
Abstract
Abrocitinib is a Janus kinase (JAK) 1-selective inhibitor approved for the treatment of moderate-to-severe atopic dermatitis (AD). Although specific dose recommendations for abrocitinib vary across regional product labels, abrocitinib 100 mg once daily is recommended as a starting and maintenance dose. This review summarizes the efficacy and safety of abrocitinib 100 mg once daily for patients with moderate-to-severe AD based on data from the pivotal phase 3 studies of the JAK1 Atopic Dermatitis Efficacy and Safety (JADE) clinical program, JADE MONO-1 (NCT03349060), JADE MONO-2 (NCT03575871), JADE COMPARE (NCT03720470), JADE TEEN (NCT03796676), and JADE REGIMEN (NCT03627767). Preliminary long-term efficacy and safety data are also summarized from the long-term extension study JADE EXTEND (NCT03422822). Expert opinion on use of abrocitinib 100 mg once daily in clinical practice is provided. In addition to efficacy, the decision to use abrocitinib for the treatment of AD should allow for individual patient factors such as age, comorbidities, previous therapy, quality of life, and treatment tolerability, and involve shared decision-making between the patient and clinician.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Plain Language Summary
Abrocitinib is an approved treatment for people with moderate or severe atopic dermatitis, also known as AD or atopic eczema. It can be taken as a pill once a day in two doses: 100 mg or 200 mg. In certain countries, doctors may prescribe abrocitinib 100 mg as a starting dose. Doctors can then adjust the dose based on how well the treatment has helped and any side effects. This review article summarizes data from people with AD who took 100 mg abrocitinib in multiple clinical trials. We found that people who took abrocitinib 100 mg were more likely to experience fast itch relief, as early as day 1 after treatment, and have clear skin than those who took placebo (a sugar pill without medicine). This benefit of treatment with abrocitinib 100 mg was maintained for a long time. In a group of people who took dupilumab (a different treatment for AD) and did or did not see improvement in their AD symptoms, switching to abrocitinib provided substantial improvement in itch and skin lesions. In general, people who took abrocitinib 100 mg tolerated it well. They had few side effects that were serious or required stopping the treatment, even with long-term use. People with AD should discuss with their doctors which dose of abrocitinib will help with symptoms based on their specific needs. This discussion should include age, history of other diseases, previous medications, quality of life, and how well they are able to tolerate the side effects.
Video Abstract (MP4 52765 KB)
Key Summary Points
Abrocitinib 100 mg once daily provides rapid itch relief as early as day 1 after treatment and skin clearance in patients with moderate-to-severe atopic dermatitis, which is sustained over the long term. |
Abrocitinib 100 mg once daily is well tolerated, and safety results were consistent for up to 72 weeks of treatment. |
Abrocitinib 100 mg once daily can be considered for long-term maintenance treatment of AD. |
The use of abrocitinib for moderate-to-severe AD in clinical practice should be a shared decision between the patient and clinician, allowing for individual patient factors such as age, comorbidities, previous therapy, quality of life, and treatment tolerability. |
Digital Features
This article is published with digital features, including a video abstract to facilitate understanding of the article. To view digital features for this article go to https://doi.org/10.6084/m9.figshare.23175839.
Introduction
Abrocitinib is an oral, once-daily (QD) Janus kinase (JAK) 1-selective inhibitor currently approved for the treatment of moderate-to-severe atopic dermatitis (AD) in the United States, European Union, Great Britain, and Japan [1,2,3,4,5]. Abrocitinib is currently approved for use in adults, as well as adolescents in certain geographic regions. Although specific dose recommendations for abrocitinib vary across regional product labels, abrocitinib 100 mg QD is recommended as a starting and maintenance dose for indicated patient populations.
This review summarizes the efficacy and safety of abrocitinib 100 mg QD for patients with moderate-to-severe AD from the JAK1 Atopic Dermatitis Efficacy and Safety (JADE) clinical development program, our expert opinion on its appropriate therapeutic use, and prescribing recommendations for clinical practice.
Clinical Development Program
Key phase 3 placebo-controlled studies in the clinical development program of abrocitinib for the treatment of moderate-to-severe AD are outlined in Table 1. Abrocitinib monotherapy was assessed in adolescents and adults in JADE MONO-1 (NCT03349060) and JADE MONO-2 (NCT03575871), which were identically designed studies that included 387 and 391 patients, respectively [6, 7]. Abrocitinib in combination with topical medications was assessed in JADE COMPARE (NCT03720470), which included 838 adults [8], and in JADE TEEN (NCT03796676), which included 285 adolescents [9]. In JADE COMPARE and JADE TEEN, patients were required to apply medium-potency topical corticosteroids daily to areas with active lesions, until the lesions were under control (clear or almost clear), followed by 7 days of further treatment; low-potency topical corticosteroids, topical calcineurin inhibitors, or crisaborole could be used in sensitive body areas (e.g., areas of thin skin such as the face) [8, 9].
JADE REGIMEN (NCT03627767) was a phase 3 induction-randomized withdrawal trial of abrocitinib in adolescent and adult patients with moderate-to-severe AD designed to assess the maintenance of clinical responses following an initial response to the 200-mg dose and the ability to recapture responses following flares [10]. After 12 weeks of open-label induction monotherapy with abrocitinib 200 mg QD, the subset of patients who responded to treatment was randomly assigned to receive double-blind abrocitinib 200 mg QD, abrocitinib 100 mg QD, or placebo for 40 weeks [10]. Patients who experienced a flare during the 40-week treatment period entered a 12-week open-label rescue period of abrocitinib 200 mg QD with medicated topical therapy [10].
