150 mg subcutaneously at weeks 0 and 4, and every 12 weeks thereafter, in patients within the upper or lower 25th percentiles of covariate distribution of baseline serum albumin, baseline hs-CRP, and baseline serum creatinine were estimated to be well within the default equivalence boundaries of 0.8–1.25 relative to patients in the middle 50% of the covariate distribution for these factors.
Only two covariates were found to have more appreciable impact on risankizumab exposures: bodyweight and high antidrug antibody (ADA) titers (≥ 128). These are discussed in detail under the assessment of intrinsic factors (see Sects. 3.5.5, 3.5.6).
In addition to the population pharmacokinetic analyses from global studies, risankizumab population pharmacokinetics were further characterized in the Asian population upon availability of the data from these trials; this analysis was an extension of the global pharmacokinetic model and confirmed the adequacy of the global population pharmacokinetic model for describing risankizumab exposure in Asian (mainly Japanese) subjects. The impact of relevant patient-specific covariates on risankizumab systemic exposures was re-assessed with the larger dataset including studies conducted in Japan, and was used to inform dosing recommendations and the comparability of risankizumab exposures across Japanese patients with plaque psoriasis, GPP, or EP, as well as between Japanese and non-Japanese patients with psoriasis enrolled across all global and Japanese phase II/III clinical trials, referred to as the updated population pharmacokinetic model [13].
Based on the Asian population pharmacokinetic analyses, risankizumab plasma CL, Vss, and t½ were estimated to be approximately 0.24 L/day, 9.12 L, and 28 days, respectively, for a typical 70 kg patient with psoriasis, GPP or EP (70 kg was selected here because it approximates the median bodyweight in Japanese psoriasis patients). Risankizumab steady-state exposures (maximum concentration [Cmax], area under the plasma concentration–time curve during a dosing interval [AUCτ], and Ctrough) in Japanese subjects with moderate-to-severe plaque psoriasis were comparable with those with GPP or EP. Consistent with the pharmacokinetic analysis from the phase I Asian pharmacokinetic study (Study 1), risankizumab steady-state exposures following administration of 150 mg subcutaneously in Japanese patients with psoriasis (moderate-to-severe plaque psoriasis, GPP, or EP) were approximately 17% higher than non-Japanese patients with moderate-to-severe plaque psoriasis, as a result of the median bodyweight difference [13].
Risankizumab mean (standard deviation) steady-state Cmax, AUCτ, and Ctrough values, estimated based on the population pharmacokinetic modeling, are summarized in Table 5 in non-Japanese and Japanese patients with psoriasis following the clinical dosing regimen of 150 mg subcutaneously at weeks 0 and 4, and every 12 weeks thereafter.
3.4 Bioavailability of Subcutaneous Risankizumab
Results from cross-study population pharmacokinetic analyses of risankizumab following intravenous and subcutaneous administrations showed that the population estimate for the absolute subcutaneous bioavailability of risankizumab was 89% for the phase III clinical regimen in a typical patient with psoriasis (150 mg subcutaneously at weeks 0 and 4, and every 12 weeks thereafter), which is on the higher side of the range of 50–100% absolute bioavailability reported for other monoclonal antibodies [12, 24].
3.5 Effect of Intrinsic Factors
3.5.1 Renal and Hepatic Impairment
As an IgG1 monoclonal antibody, risankizumab is expected to be degraded into small peptides and amino acids via catabolic pathways, in a manner similar to endogenous IgG, and thus is not expected to undergo metabolism by hepatic metabolic enzymes or renal elimination. Therefore, no dedicated studies were conducted to evaluate risankizumab pharmacokinetics in patients with hepatic or renal impairment. Based on cross-study population pharmacokinetic analyses, hepatic function markers, including total bilirubin, aspartate aminotransferase (AST), and alanine aminotransferase (ALT) levels were not correlated with risankizumab CL, and serum creatinine level or creatinine CL had no meaningful impact on risankizumab exposure [12].
