
Abstract
Introduction
Although many biologic therapies are effective for clearing skin of patients with psoriasis, some lose effectiveness over time. This phase 2 open-label extension (OLE) trial was designed to investigate the long-term safety and efficacy of risankizumab.
Methods
In the phase 2, double-blind, active comparator, predecessor trial (NCT02054481), patients with moderate-to-severe chronic plaque psoriasis were treated for 24 weeks with subcutaneous (SC) risankizumab or ustekinumab, followed by a 24-week follow-up without treatment administration. Patients could enroll in the OLE (NCT02203851) when they experienced loss of treatment response (< 50% improvement in the Psoriasis Area Severity Index [PASI 50]) during follow-up) or at the end of follow-up if treatment response was ongoing. In the OLE, patients were treated every 12 weeks for at least 48 weeks with SC risankizumab 90 or 180 mg, beginning at week 12 (OLE visit 2), if the patient had not achieved PASI 90. Efficacy endpoints included the proportions of patients who achieved PASI 50/75/90/100 and static Physician’s Global Assessment (sPGA) of clear or almost clear skin at week 48 (sPGA 0/1; OLE visit 5).
Results
Of the 110 enrolled patients, 99 (90.0%) completed the OLE. No patients discontinued the study because of adverse events. At week 48, 74.1% of patients achieved PASI 90, whereas 98.1, 91.7, 53.7, and 67.6% achieved PASI 50/75/100 and sPGA 0/1, respectively. All efficacy results were consistent or slightly increased at OLE week 48 compared with week 12. No new safety findings were observed.
Conclusion
Risankizumab treatment was well tolerated with sustained clinical efficacy for at least 48 weeks.
Trial Registration
ClinicalTrials.gov identifier; NCT02203851.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Why carry out this study? |
Psoriasis is associated with increased risk of morbidity, mortality, disability, negative psychological stress, and reduced quality of life. |
The identification of new therapeutic targets has led to the discovery of several new compounds effective at clearing skin lesions in patients with psoriasis; however, many patients experience loss of treatment response over time. |
This phase 2 open-label extension (OLE) trial investigated whether risankizumab, a humanized immunoglobulin G1 monoclonal antibody that inhibits the proinflammatory cytokine interleukin-23 by binding to its p19 subunit, is effective for long-term treatment of moderate-to-severe plaque psoriasis. |
What was learned from the study? |
Risankizumab 90 or 180 mg treatment administered every 12 weeks resulted in sustained clinical efficacy over 48 weeks following a 48-week predecessor study (24 weeks of active treatment plus up to 24 weeks of follow-up without treatment). |
Over 48 weeks, risankizumab was well tolerated, with only 10% of patients discontinuing the study, none of whom discontinued because of adverse events, and no new or unexpected safety signals observed. |
Digital Features
This article is published with digital features, including a summary slide, to facilitate understanding of the article. To view digital features for this article go to https://doi.org/10.6084/m9.figshare.13582640.
Introduction
Psoriasis is a chronic, immune-mediated, inflammatory skin disease with a global prevalence range of 1–4% [1,2,3]. Patients with psoriasis are at greater risk of morbidity, mortality, disability, negative psychological stress, and reduced quality of life than are unaffected individuals [4,5,6].
The identification of specific immune pathways that are involved in psoriasis have provided important therapeutic targets and led to the discovery of several new compounds with clinical efficacy in clearing skin lesions in people with psoriasis [7]; however, many patients experience loss of treatment response over time, often resulting in stopping or switching treatment [8,9,10,11]. The proinflammatory cytokine interleukin-23 (IL-23) plays an important role in the pathogenesis of psoriasis because it stimulates the production of inflammatory cytokines, such as IL-17, by T-helper 17 (Th17) and innate immune cells [12,13,14]. Targeting upstream mediators in the IL-23/IL-17 axis, such as IL-23, may translate into prolonged efficacy because of reduced Th17 cell survival or changes in phenotype, restoration of regulatory T (Treg) cell function, or inhibition of IL-22 production [13].