Adolescent and adult patients with moderate-to-severe AD who completed the full treatment period of a qualifying phase 3 study and who remained eligible to receive abrocitinib could enter a long-term, phase 3 extension trial to receive abrocitinib 200 mg QD or 100 mg QD (JADE EXTEND; NCT03422822) [11]. Patients who completed the 12-week open-label induction period of JADE REGIMEN without meeting the protocol-defined response criteria at week 12, or after completing the full 12-week open-label rescue treatment period in that study, were also eligible to enter JADE EXTEND. JADE EXTEND is ongoing; data from the April 22, 2020, cutoff date are available [11, 12]. Medicated and nonmedicated topical treatments for AD are permitted throughout JADE EXTEND, by investigator discretion. The primary endpoint of JADE EXTEND is safety; including the incidence of treatment-emergent adverse events (TEAEs), serious TEAEs, TEAEs leading to discontinuation, and clinical abnormalities.
Compliance with Ethics Guidelines
This article is based on previously conducted studies and does not contain any new studies with human participants or animals performed by any of the authors.
Efficacy
Up to Week 12 Efficacy: Skin Clearance and Itch Response
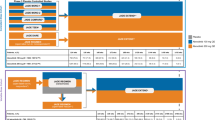
In key phase 3 trials of abrocitinib, significantly more patients who received abrocitinib 100 mg than placebo achieved the co-primary endpoints, which focused on skin clearance: an Investigator’s Global Assessment (IGA) score of 0 (clear) or 1 (almost clear) with a ≥ 2-point improvement from baseline (IGA 0/1) at week 12; and a ≥ 75% improvement from baseline in Eczema Area and Severity Index (EASI-75) at week 12 (Fig. 1). The proportions of patients who attained ≥ 90% improvement from baseline in Eczema Area and Severity Index (EASI-90) were also higher with abrocitinib 100 mg than with placebo at week 12 in these trials. The proportions of patients achieving these skin clearance endpoints in both the abrocitinib 100 mg and placebo arms appeared to be higher in JADE COMPARE and JADE TEEN compared with the JADE MONO studies, likely because of the concomitant use of medicated topical therapy [13].

Summary of short-term efficacy in JADE MONO-1, JADE MONO-2, JADE COMPARE, and JADE TEEN: the proportion of patients at week 12 achieving IGA 0/1, EASI-75, EASI-90, and PP-NRS4 and the number needed to treat (NNT) with abrocitinib 100 mg once daily. The icon View indicates that patients received concomitant topical medications in JADE COMPARE and JADE TEEN. EASI-75 ≥ 75% improvement from baseline in Eczema Area and Severity Index, EASI-90 ≥ 90% improvement from baseline in Eczema Area and Severity Index, IGA 0/1 Investigator’s Global Assessment score of 0 (clear) or 1 (almost clear) and a ≥ 2-point improvement from baseline, NNT number needed to treat, PP-NRS4 ≥ 4-point improvement from baseline in Peak Pruritus Numerical Rating Scale
More patients who received abrocitinib 100 mg, either as monotherapy or in combination with topical therapy, had a clinically meaningful reduction in pruritus intensity compared with placebo at week 12 (Fig. 1). Pruritus intensity in key phase 3 trials of abrocitinib was measured with the Peak Pruritus Numerical Rating Scale (PP-NRS; used with permission from Regeneron Pharmaceuticals, Inc., and Sanofi), an 11-point scale, with 0 representing no itch and 10 representing worst imaginable itch; a ≥ 4-point improvement in PP-NRS score (PP-NRS4) is considered to represent clinically meaningful improvement [14]. A key secondary endpoint of JADE MONO-1, JADE MONO-2, and JADE TEEN was PP-NRS4 at weeks 2, 4, and 12; PP-NRS4 at week 2 was a key secondary endpoint of JADE COMPARE. PP-NRS4 response rates with abrocitinib 100 mg were higher than with placebo after 2 weeks of treatment; differences between abrocitinib monotherapy and placebo at week 2 were 18.0% (95% confidence interval [CI] 10.2–25.8; p = 0.0004) in JADE MONO-1 and 19.2% (95% CI 11.0–27.4; p = 0.0002) in JADE MONO-2 [6, 7]. For patients who received concomitant topical therapy, the difference in the proportion of patients who achieved PP-NRS4 with abrocitinib compared with placebo at week 2 was 14.7% (95% CI 3.5–25.9; p = 0.0119) in adolescents in JADE TEEN and 17.9% (95% CI 9.5–26.3; p < 0.001) in adults in JADE COMPARE [8, 9]. In the pooled monotherapy analysis of adults and adolescents (phase 2b [NCT02780167], JADE MONO-1, and JADE MONO-2) and adolescents receiving topical therapy in JADE TEEN, the mean percentage reduction in PP-NRS score was greater with abrocitinib 100 mg than with placebo at 24–48 h after receiving the first dose of study treatment [9, 15], indicating abrocitinib’s very rapid onset of action in reducing itch.