3.5.2 Psoriasis Disease (Disease State)
From the cross-study population pharmacokinetic analyses, risankizumab pharmacokinetic parameters were similar in healthy subjects and patients with moderate-to-severe plaque psoriasis [12]. Risankizumab plasma exposures in Japanese patients with moderate-to-severe plaque psoriasis with concomitant psoriatic arthritis were comparable with those without concomitant psoriatic arthritis, while plasma exposures in Japanese patients with moderate-to-severe plaque psoriasis (with or without psoriatic arthritis) were comparable with those with GPP or EP [13].
3.5.3 Race/Ethnicity
After accounting for bodyweight differences, race/ethnicity did not have an impact on risankizumab CL [13]. This was demonstrated in the phase I study of risankizumab in Asian versus Caucasian subjects (bodyweight rather than race accounted for the apparent small difference in exposure), as well as in the population pharmacokinetic analyses (race or region/country was not a significant covariate).
Moreover, comparisons of risankizumab plasma exposures in psoriasis patients across various ethnic/racial groups, including Korean, Taiwanese, Japanese, and non-Asian patients, were conducted based on data from phase III studies following the proposed clinical regimen of 150 mg subcutaneously at weeks 0 and 4, and every 12 weeks thereafter. In general, exposures to risankizumab were comparable across all groups, and Asian race or ethnicity had no clinically relevant impact on risankizumab pharmacokinetics (Table 5).
3.5.4 Age and Sex
Based on the cross-study population pharmacokinetic analyses, risankizumab pharmacokinetic parameters were not impacted by age (range 18–85 years) or sex in adult patients with plaque psoriasis [12].
3.5.5 Body Weight
Similar to other IgG1 monoclonal antibodies, bodyweight was correlated with risankizumab CL, with an increase in bodyweight associated with an increase in CL [12]. Patients with psoriasis with a bodyweight > 100 kg are estimated to have approximately 30% lower exposures compared with subjects weighing ≤ 100 kg; however, this difference is not clinically relevant for any major impact on efficacy or safety (see Sect. 4) [14].
3.5.6 Antidrug Antibodies and Neutralizing Antibodies
As summarized later in Sect. 5, the potential impact of ADAs on risankizumab exposure, efficacy, and safety were extensively evaluated.
Overall, immunogenicity to risankizumab did not have a clinically relevant impact on risankizumab plasma exposure, efficacy (as assessed by PASI 90 at week 16), or safety (as assessed for hypersensitivity and injection-site reactions) in psoriasis patients. Only a very small fraction of psoriasis patients who received risankizumab in the phase II/III clinical trials (approximately 1% [6/598] of ADA-evaluable subjects who received the phase III clinical regimen over a 52-week period in the UltIMMa-1 and UltIMMa-2 trials, or approximately 1.5% [28/1807] of ADA-evaluable subjects who received any dose of risankizumab in all phase II/III trials) developed high titer ADAs (≥ 128), and, only in these subjects with high ADA titers, risankizumab exposures were estimated, on average, to be 30% lower than ADA-negative subjects or subjects with relatively lower ADA titers (< 128) [12]. This modest difference in risankizumab exposure is not considered to have a major impact on efficacy because the 150 mg dosing regimen provides exposures that are well within the plateau of response (see Sects. 4, 5) [14].