Risankizumab is a humanized immunoglobulin G1 (IgG1) monoclonal antibody that inhibits IL-23 activity by binding to its p19 subunit [15, 16]. Risankizumab was investigated as a treatment for moderate-to-severe plaque psoriasis in four phase 3 multicenter, randomized, double-blind studies (UltIMMa-1, UltIMMa-2, IMMhance, and IMMvent [17,18,19]) and was subsequently approved for this indication in the USA and several other countries in 2019 [20]. In a 48-week, phase 2 study, treatment with risankizumab led to superior clinical responses compared with ustekinumab [21].
The study reported here is an open-label extension (OLE) trial of the phase 2 study designed to investigate the long-term safety and efficacy of risankizumab for the treatment of patients with moderate-to-severe chronic plaque psoriasis.
Methods
Study Design and Treatment
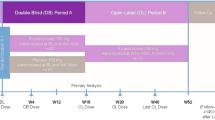
This was a phase 2, multicenter OLE trial (Clinicaltrials.gov identifier: NCT02203851) for patients with moderate-to-severe chronic plaque psoriasis who had successfully completed the 48-week, phase 2, double-blind, active comparator trial of subcutaneous (SC) risankizumab versus ustekinumab (Clinicaltrials.gov identifier: NCT02054481). The full details of the predecessor study have been published elsewhere [21] and are summarized in Fig. 1. Briefly, during a 24-week treatment period, patients received double-blind risankizumab as a single 18 mg dose at week 0, or as fixed doses of 90 or 180 mg at weeks 0, 4, and 16; in contrast, patients in the active comparator arm received open-label ustekinumab at doses of 45 or 90 mg, based on body weight (< 100 vs. ≥ 100 kg, respectively), at weeks 0, 4, and 16. At the end of the treatment period, patients were monitored for another 24 weeks without treatment administration [21]. Any patient who demonstrated a loss of response (< 50% improvement from baseline in the Psoriasis Area Severity Index [PASI 50]) during the follow-up period could immediately switch to the OLE, whereas any patient who maintained a clinical response (≥ PASI 50) could enter the OLE at week 48. After completion of this OLE study, patients who chose to continue treatment were transferred to a phase 3 OLE study (Clinicaltrials.gov identifier: NCT03047395).

Patient disposition. OLE Open-label extension, UST ustekinumab. Full details on patient disposition in the double-blind portion have been published elsewhere [21]

Efficacy outcomes. Graphs show results for PASI 90 (a), PASI 100 (b), PASI 75 (c), PASI 50 (d), and sPGA of clear or almost clear skin (sPGA 0/1) (e) over time and by treatment received in the predecessor study. PASI 50, 75, 90, 100 Psoriasis Area and Severity Index ≥ 50, 75, 90 and 100%, respectively, improvement from baseline, sPGA static Physician’s Global Assessment. Of the 110 patients, 23 switched to risankizumab 180 mg at week 12 (OLE visit 2)
In the OLE, patients were treated with SC risankizumab 90 mg every 12 weeks (q12w) for at least 48 weeks. If at week 12 (OLE visit 2) a patient demonstrated an inadequate response (< PASI 90), the dose could be increased to 180 mg q12w for the remainder of the trial.
This study was conducted in accordance with the Good Clinical Practice Guideline as defined by the International Conference on Harmonisation, the Declaration of Helsinki, and/or all applicable federal and local regulations and the following institutional review boards: Institutional Review Board Services, Aurora, Ontario, Canada; Research Review Board Inc., Richmond Hill, Ontario, Canada; CPP- Ile de France IV, Hôpital Saint-Louis, Paris, France; Ethikkommission des Landes Berlin, Landesamt fur Gesundheit und Soziales, Berlin, Germany; Chesapeake IRB, Columbia, Maryland, USA. All patients provided informed consent and signed approved consent forms prior to any study procedures in accordance with Good Clinical Practice and local legislation.