In JADE COMPARE, the proportions of dupilumab-treated patients who achieved IGA 0/1, EASI-75, EASI-90, and PP-NRS4 at week 12 were 36.5, 58.1, 34.9, and 54.5%, respectively; these proportions were similar to those with abrocitinib 100 mg QD (Fig. 1).
Long-Term Efficacy: Skin Clearance and Itch Response
Among patients from JADE MONO-1, JADE MONO-2, and JADE COMPARE who received treatment with abrocitinib 100 mg in JADE EXTEND, efficacy at week 48 was comparable to short-term efficacy at week 12 (time after first dose of abrocitinib, either in the parent study or in JADE EXTEND) [12]. After 48 weeks of treatment with abrocitinib 100 mg, IGA 0/1, EASI-75, EASI-90, and PP-NRS4 responses were achieved by 29.1, 49.5, 29.2, and 37.6% of patients, compared with 34.2, 55.7, 31.6, and 49.5% of patients at week 12, respectively [12]. Most patients who attained a short-term efficacy response at week 12 maintained the response over the longer term (to week 48) following treatment with abrocitinib 100 mg (IGA 0/1, 53.3%; EASI-75, 69.3%; PP-NRS4, 51.6%) [12]. After continued treatment with abrocitinib 100 mg, a substantial proportion of abrocitinib nonresponders at week 12 attained a clinically meaningful response at week 24 (IGA 0/1, 22.4%; EASI-75, 44.8%; PP-NRS4, 32.3%) and week 48 (IGA 0/1, 21.9%; EASI-75, 35.2%; PP-NRS4, 26.8%) [12]. Among patients who received dupilumab for 16 weeks in JADE COMPARE and switched to abrocitinib 100 mg treatment for 12 weeks in JADE EXTEND, clinically meaningful efficacy responses were achieved by a substantial proportion of patients who were prior dupilumab responders (IGA 0/1, 76.9%; EASI-75, 90.2%; PP-NRS4, 81.6%) and dupilumab nonresponders (IGA 0/1, 35.2%; EASI-75, 67.7%; PP-NRS4, 37.8%) [16].
In the phase 3 randomized withdrawal study JADE REGIMEN, after 12 weeks of treatment with open-label abrocitinib 200 mg QD monotherapy, a subset of patients who responded to treatment switched to receiving double-blind abrocitinib 100 mg QD for 40 weeks [10]. At week 40, a larger proportion of patients who received abrocitinib 100 mg QD vs. placebo had skin clearance and an itch response (IGA 0/1, 42.3 vs. 10.6%; EASI-75, 54.2 vs. 15.1%; PP-NRS4, 39.4 vs. 10.1%) [10].
Efficacy by Age Group
The clinical effects of abrocitinib in adolescents were evaluated in clinical trials, although regional/country approvals of abrocitinib in adolescent patients currently vary. The efficacy of abrocitinib 100 mg in adolescent patients in JADE TEEN is reported above. Although the number of adolescents included in JADE MONO-1 and JADE MONO-2 was limited, IGA 0/1 and EASI-75 responses to abrocitinib 100 mg monotherapy at week 12 were higher than with placebo in the adolescent subgroup [6, 7], which is consistent with findings from abrocitinib in combination with topical therapy in JADE TEEN.
Based on a pooled analysis of phase 2b (NCT02780167), JADE MONO-1, and JADE MONO-2, the efficacy of abrocitinib 100 mg monotherapy vs. placebo among older adults (aged ≥ 51 years) was generally comparable to the response seen in younger patient age groups: IGA 0/1 responses with abrocitinib 100 mg occurred in 40.8% compared with 10.0% with placebo, EASI-75 responses were achieved in 54.9 vs. 20.0%, and PP-NRS4 responses were achieved in 55.0 vs. 17.1% [17]. Similarly in older adults who received abrocitinib with concomitant topical therapy (JADE COMPARE), both IGA 0/1 and EASI-75 responses at week 16 were higher with abrocitinib 100 mg than with placebo: IGA 0/1, 26.1 vs. 16.7%; EASI-75, 58.7 vs. 45.8%; PP-NRS4, 50.0 vs. 36.8% [17].
Patient-Reported Outcomes
Clinically meaningful improvements in patient quality of life (QoL) were attained with abrocitinib 100 mg when administered with or without concomitant topical therapy in adults and adolescents. In abrocitinib clinical trials, QoL was evaluated with the Dermatology Quality of Life Index (DLQI) and Children’s DLQI (CDLQI), which assess the effect of skin disease on the patients’ QoL. Scores on DLQI and CDLQI range from 0 to 30. For DLQI, scores of 0–1 represent no effect of AD on QoL, 2–5 represent a small effect, 6–10 represent a moderate effect, 11–20 represent a very large effect, and 21–30 represent an extremely large effect [18]; for CDLQI, scores of 0–1 represent no effect, 2–6 represent a small effect, 7–12 represent a moderate effect, 13–18 represent a very large effect, and 19–30 represent an extremely large effect [19]. In a pooled analysis of three monotherapy studies (phase 2b, JADE MONO-1, and JADE MONO-2), the proportion of patients who achieved DLQI scores of 0–1 (i.e., no effect of AD on QoL) after 12 weeks of treatment with abrocitinib 100 mg was 25.1% compared with 14.9% in the placebo group [20]. The proportion of patients who achieved DLQI scores 2–5 (i.e., a small effect of AD on QoL) at week 12 were 27.6% with abrocitinib 100 mg compared with 23.1% in the placebo group [20]. CDLQI scores 0–1 (i.e., no effect of AD on QoL) were observed in 16.3% of adolescent patients treated with abrocitinib 100 mg monotherapy compared with 0% treated with placebo. CDLQI scores of 2–6 (i.e., a small effect of AD on QoL) were observed in 38.3% of adolescents treated with abrocitinib 100 mg compared with 35.0% in the placebo group. In adolescent patients who received concomitant medicated topical therapy in JADE TEEN, CDLQI score of 0–6 (i.e., no effect or a small effect of AD on QoL) was achieved at week 12 in 59.0% of patients treated with abrocitinib 100 mg versus 42.2% of those who received placebo (p = 0.0345) [21]. In adult patients who received concomitant medicated topical therapy in JADE COMPARE, a clinically meaningful improvement in DLQI (defined as a ≥ 4-point improvement [22]) was achieved at week 12 by 74.7% of patients who received abrocitinib 100 mg versus 56.5% of those who received placebo (p = 0.0004) [23]; a DLQI score of 0–1 signifying no effect of AD on QoL was achieved in 21.9% of patients treated with abrocitinib 100 mg versus 8.6% treated with placebo at week 12 [8].