3.6 Effect of Extrinsic Factors
3.6.1 Risankizumab as a Perpetrator for Drug–Drug Interaction (DDI)
Patients with psoriasis and other immune-mediated inflammatory diseases may have comorbidities that require treatment with other medications that are metabolized through cytochrome P450 (CYP) enzymes. Therapeutic proteins that modulate cytokine levels are suggested to potentially result in interactions with drugs that are sensitive substrates of CYP enzymes because some elevated cytokines can downregulate the transcription of CYP genes and reduce the metabolism of such drugs. As such, certain therapeutic proteins impacting cytokine levels can reverse cytokine-mediated suppression of CYPs, resulting in increased CL and decreased exposures of sensitive CYP substrates compared with prior treatment. A dedicated drug interaction study (Study 8) was conducted for risankizumab in patients with moderate-to-severe plaque psoriasis to assess the effect of repeated administration of risankizumab on the pharmacokinetics of CYP-sensitive probe substrates. Repeated administration of risankizumab 150 mg subcutaneously at weeks 0, 4, 8, and 12 (more frequent administration resulting in higher risankizumab exposure than the clinical regimen) had no effect on the exposures of probe substrates of CYP1A2 (caffeine 100 mg), CYP2C9 (warfarin 10 mg), CYP2C19 (omeprazole 20 mg), CYP2D6 (metoprolol 50 mg), and CYP3A (midazolam 2 mg) in patients with moderate-to-severe plaque psoriasis (n = 21) [25]. The 90% confidence intervals for the ratios of the probe substrates Cmax and AUC when administered 6 days following the fourth dose of risankizumab 150 mg subcutaneously every 4 weeks versus when these substrates were administered prior to initiating risankizumab treatment were within the default no-effect boundaries of 0.8–1.25 (with the exception of the lower 90% confidence bound for omeprazole Cmax ratio, which extended slightly below 0.8). Therefore, no dosage adjustments are needed for drugs that are substrates of these CYP enzymes when administered concomitantly with risankizumab in patients with moderate-to-severe psoriasis [25].
3.6.2 Risankizumab as a Victim for DDIs
Because risankizumab is not expected to undergo metabolism by CYP enzymes, it is not expected to be a victim of drug–drug interactions (DDIs) with substrates/inhibitors/inducers of drug metabolizing enzymes. Consistent with this, in the cross-study population pharmacokinetic analyses, none of the evaluated medications (metformin, atorvastatin, lisinopril, amlodipine, ibuprofen, acetylsalicylate, and levothyroxine) that were concomitantly administered by some patients during risankizumab phase III clinical trials were found to significantly affect risankizumab CL [12].
4 Exposure Response Analyses for Efficacy
4.1 Phase I and II Studies
The exposure–response relationships between model-estimated risankizumab weeks 0–16 average plasma concentrations (Cavg) and the observed percentage of subjects achieving PASI 90, PASI 100, or static Physicians Global Assessment (sPGA) clear or almost clear (sPGA 0/1) responses at week 16 in the phase II study (Study 3) were evaluated using quartile plots in which subjects were binned into quartiles based on risankizumab plasma Cavg levels [14]. The analyses indicated that while the percentage of subjects achieving PASI 90 or sPGA 0/1 at week 16 appeared to have plateaued at the third quartile of risankizumab exposure (median Cavg of 3.97 μg/mL), the highest quartile of exposure (median Cavg of 7.88 μg/mL) clearly showed a higher percentage of subjects achieving the complete CL of PASI score (i.e. PASI 100). The median weeks 0–16 Cavg value with the proposed phase III clinical dosing regimen (150 mg subcutaneously at weeks 0 and 4, and every 12 weeks thereafter) was 7.32 μg/mL, which is similar to the median risankizumab Cavg value in the fourth exposure quartile in the phase II study. These results support that the regimen selected for phase III trials was adequate to maximize risankizumab efficacy in patients with psoriasis.
4.2 Phase II and III Studies
Non-linear regression analyses were conducted using maximum effect (Emax) models to describe the relationships between risankizumab plasma exposure (Cavg) and the probability of achieving PASI 75, PASI 90, PASI 100, and sPGA 0/1 responses at weeks 16 and 52 using combined data from phase II (Study 3) and phase III studies (UltIMMa-1, UltIMMa-2, IMMhance, and IMMvent) [14].