Patients
To enroll in the predecessor study, patients were required to have moderate-to-severe plaque psoriasis for at least 6 months prior to screening (including ≥ 10% body surface area [BSA] affected by psoriasis; PASI ≥ 12; and static Physician Global Assessment [sPGA] ≥ 3) and had to be a candidate for systemic psoriasis treatment or phototherapy. To enroll in the OLE, patients were required to have completed the double-blind predecessor study, defined as completing the full 48 weeks or demonstrating a loss of response during the follow-up period. Patients could not enroll in the OLE if they had experienced any drug-related serious adverse events (SAEs) or developed guttate, erythrodermic, pustular, or drug-induced psoriasis during the predecessor study or had any other condition that may have compromised safety or data quality.
Assessments
The key efficacy endpoints were the proportions of patients who achieved PASI 50, PASI 75, PASI 90, and PASI 100 (as measured from baseline in the predecessor study) and sPGA scores of clear (0) or almost clear skin (1) at week 48 (OLE visit 5).
Safety assessments were based on treatment-emergent adverse events (TEAEs), which were monitored from the first day of study drug administration in the OLE through 105 days after the last dose in the analysis period. Clinical laboratory parameters, local tolerability, and vital sign measurements were also recorded.
Statistical Analysis
All efficacy and safety analyses were performed for patients who received at least one dose of study drug (intent-to-treat/safety population). Since this was a single-arm OLE study, no statistical tests were performed, and only summary statistics (number, percentage, mean, standard deviation [SD], median, 95% confidence interval, and range) are provided. All efficacy results are based on observed cases and no imputation for missing data was applied.
Results
Patients
Of the 166 patients who were initially randomly assigned to the treatment group in the double-blind predecessor study, 110 (66.3%) entered the OLE. A total of 78 (70.9%) patients across all treatment groups completed the initial study without a loss of clinical response during the follow-up period (Fig. 1). Of the 83 patients who entered the OLE following risankizumab treatment, 62 (74.7%) completed the predecessor study follow-up period without losing clinical response and therefore had initiated risankizumab treatment at least 48 weeks prior to entering the OLE.
Of the 110 patients who entered the OLE, 99 (90.0%) completed the study. The reasons for discontinuation were withdrawal of consent (n = 6; 5.5%), loss to follow-up (n = 4; 3.6%) and other (n = 1; 0.9%). A total of 23 patients switched to 180 mg risankizumab at week 12 (OLE visit 2) because of a lack of PASI 90 response. The mean (SD) duration of study drug exposure for the combined predecessor and OLE studies was 3.69 (0.66) years and the range was 48.1 weeks–4.34 years.
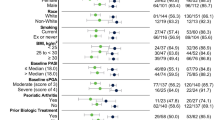
Most patients were white (91.8%) and male (60%), with a mean (SD) age of 49.0 (13.0) years at OLE baseline (Table 1). Key disease characteristics at baseline included a mean (SD) BSA affected by psoriasis of 23.9 (16.2) m2, mean PASI of 19.4 (7.2), and mean sPGA of 3.4 (0.5) (Table 1).
Efficacy
The overall proportion of patients with PASI 90 responses at week 48 (OLE visit 5) was 74.1%, which was greater than that observed at the first OLE efficacy assessment at week 12 (OLE visit 2), 67.0% (Fig. 2a). PASI 90 response rates at week 48 (OLE visit 5) were similar for all patients, regardless of which treatment they had received in the predecessor study (range 71.9–77.8%). The proportions of patients who achieved PASI 50/75/100 and sPGA 0/1 were also similar between all the patient groups, regardless of the initial treatment they received (Fig. 2b–e). Responder rates for all endpoints were slightly increased at week 48 (OLE visit 5) compared with week 12 (OLE visit 2), except for sPGA, whose responder rates remained consistent between week 48 (OLE visit 5) and week 12 (OLE visit 2).
Safety
A total of 85 (77.3%) patients reported at least one TEAE during the OLE study (Table 2). The most frequently reported TEAEs were nasopharyngitis (n = 19; 17.3%), upper respiratory tract infection (n = 15; 13.6%), and arthralgia (n = 11; 10.0%). Overall, most TEAEs were mild or moderate, and most were not considered to be related to the study drug. No deaths occurred during the study.