In abrocitinib clinical trials, the peak intensity of nighttime itch was evaluated by a scale that ranges from 0 (no itch) to 10 (worst itch imaginable). Treatment with abrocitinib 100 mg QD, with or without topical therapy, in adults and adolescents reduced nighttime itch severity. A rapid onset of action was evident based on clinically meaningful itch reduction at week 2, and the benefit appeared to be enhanced at week 12. In the pooled monotherapy analysis (phase 2b, JADE MONO-1, and JADE MONO-2), a higher proportion of patients treated with abrocitinib 100 mg achieved a clinically meaningful ≥ 4-point improvement on the Night Time Itch Scale compared with placebo at weeks 2 (25.3 vs. 6.4%) and 12 (42.7 vs. 12.7%) [20]. Among adolescents receiving background topical therapy in JADE TEEN, ≥ 4-point improvement in the Night-Time Itch Scale was achieved by 25.9 and 51.5% of patients treated with abrocitinib 100 mg at weeks 2 and 12 compared with 13.8 and 35.7% of placebo-treated patients [21]. Among adults receiving background topical therapy in JADE COMPARE, ≥ 4-point improvement in the night-time itch scale was achieved by 37.7 and 51.6% of patients treated with abrocitinib 100 mg at weeks 2 and 12 compared with 15.5 and 29.2% of placebo-treated patients [23].
Treat-to-Target Efficacy Goals
A treat-to-target framework, based on expert opinion, clinical experience, and consensus agreement, was recently developed to guide decisions for treatment with systemic therapies in patients with AD [24]. Guideline-recommended treatment target goals at 3 months and 6 months after start of treatment consisted of specified improvements in Patient Global Assessment, Eczema Area and Severity Index, SCORing Atopic Dermatitis, PP-NRS, DLQI, and Patient-Oriented Eczema Measure (see the guideline for specific goals) [24]. Based on pooled analyses of data from adolescent and adult patients with moderate-to-severe AD who received abrocitinib 100 mg monotherapy for 12 weeks in the phase 2b, JADE MONO-1, and JADE MONO-2 studies, 88.9% of patients achieved at least one 3-month target with a median time of 12 days (95% CI 9–14), and 70.8% achieved at least one 6-month target at week 12, with a median time of 15 days (95% CI 14–15) [25].
Safety and Tolerability
Short-Term Data up to 16 Weeks
The proportion of patients who experienced TEAEs across the week 12/16 trials (up to 28 days after last dose of study treatment) with abrocitinib 100 mg (with or without concomitant topical therapy) ranged from 50.8 to 69.2% and ranged from 52.1 to 57.1% with placebo (Table 2) [6,7,8]. Serious TEAEs occurred infrequently in both groups and were not elevated in the abrocitinib group (≤ 3.2% of patients who received abrocitinib 100 mg and ≤ 3.9% who received placebo). Rates of TEAEs leading to treatment discontinuation were low overall and were higher in the placebo group (≤ 5.8% of patients who received abrocitinib 100 mg and ≤ 12.8% who received placebo). Only one death was reported in the JADE MONO-2 study, which was adjudicated as unrelated to treatment in the abrocitinib 100-mg group. Sudden cardiac death occurred in a woman aged 73 years, which occurred 3 weeks after discontinuation of abrocitinib 100 mg. The patient had a history of cardiovascular disease [7].
Nausea appeared to be a dose-dependent TEAE in key phase 3 trials of abrocitinib, with a lower proportion of patients experiencing this event with abrocitinib 100 mg QD vs. 200 mg QD dosing. Nausea was reported in 4.2–9.0% of patients treated with abrocitinib 100 mg compared with abrocitinib 200 mg in 11.1–20.1% of patients and placebo in 1.0–2.6% of patients [6,7,8,9]. The median time to resolution of nausea in the abrocitinib 100 mg group was 13.0 days in JADE MONO-1, 2.5 days in JADE MONO-2, and 23.0 days in JADE TEEN; two patients treated with abrocitinib 100 mg (one each in JADE MONO-1 and JADE COMPARE) discontinued treatment because of nausea [6,7,8,9].