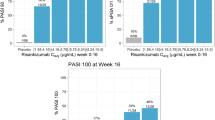
The estimated half maximal effective concentration (EC50) values to achieve PASI 75, PASI 90, PASI 100, and sPGA 0/1 responses at week 16 (0.20, 0.44, 2.4, and 0.43 µg/mL, respectively) and week 52 (0.30, 0.96, 2.88, and 0.71 µg/mL, respectively) were significantly lower than the estimated Cavg value over weeks 0–16 (median value: 7.32 µg/mL) and weeks 40–52 (median value: 5.42 µg/mL) for the risankizumab clinical regimen (150 mg subcutaneously at weeks 0 and 4, and every 12 weeks thereafter). At the median risankizumab Cavg associated with the proposed clinical regimen, the estimated probabilities for PASI 75, PASI 90, PASI 100, and sPGA 0/1 responses were 92%, 77%, 49%, and 87%, respectively, at week 16, and 95%, 85%, 61%, and 88%, respectively, at week 52 based on the exposure–response models [14]. These probabilities were at the estimated plateau for each response. These results confirmed that the clinical regimen maximized the efficacy of risankizumab, as measured by the PASI and sPGA endpoints in patients with psoriasis.
In the covariate analyses, hs-CRP was a statistically significant covariate for risankizumab EC50, but had a negligible estimated effect on efficacy at the clinical regimen. Subjects with higher baseline hs-CRP levels were predicted to result in a slightly lower (≤ 6%) probability of achieving PASI or sPGA endpoints at week 16, which could be related to the higher inflammatory burden in subjects with higher values of baseline hs-CRP. Asian race was a statistically significant covariate for risankizumab EC50 for PASI 100 response at week 16, but not for any other response level. No covariates were statistically significant at week 52 [14].
Other covariates, including demographic variables (body weight, BMI, age, sex and race), baseline disease characteristics (baseline PASI score) and comorbidities (presence of psoriatic arthritis), immunogenicity (treatment-emergent ADA-or NAb-positive status), and prior therapies were not statistically significant covariates for the relationship between risankizumab exposure and efficacy in psoriasis [14].
The model-estimated exposure–response relationships for PASI 90, PASI 100, and sPGA 0/1 responses at week 16 were at the plateau of response for exposures observed with subjects with bodyweight ≤ 100 kg and those with bodyweight > 100 kg or > 120 kg following administration of risankizumab 150 mg subcutaneously at weeks 0 and 4, and every 12 weeks thereafter [14]. This further supported the lack of differences in efficacy in subjects with higher bodyweight who experienced relatively lower risankizumab exposure levels. These results were consistent with observed efficacy data in phase III trials; the placebo-adjusted PASI 90 responses were 72.1% and 69.2%, while the sPGA0/1 responses were 77.5% and 80.0%, in patients with bodyweight ≤ 100 kg and > 100 kg, respectively [14].
Overall, these analyses indicated that the 150 mg subcutaneous dose of risankizumab at weeks 0 and 4, and every 12 weeks thereafter, maximized the efficacy in patients with psoriasis across the entire expected bodyweight range in this population [14].
The relationship between risankizumab exposures and key efficacy variables in Japanese patients with moderate-to-severe plaque psoriasis following treatment with either 75 or 150 mg subcutaneous doses at weeks 0 and 4, and every 12 weeks thereafter, were also characterized [15]. The exposure–efficacy relationships in Japanese patients were consistent with the population enrolled in global phase III trials. A plateau of efficacy at week 16 was demonstrated with a risankizumab 150 mg subcutaneous dose providing PASI 90 and sPGA 0/1 response probabilities of 77%, and 88%, respectively. These exposure–response analyses indicated that a risankizumab 150 mg dose maximized efficacy, including PASI 100 response (probability of 31%) in Japanese patients with plaque psoriasis [15]. Risankizumab exposures for the 75 mg dose were suboptimal for PASI 100 response (probability of 21%) and other efficacy responses, particularly in heavier patients.