Acne and headache also appear to be dose-dependent TEAEs associated with abrocitinib, with a lower proportion of patients experiencing this event with abrocitinib 100 mg QD vs. 200 mg QD dosing. Acne was reported in 0.6–3.2% of patients treated with abrocitinib 100 mg compared with 2.6–6.6% of patients treated with abrocitinib 200 mg, and 0–1.0% of the placebo group [6,7,8,9]. Headache was reported in 4.2–7.7% of patients treated with abrocitinib 100 mg, 6.6–9.7% treated with abrocitinib 200 mg, and 2.6–7.3% of the placebo group [6,7,8,9].
Long-Term Data up to 72 Weeks
An integrated safety analysis of patients who received abrocitinib in phase 2b, JADE MONO-1, JADE MONO-2, JADE COMPARE, or JADE REGIMEN, and JADE EXTEND (data cutoff date April 22, 2020), included 885 patients treated with abrocitinib 100 mg QD and 1971 patients treated with abrocitinib 200 mg QD [11]. Cumulative exposure to abrocitinib was ≥ 24 weeks in 62.5% (100 mg QD group) and 35.3% (200 mg QD group), ≥ 36 weeks in 46.8% (100 mg) and 29.5% (200 mg), ≥ 48 weeks in 29.9% (100 mg) and 17.3% (200 mg), and ≥ 72 weeks in 9.0% (100 mg) and 6.0% (200 mg) of patients [11]. TEAEs were reported in 70.8%, serious TEAEs in 5.4%, and severe TEAEs (interferes significantly with patient’s usual function) in 7.8% of patients treated with abrocitinib 100 mg [11]. For abrocitinib 200 mg, TEAEs were reported in 72.0%, serious TEAEs in 3.8%, and severe TEAEs in 5.2% of patients [11]. The incidence rate (i.e., the number of patients with events per 100 patient-years) of serious infections was 2.65 for 100 mg and 2.33 for 200 mg; and incidence rates for herpes zoster, herpes simplex, and eczema herpeticum were 2.04 (100 mg) and 4.34 (200 mg), 8.73 (100 mg) and 11.83 (200 mg), and 2.34 (100 mg) and 0.78 (200 mg), respectively [11]. Of 15 cases of eczema herpeticum reported in patients treated with abrocitinib 100 mg, four events were serious and four led to study discontinuation [11]. Eczema herpeticum was more likely to occur in patients with uncontrolled AD (IGA > 1). Nonmelanoma skin cancer was reported in three patients treated with abrocitinib 100 mg with no known prior exposure to phototherapy and with unknown amount of exposure to sunlight (two cases of basal cell carcinoma and one case of T-cell cutaneous lymphoma); an event of adjudicated malignancy was prostate cancer in a patient with a history of an enlarged prostate [11]. Four patients treated with abrocitinib 200 mg who reported no prior exposure to phototherapy and with unknown amount of exposure to sunlight had nonmelanoma skin cancer (all cases were squamous cell carcinoma); and two events of adjudicated malignancy were prostate cancer and gastric adenocarcinoma [11]. All cases of nonmelanoma skin cancer were considered unrelated to the study drug by the investigators. As noted above for JADE MONO-2, one major adverse cardiovascular event (sudden cardiac death in a patient with a previous history of cardiovascular disease) occurred in a patient treated with abrocitinib 100 mg [11]. Two patients treated with abrocitinib 200 mg had major adverse cardiovascular events (myocardial infarctions) [11]. In two patients treated with abrocitinib 100 mg, events were adjudicated as transient ischemic attacks [11]. There were no venous thromboembolism events in patients treated with abrocitinib 100 mg, and five events in patients treated with abrocitinib 200 mg (three pulmonary embolism and two deep venous thrombosis events) [11].
Safety by Age Group
Among 51 older adults (aged ≥ 65 years) who received abrocitinib 100 mg QD (pooled phase 2b, JADE MONO-1, JADE MONO-2, JADE COMPARE, JADE TEEN, JADE REGIMEN, and JADE EXTEND), incidence of serious TEAEs (13.7%) and TEAEs leading to discontinuation (17.6%) were higher than in 972 younger adolescents/adults [26]. For comparison, serious TEAEs occurred in 4.0, 5.7, and 5.1% of patients aged 12–17, 18–39, and 40–64 years, respectively, who were treated with abrocitinib 100 mg QD. TEAEs leading to discontinuation occurred in 6.5, 9.1, and 9.4% of patients aged 12–17, 18–39, and 40–64 years, respectively, who were treated with abrocitinib 100 mg QD.
Expert Opinion on Appropriate Use in Clinical Practice
Abrocitinib is approved for the treatment of moderate-to-severe AD at QD doses of 200 mg, 100 mg, and 50 mg in different countries and regions, and dosing recommendations vary [2,3,4,5]. Currently, a starting dose of 100 mg QD is recommended for adults of all ages in the United States and Japan, adults aged ≥ 65 years in the European Union and Great Britain, and adolescents in Great Britain and Japan [2,3,4,5]. It is also the recommended starting dose for patients with moderate renal impairment (estimated glomerular filtration rate 30–59 mL/min) in some instances [3, 4]. The dose can subsequently be adjusted up or down based on efficacy and tolerability [2,3,4,5].
Phase 3 studies clearly demonstrate the efficacy and safety of the 100-mg QD dose of abrocitinib when compared with placebo (Fig. 1). The number needed to treat with abrocitinib 100 mg QD for an additional patient to meet each of the efficacy response criteria at week 12 in the pivotal trials is summarized in Fig. 1, with consistent and reproducible responses observed with abrocitinib 100 mg vs. placebo for measures of skin clearance and clinically meaningful itch improvement. Findings from JADE EXTEND and JADE REGIMEN provide support for the use of 100 mg QD as the long-term maintenance dose of abrocitinib. Results from the JADE REGIMEN trial suggest the majority of patients who have well-controlled AD with abrocitinib 200 mg QD will continue to have well-controlled AD with the 100 mg QD dose [10]. For patients on a stable regimen of abrocitinib 100 mg QD who experience seasonal/episodic flares, there is the possibility of increasing the dose to 200 mg QD episodically as needed.
In addition to efficacy, the decision to commence and continue the use of a particular systemic agent for the treatment of AD should allow for individual patient factors, such as age, comorbidities, previous therapy, QoL, and treatment tolerability, and involve shared decision-making between the patient and clinician. Abrocitinib was proven to be effective in patients previously treated with dupilumab; regardless of prior response to dupilumab [16]. In phase 3 studies, clinically meaningful improvements in QoL were attained with abrocitinib 100 mg QD when administered with or without concomitant topical therapy in adults and adolescents. We recommend that clinicians continue to monitor the QoL of their patients during treatment with abrocitinib.
Regarding safety, the available evidence suggests that abrocitinib 100 mg QD is well tolerated with an acceptable safety profile for up to 72 weeks of treatment, with low rates of serious TEAEs or TEAEs leading to discontinuation (Table 2), although rates of these events were higher in those aged ≥ 65 years. Nausea, acne, and headache observed in patients treated with abrocitinib in key phase 3 studies were less frequently observed in patients treated with 100 mg compared with 200 mg QD. If patients experience nausea, they may consider taking the oral tablets with food because some evidence suggests that a fed state can lower nausea with abrocitinib [3, 4]. The incidence rates of herpes zoster and herpes simplex were lower in patients treated with 100 mg compared with 200 mg QD, but the rates of serious infections were similar.
The comparable short-term efficacy of abrocitinib 100 mg QD and dupilumab suggests that abrocitinib 100 mg QD may be an appropriate treatment option for many patients with moderate-to-severe AD who prefer to receive treatment by oral administration rather than subcutaneous injection, are not able to tolerate dupilumab (e.g., due to conjunctivitis), or have an inadequate response to dupilumab therapy. In terms of the tolerability profile, 50.0% of dupilumab-treated patients experienced TEAEs in JADE COMPARE; the incidence of nasopharyngitis (9.5%), upper respiratory tract infection (3.7%), nausea (2.9%), and headache (5.4%) in dupilumab-treated patients was similar to that in patients treated with abrocitinib 100 mg QD (see Table 2). Although acne was more frequently reported in patients treated with abrocitinib 100 mg than dupilumab (2.9 vs. 1.2%), conjunctivitis was more frequently reported in patients treated with dupilumab than abrocitinib 100 mg (6.2 vs. 0.8%).
As with all studies, there are limitations to the data presented, including that the data were obtained in a clinical trial setting rather than in the real world, and therefore, patient characteristics may differ; that some analyses summarized in this review were post hoc analyses that were not prespecified; and that criteria for the conclusions of this review were not prespecified.
Conclusions
In conclusion, abrocitinib 100 mg QD has demonstrated a rapid onset of efficacy, safety, and a positive benefit–risk profile for patients who are considering long-term use of oral therapy, patients who may need flexible treatment with different dose options (dose escalation and de-escalations), and patients who were treated previously with biologics. This article highlights the scope of therapeutic use of abrocitinib 100 mg QD for patients with moderate-to-severe AD based on efficacy and safety data from the JADE clinical development program. The decision to use abrocitinib for the treatment of AD should allow for individual patient factors such as age, comorbidities, previous therapy, QoL, and treatment tolerability and involve shared decision-making between the patient and clinician.
References
Vazquez ML, Kaila N, Strohbach JW, Trzupek JD, Brown MF, Flanagan ME, et al. Identification of N-{cis-3-[methyl(7H-pyrrolo[2,3-d]pyrimidin-4-yl)amino]cyclobutyl}propane-1-sulfo namide (PF-04965842): a selective JAK1 clinical candidate for the treatment of autoimmune diseases. J Med Chem. 2018;61:1130–52.
Cibinqo (abrocitinib). Prescribing information. Pfizer Labs; 2022.
European Medicines Agency. Cibinqo® (abrocitinib). Summary of product characteristics (SmPC). Pfizer Europe MA EEIG; 2021.
Cibinqo 100 mg film-coated tablets. Summary of product characteristics. Pfizer Ltd.; 2021.
Cibinqo™ (abrocitinib) tablets, for oral use 50 mg 100 mg 200 mg. Prescribing information. Pfizer Japan Inc.; 2021.
Simpson EL, Sinclair R, Forman S, Wollenberg A, Aschoff R, Cork M, et al. Efficacy and safety of abrocitinib in adults and adolescents with moderate-to-severe atopic dermatitis (JADE MONO-1): a multicentre, double-blind, randomised, placebo-controlled, phase 3 trial. Lancet. 2020;396:255–66.