4.3 Exposure–Response Analyses for Safety
The exposure–response relationships for safety were performed using pooled data from subjects recruited across the four phase III studies (UltIMMa-1, UltIMMa-2, IMMhance, and IMMvent). The exposure–response relationships were evaluated for the safety events of interest (any adverse event [AE], serious AE [SAE], infection, and serious infection) through the first 16 weeks (weeks 0–16; placebo-controlled period), as well as through the first 52 weeks (weeks 0–52 for UltIMMa-1, UltIMMa-2 and IMMhance, and weeks 0–44 for IMMvent).
There was no apparent relationship between risankizumab exposure and any AE, SAE, infections, or any serious infections across the first 16 weeks or up to 52 weeks’ duration using pooled data from all four phase III trials of risankizumab in subjects with plaque psoriasis [14]. Extension of the analyses later with the Japanese phase II/III studies in patients with plaque psoriasis, GPP, and EP provided consistent results [15].
5 Immunogenicity
Immunogenicity of risankizumab was assessed across all phase I, II, and III studies. Plasma samples were analyzed for the presence of ADAs and NAbs to risankizumab using validated assays. Briefly, immunogenicity of risankizumab was first assessed using a three-tiered approach, including screening, confirmation, and titration. The confirmed positive samples were then evaluated in the NAb assay to detect the presence of NAb. The incidence of ADAs (treatment-emergent) to risankizumab was defined when a subject was (1) ADA-negative or missing assessment at baseline (prior to the first risankizumab dose) and became ADA-positive at one or more time points post baseline; or (2) ADA-positive at baseline and showed a fourfold (based on two dilution steps at the titer determination stage) or greater increase in titer values relative to baseline, or a titer value of ≥ 2 in at least one post-dose sample if the baseline titer value was < 1 (in this case, a fourfold increment over the midpoint of 0.5 was used).
It is noteworthy that the detection of ADA formation is highly dependent on the sensitivity and specificity of the assay. Additionally, the observed incidence of ADA (including NAb) positivity in an assay may be influenced by several factors, including assay methodology, sample handling, timing of sample collection, concomitant medications, and underlying disease. For these reasons, comparison of the ADA incidence to risankizumab in the clinical studies listed above, with the incidence of antibodies for other products, can be misleading [26].
For subjects who received the clinical regimen of risankizumab in psoriasis (150 mg at weeks 0 and 4, and every 12 weeks thereafter) in phase III trials, the ADA and NAb incidence (treatment-emergent) to risankizumab was 24% and 14%, respectively, over 52 weeks’ duration (based on 1079 evaluable subjects). The median time to appearance of ADA was 16 weeks across studies. The median ADA titer values were low and ranged from 1 to 4.5 across visits and studies; a very limited number of subjects (approximately 1% of ADA-evaluable subjects over a 52-week period of risankizumab treatment in phase III trials) had ADA titer values ≥ 128.
Across all global phase III studies of risankizumab in subjects with moderate-to-severe psoriasis, the ADA and NAb incidence to risankizumab were comparable among subjects who received risankizumab from week 0, or those who underwent placebo withdrawal after an initial treatment with risankizumab (with or without retreatment with risankizumab). Additionally, the ADA or NAb incidence to risankizumab was comparable across Japanese and non-Japanese subjects with psoriasis (plaque psoriasis, GPP, or EP) or psoriatic arthritis (Table 6).
Across phase II and III studies, immunogenicity to risankizumab did not have a clinically relevant impact on the risankizumab plasma exposures, efficacy (as assessed by sPGA 0/1 and PASI 90 response at weeks 16 and 52), or safety (as assessed for hypersensitivity and injection-site reactions) of risankizumab [12,13,14,15]. The analyses of the clinical impact of immunogenicity based on global data are further summarized below.
5.1 Assessment of the Effect of Immunogenicity on Risankizumab Plasma Exposures
Based on intersubject comparison using pooled data across all four phase III studies, risankizumab Ctrough (central tendency and distribution) were comparable between subjects who were ADA-positive and those who were ADA-negative over time (Fig. 2). From the intrasubject comparison across all phase III studies; the median value of the ratio of risankizumab steady-state Ctrough after versus before the appearance of ADA was 0.98 for subjects who received risankizumab from week 0, and 1.18 for subjects who switched from placebo to risankizumab at week 16.