Silverberg JI, Simpson EL, Thyssen JP, Gooderham M, Chan G, Feeney C, et al. Efficacy and safety of abrocitinib in patients with moderate-to-severe atopic dermatitis: a randomized clinical trial. JAMA Dermatol. 2020;156:863–73.
Bieber T, Simpson EL, Silverberg JI, Thaçi D, Paul C, Pink AE, et al. Abrocitinib versus placebo or dupilumab for atopic dermatitis. N Engl J Med. 2021;384:1101–12.
Eichenfield LF, Flohr C, Sidbury R, Siegfried E, Szalai Z, Galus R, et al. Efficacy and safety of abrocitinib in combination with topical therapy in adolescents with moderate-to-severe atopic dermatitis: the JADE TEEN randomized clinical trial. JAMA Dermatol. 2021;157:1165–73.
Blauvelt A, Silverberg JI, Lynde CW, Bieber T, Eisman S, Zdybski J, et al. Abrocitinib induction, randomized withdrawal, and retreatment in patients with moderate-to-severe atopic dermatitis: results from the JAK1 Atopic Dermatitis Efficacy and Safety (JADE) REGIMEN phase 3 trial. J Am Acad Dermatol. 2022;86:104–12.
Simpson EL, Silverberg JI, Nosbaum A, Winthrop KL, Guttman-Yassky E, Hoffmeister KM, et al. Integrated safety analysis of abrocitinib for the treatment of moderate-to-severe atopic dermatitis from the phase II and Phase III clinical trial program. Am J Clin Dermatol. 2021;22:693–707.
Reich K, Silverberg JI, Papp K, Deleuran M, Katoh N, Strober B, et al., editors. Long-term management of moderate-to-severe atopic dermatitis with abrocitinib: a phase 3 extension study (JADE EXTEND). Revolutionizing Atopic Dermatitis (RAD); Virtual; June 13, 2021.
Andreasen TH, Christensen MO, Halling AS, Egeberg A, Thyssen JP. Placebo response in phase 2 and 3 trials of systemic and biological therapies for atopic dermatitis-a systematic review and meta-analysis. J Eur Acad Dermatol Venereol. 2020;34:1143–50.
Yosipovitch G, Reaney M, Mastey V, Eckert L, Abbe A, Nelson L, et al. Peak Pruritus Numerical Rating Scale: psychometric validation and responder definition for assessing itch in moderate-to-severe atopic dermatitis. Br J Dermatol. 2019;181:761–9.
Kim BS, Silverberg JI, Ständer S, Yosipovitch G, Simpson EL, DiBonaventura M, et al. Rapid improvement of itch associated with atopic dermatitis with abrocitinib is partially independent of overall disease improvement: results from pooled phase 2b and 3 monotherapy studies. Dermatitis. 2021;32:S39–44.
Shi VY, Bhutani T, Fonacier L, Deleuran M, Shumack S, Valdez H, et al. Phase 3 efficacy and safety of abrocitinib in adults with moderate-to-severe atopic dermatitis after switching from dupilumab (JADE EXTEND). J Am Acad Dermatol. 2022;87:351–8.
Alexis A, Deleuran M, Silverberg JI, Gooderham M, Johnson SL, Chan G, et al., editors. Short-term efficacy and safety of abrocitinib in patients with moderate-to-severe atopic dermatitis stratified by age. Revolutionizing Atopic Dermatitis (RAD); Virtual; December 11–13, 2021.
Basra MK, Fenech R, Gatt RM, Salek MS, Finlay AY. The Dermatology Life Quality Index 1994–2007: a comprehensive review of validation data and clinical results. Br J Dermatol. 2008;159:997–1035.
Salek MS, Jung S, Brincat-Ruffini LA, MacFarlane L, Lewis-Jones MS, Basra MK, et al. Clinical experience and psychometric properties of the Children’s Dermatology Life Quality Index (CDLQI), 1995–2012. Br J Dermatol. 2013;169:734–59.
Silverberg JI, Thyssen JP, Simpson EL, Yosipovitch G, Ständer S, Valdez H, et al. Impact of oral abrocitinib monotherapy on patient-reported symptoms and quality of life in adolescents and adults with moderate-to-severe atopic dermatitis: a pooled analysis of patient-reported outcomes. Am J Clin Dermatol. 2021;22:541–54.
Cork MJ, McMichael A, Teng J, Valdez H, Rojo R, Chan G, et al. Impact of oral abrocitinib on signs, symptoms and quality of life among adolescents with moderate-to-severe atopic dermatitis: an analysis of patient-reported outcomes. J Eur Acad Dermatol Venereol. 2022;36:422–33.
Basra MK, Salek MS, Camilleri L, Sturkey R, Finlay AY. Determining the minimal clinically important difference and responsiveness of the Dermatology Life Quality Index (DLQI): further data. Dermatology. 2015;230:27–33.
Thyssen JP, Yosipovitch G, Paul C, Kwatra SG, Chu CY, DiBonaventura M, et al. Patient-reported outcomes from the JADE COMPARE randomized phase 3 study of abrocitinib in adults with moderate-to-severe atopic dermatitis. J Eur Acad Dermatol Venereol. 2022;36:434–43.