Comparison of risankizumab plasma concentrations (µg/mL) by antidrug antibody status. SC subcutaneous. In the box plots, the solid square and the horizontal bar within each box represents the geometric mean and the median values, respectively. The other symbols (*, +, x, #) represent risankizumab concentration values for each individual subject. The risankizumab 150 mg SC group included subjects who received risankizumab from week 0, i.e. 150 mg SC at weeks 0 and 4, and every 12 weeks thereafter, using pooled data across all four phase III studies. The placebo to risankizumab 150 mg SC group included subjects who received placebo at weeks 0 and 4, and then switched to risankizumab at week 16
From the population pharmacokinetic analyses [12], ADAs to risankizumab did not have an effect on risankizumab CL or exposure in the majority of ADA-positive subjects, with the exception of a few subjects (approximately 1% of ADA-evaluable subjects over a 52-week period of risankizumab treatment in phase III studies) who developed an ADA titer ≥ 128 titer units, in which risankizumab CL was estimated to increase by 43% and steady-state AUCτ was estimated to decrease by 30%, on average.
Overall, these results indicate that the development of ADAs to risankizumab does not result in a clinically relevant impact on risankizumab exposure.
5.2 Assessment of the Effect of Immunogenicity on Risankizumab Efficacy
The PASI 90 and sPGA 0/1 responses were compared between ADA-positive and ADA-negative subjects, as well as NAb-positive and NAb-negative subjects, using pooled data across phase III studies (Table 7; with week 16 comparisons using a placebo-controlled population and an ustekinumab-controlled population, and week 52 comparisons using an ustekinumab-controlled population). PASI 90 and sPGA 0/1 responses at weeks 16 and 52 were comparable between subjects who were ADA- or NAb-positive and those who were ADA- or NAb-negative. Additionally, in exposure–response analyses, the ADA and NAb status (positive or negative) was not found to be a significant covariate for the efficacy endpoints PASI 75, PASI 90, PASI 100, or sPGA0/1 at week 16 or 52 [14, 15]. While the percentages of responders appeared to be numerically lower in subjects with high ADA titers (> 128) than the rest of the evaluated population (with the limitation of the extremely low sample size of patients with high ADA titer that precludes meaningful assessment of a percentage of responders), some subjects with high ADA titers still showed and maintained PASI 75, PASI 90, or sPGA 0/1 responses (Table 7).
5.3 Effect of Immunogenicity on Risankizumab Safety
The incidence of hypersensitivity reaction and injection site reaction across up to 52 weeks of risankizumab exposure were comparable among ADA-positive and ADA-negative subjects (Table 8). These results indicate that immunogenicity to risankizumab has no clinically relevant impact on the safety of risankizumab assessed by hypersensitivity reaction and injection-site reaction.
6 Conclusions
The clinical pharmacology of risankizumab has been well-characterized using data from phase I, II, and III studies in healthy subjects and psoriasis patients following single- and multiple-dose administrations. These data provided quantitative understanding of the in vivo disposition of risankizumab, the exposure–response relationship between risankizumab and downstream efficacy and safety, the intrinsic and extrinsic factors that had no major clinically meaningful impact regardless of their statistical significance, the lack of DDI potential of risankizumab and concomitant medications, and the lack of clinical impact by ADAs that developed in subjects who received risankizumab. Together, these clinical pharmacology data support the risankizumab dosing regimen of 150 mg subcutaneously at weeks 0 and 4, and every 12 weeks thereafter, for the treatment of moderate-to-severe plaque psoriasis.
References
Iwakura Y, Nakae S, Saijo S, Ishigame H. The roles of IL-17A in inflammatory immune responses and host defense against pathogens. Immunol Rev. 2008;226:57–79.