De Bruin-Weller M, Biedermann T, Bissonnette R, Deleuran M, Foley P, Girolomoni G, et al. Treat-to-target in atopic dermatitis: an international consensus on a set of core decision points for systemic therapies. Acta Derm Venereol. 2021;101:adv00402.
de Bruin-Weller M, Weidinger S, Gooderham M, Pink A, Silverberg J, DiBonaventura M, et al., editors. Efficacy of abrocitinib in adults and adolescents with moderate-to-severe atopic dermatitis using treat-to-target goals. Revolutionizing Atopic Dermatitis (RAD); 2021; Virtual: December 11–13, 2021.
Cork MJ, Deleuran M, Geng B, Silverberg JI, Simpson EL, Stein Gold LF, et al. Abrocitinib treatment in patients with moderate-to-severe atopic dermatitis: safety of abrocitinib stratified by age. European Academy of Dermatology and Venereology (EADV); Berlin, Germany; September 29–October 2, 2021.
Acknowledgements
Funding
This study was funded by Pfizer Inc., New York, NY, USA. Pfizer Inc. also funded the Rapid Service Fee for this article.
Authorship
All authors met the International Committee of Medical Journal Editors (ICMJE) criteria for authorship, take responsibility for the integrity of the work, and have given their approval to submit the manuscript for publication.
Medical Writing and Editorial Assistance
Editorial/medical writing support under the guidance of the authors was provided by Renee Gordon, PhD, at ApotheCom, San Francisco, CA, USA, and was funded by Pfizer Inc., New York, NY, USA, in accordance with Good Publication Practice (GPP 2022) guidelines (Ann Intern Med. 2022; 10.7326/M22-1460).
Author Contributions
Melissa Watkins conceived the original idea; Melissa Watkins, Erman Güler, and Melinda J. Gooderham made substantial contributions to the conception of the work. All authors contributed to the construction of the article.
Prior Publication
This review article contains data from previously published studies, and these studies have been cited as appropriate throughout the article.
Disclosures
Melinda J. Gooderham has received grants, personal fees, honoraria, and/or nonfinancial support from Pfizer Inc., AbbVie, Amgen, Akros Pharma, Arcutis, Aristea, ASLAN Pharmaceuticals, Bausch Health (Valeant), Bristol Myers Squibb, Boehringer Ingelheim, Celgene, Dermira, Dermavant, Eli Lilly and Company, Galderma, Incyte, Janssen, Kyowa Kirin, LEO Pharma, MedImmune, Meiji, Merck, Moonlake, Nimbus, Novartis, Regeneron, Reistone, Roche, Sanofi Genzyme, Sun Pharma, and UCB. Andrew E. Pink has acted as an advisor or speaker for Pfizer Inc., AbbVie, Almirall, Eli Lilly and Company, Janssen, La Roche-Posay, LEO Pharma, Novartis, Sanofi, and UCB. Eric L. Simpson has received grants from Pfizer Inc., AbbVie, Amgen, Arcutis, ASLAN Pharmaceuticals, Castle BioSciences, CorEvitas, Dermavant, Dermira, Eli Lilly and Company, Incyte, Kymab, Kyowa Kirin, LEO Pharma, Merck, National Jewish Health, Regeneron, Sanofi Genzyme, and Target RWE; and personal fees from Pfizer Inc., Advances in Cosmetic and Medical Dermatology Hawaii, AbbVie, Amgen, AOBiome, Arcutis, ASLAN Pharmaceuticals, Bausch Health (Valeant), Boehringer Ingelheim, Boston Consulting Group, Bristol Myers Squibb, Collective Acumen, CorEvitas, Dermira, Eli Lilly and Company, Evelo Biosciences, Evidera, Exerpta Medica, FIDE, Forte Bio RX, Galderma, Gesellschaft Z, GlaxoSmithKline, Incyte, Janssen, Johnson & Johnson, Kyowa Kirin, LEO Pharma, MauiDerm, Medscape, Menlo Therapeutics, Merck, MLG Operating, MJH Holdings, Physicians World, PRImE, Regeneron, Revolutionizing Atopic Dermatitis, Roivant, Sanofi Genzyme, Trevi Therapeutics, Valeant, Vindico Medical Education, and WebMD. Jonathan I. Silverberg has served as an investigator for Celgene, Eli Lilly and Company, F. Hoffmann-LaRoche, Menlo Therapeutics, Realm Therapeutics, Regeneron, and Sanofi; as a consultant for Pfizer Inc., AbbVie, Anacor, AnaptysBio, Arena Pharmaceuticals, Dermavant, Dermira, Eli Lilly and Company, Galderma, GlaxoSmithKline, Glenmark, Incyte, Kiniksa Pharmaceuticals, LEO Pharma, Menlo Therapeutics, Novartis, Realm Therapeutics, Regeneron, and Sanofi; and as a speaker for Regeneron and Sanofi. Erman Güler and Melissa Watkins are employees and shareholders of Pfizer Inc.
Compliance with Ethics Guidelines
This article is based on previously conducted studies and does not contain any new studies with human participants or animals performed by any of the authors.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Gooderham, M.J., Pink, A.E., Simpson, E.L. et al. Abrocitinib 100 mg Once Daily for Moderate-to-Severe Atopic Dermatitis: A Review of Efficacy and Safety, and Expert Opinion on Use in Clinical Practice. Dermatol Ther (Heidelb) 13, 1893–1907 (2023). https://doi.org/10.1007/s13555-023-00948-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13555-023-00948-6