Griffiths CE, Barker JN. Pathogenesis and clinical features of psoriasis. Lancet. 2007;370(9583):263–71.
Christophers E. Psoriasis: epidemiology and clinical spectrum. Clin Exp Dermatol. 2001;26(4):314–20.
Menter A, Gottlieb A, Feldman SR, Van Voorhees AS, Leonardi CL, Gordon KB, et al. Guidelines of care for the management of psoriasis and psoriatic arthritis: Section 1. Overview of psoriasis and guidelines of care for the treatment of psoriasis with biologics. J Am Acad Dermatol. 2008;58(5):826–50.
Hoegler KM, John AM, Handler MZ, Schwartz RA. Generalized pustular psoriasis: a review and update on treatment. J Eur Acad Dermatol Venereol. 2018;32(10):1645–51.
Singh RK, Lee KM, Ucmak D, Brodsky M, Atanelov Z, Farahnik B, et al. Erythrodermic psoriasis: pathophysiology and current treatment perspectives. Psoriasis (Auckl). 2016;6:93–104.
Stelara (Ustekinumab) [US prescribing information]. Horsham: Janssen Biotech, Inc.; 2018.
Tremfya (Guselkumab) [US prescribing information]. Horsham: Janssen Biotech, Inc.; 2017.
ILUMYA (tildrakizumab) [US prescribing information]. Whitehouse Station: Merck Sharp & Dohme Corp.; 2018.
Wasilewska A, Winiarska M, Olszewska M, Rudnicka L. Interleukin-17 inhibitors. A new era in treatment of psoriasis and other skin diseases. Postepy Dermatol Alergol. 2016;33(4):247–52.
Singh S, Kroe-Barrett RR, Canada KA, Zhu X, Sepulveda E, Wu H, et al. Selective targeting of the IL23 pathway: generation and characterization of a novel high-affinity humanized anti-IL23A antibody. mAbs. 2015;7(4):778–91.
Suleiman AA, Minocha M, Khatri A, Pang Y, Othman AA. Population pharmacokinetics of risankizumab in healthy volunteers and subjects with moderate to severe plaque psoriasis: integrated analyses of phase I–III clinical trials. Clin Pharmacokinet. 2019;58(10):1309–21.
Khatri A, Eckert D, Oberoi R, Suleiman A, Pang Y, Cheng L, et al. Pharmacokinetics of risankizumab in Asian Healthy subjects and patients with moderate-to-severe plaque psoriasis, generalized pustular psoriasis, and erythrodermic psoriasis. J Clin Pharmacol. 2019;59(12):1656–68.
Khatri A, Suleiman A, Polepally AR, Othman AA. Exposure–response relationships for efficacy and safety of risankizumab in phase 2 and 3 trials in psoriasis patients. Clin Pharmacol Ther. 2019. https://doi.org/10.1002/cpt.1594(Epub 29 Jul 2019).
Suleiman AA, Khatri A, Oberoi RK, Othman AA. Exposure–Response relationships for the efficacy and safety of risankizumab in Japanese subjects with psoriasis. Clin Pharmacokinet. 2019. https://doi.org/10.1007/s40262-019-00829-2(Epub 31 Oct 2019).
Krueger JG, Ferris LK, Menter A, Wagner F, White A, Visvanathan S, et al. Anti-IL-23A mAb BI 655066 for treatment of moderate-to-severe psoriasis: safety, efficacy, pharmacokinetics, and biomarker results of a single-rising-dose, randomized, double-blind, placebo-controlled trial. J Allergy Clin Immunol. 2015;136(1):116–24 e7.
Papp KA, Blauvelt A, Bukhalo M, Gooderham M, Krueger JG, Lacour JP, et al. Risankizumab versus Ustekinumab for moderate-to-severe plaque psoriasis. N Engl J Med. 2017;376(16):1551–60.
Gordon K, Strober B, Lebwohl M, Augustin M, Blauvelt A, Poulin Y, et al. Efficacy and safety of risankizumab: results from two double-blind, randomised, placebo- and Ustekinumab-controlled, phase 3 trials in moderate-to-severe plaque psoriasis (UltIMMa-1 and UltIMMa-2). Lancet. 2018;392(10148):650–61.
Ohtsuki M, Fujita H, Watanabe M, Suzaki K, Flack M, Huang X, et al. Efficacy and safety of risankizumab in Japanese patients with moderate to severe plaque psoriasis: results from the SustaIMM phase 2/3 trial. J Dermatol. 2019;46(8):686–94.
Witjes H, Khatri A, Diderichsen PM, Mandema J, Othman AA. Meta-analyses of clinical efficacy of risankizumab in chronic plaque psoriasis: supporting evidence of Risannkizumab superiority. Clin Pharmacol Ther. 2019. https://doi.org/10.1002/cpt.1624(Epub 10 Sep 2019).
Langley RG, Blauvelt A, Gooderham M, Papp K, Philipp S, Wu JJ et al. Efficacy and safety of continuous Q12W risankizumab versus treatment withdrawal: results from the phase 3 IMMhance trial. American Academy of Dermatology Annual Meeting; 1–5 March 2019; Washington, DC.
Reich K, Gooderham M, Thaci D, Crowley JJ, Ryan C, Krueger JG, et al. Risankizumab compared with adalimumab in patients with moderate-to-severe plaque psoriasis (IMMvent): a randomised, double-blind, active-comparator-controlled phase 3 trial. Lancet. 2019;394(10198):576–86.
Suleiman AA, Khatri A, Minocha M, Othman AA. Population pharmacokinetics of the interleukin-23 inhibitor risankizumab in subjects with psoriasis and Crohn’s disease: analyses of Phase I and II Trials. Clin Pharmacokinet. 2019;58(3):375–87.
Dirks NL, Meibohm B. Population pharmacokinetics of therapeutic monoclonal antibodies. Clin Pharmacokinet. 2010;49(10):633–59.
Khatri A, Cheng L, Camez A, Ignatenko S, Pang Y, Othman AA. Lack of effect of 12-week treatment with risankizumab on the pharmacokinetics of cytochrome P450 probe substrates in patients with moderate to severe chronic plaque psoriasis. Clin Pharmacokinet. 2019;58(6):805–14.
US Department of Health and Human Services, Food and Drug Administration, Center for Drug Evaluation and Research, Center for Biologics Evaluation and Research (CBER), Center for Devices and Radiological Health. Assay development and validation for immunogenicity testing of therapeutic protein products. 1st ed. Silver Spring: Center for Drug Evaluation and Research; 2016. p. 35.
Acknowledgements
The authors thank Mia DeFino, MS, ELS, a freelance medical writer under contract with AbbVie, for medical writing support.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Ethical standards
The authors were employees of AbbVie at the time the manuscript was written and may hold AbbVie stocks or stock options. Mia DeFino was paid by AbbVie for medical writing support.
Conflicts of interest
Yinuo Pang and Ahmed Suleiman are employees of AbbVie Inc. and may hold AbbVie stock or stock options. Ahmed A. Othman and Amit Khatri are former employees of AbbVie and may hold AbbVie stocks or stock options.
Financial disclosures
This review was supported by AbbVie. The studies reviewed were supported by Boehringer Ingelheim and AbbVie. Boehringer Ingelheim and AbbVie contributed to the study designs, and AbbVie contributed to the analysis and interpretation of the data and the writing, review, and approval of the manuscript.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution-NonCommercial 4.0 International License (http://creativecommons.org/licenses/by-nc/4.0/), which permits any noncommercial use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Pang, Y., Khatri, A., Suleiman, A.A. et al. Clinical Pharmacokinetics and Pharmacodynamics of Risankizumab in Psoriasis Patients. Clin Pharmacokinet 59, 311–326 (2020). https://doi.org/10.1007/s40262-019-00842-5
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40262-019-00842-5