The SAEs reported by more than one patient were basal cell carcinoma and transient ischemic attack (each n = 2; 1.8%). The SAEs that were considered possibly related to the study drug were basal cell carcinoma and mild squamous cell carcinoma in one patient and moderate cystitis, moderate pyelonephritis, and moderate sepsis in another patient. Neither patient discontinued the study treatment because of these events.
Two patients (1.8%) reported serious infections during the study: one patient had cystitis, pyelonephritis, and sepsis, all of which were considered serious and possibly related to the study treatment, whereas the other patient had serious pneumonia that was not considered to be related to the study drug. A total of five fungal infections were reported, only one of which (vaginal mycosis) was considered possibly related to the study drug. Although 13 (11.8%) patients reported hypersensitivity reactions, none of the reactions were considered to be serious and no anaphylactic reactions were reported.
No notable changes in laboratory, vital sign, or local tolerability assessments were detected during the study.
Discussion
The results of this phase 2, multicenter OLE trial confirm that risankizumab administered q12w resulted in sustained clinical efficacy and tolerability in patients with moderate-to-severe chronic plaque psoriasis over 48 weeks in addition to 24 weeks of active treatment (plus up to 24 weeks of follow-up without treatment administration) in the predecessor study. The proportions of patients who achieved PASI 50/75/90/100 were slightly greater at week 48 (OLE visit 5) than at week 12 (OLE visit 2). Only 10% of patients discontinued the trial, none of whom reported AEs as the cause.
The availability of biologic therapies was a major advancement for patients seeking safe and effective treatment for moderate-to-severe plaque psoriasis; however, over the nearly two decades since the first biologic treatment was approved for this condition, evidence has revealed that although such biologics are initially effective, many lose effectiveness over time [9,10,11]. This has been attributed to several causes, including the development of anti-drug antibodies [22]. Additionally, some treatments are intolerable for some patients, resulting in discontinuation [23]. Therefore, there has been an ongoing unmet need for treatments that provide sustained efficacy and are well tolerated for an extended duration. The results from this study indicate that risankizumab provided sustained efficacy over 48 weeks with few patient withdrawals, none of which were due to TEAEs. It is also important to note that half of the patients who entered the OLE had been treated with risankizumab 90 or 180 mg for 24 weeks in the double-blind predecessor study and had sustained treatment responses over the 24-week follow-up period prior to re-starting treatment with risankizumab in the OLE. In sequence, these studies covered a period of up to almost 2 years of treatment with risankizumab (96 weeks, less the follow-up period when the study drug was not administered), during which the clinical efficacy demonstrated an overall trend toward improvement over time. Notably, 25% of the patients in the OLE had been treated with ustekinumab during the predecessor study, and this group’s improvements in efficacy endpoints over time were similar to those of the patients who received risankizumab during the initial trial. These findings are similar to those from a recent report showing a mean (SD) improvement in PASI from 11.9 (5.5) to 3.3 (1.7) after 16 weeks of risankizumab treatment in a study of 8 patients who failed anti–IL-17, anti–IL-12/-23, or anti–IL-23 therapy [24].
These results are supported by data from other recent trials in patients with moderate-to-severe plaque psoriasis and are consistent with findings from phase 3 studies assessing the efficacy and safety of 150 mg risankizumab treatment for up to 88 weeks [19, 25, 26]. During a randomized, open-label, efficacy assessor-blinded study in 164 patients treated with risankizumab 150 mg q12w, clinical efficacy was sustained through 52 weeks, during which only two (1.2%) patients discontinued treatment because of an AE [25]. In a double-blind, placebo-controlled, phase 2/3 trial in 113 Japanese patients, treatment with risankizumab 75 or 150 mg q12w resulted in sustained efficacy over 52 weeks, during which only three (2.7%) patients discontinued treatment with risankizumab due to AEs [26]. In a multinational, phase 3, double-blind, placebo-controlled trial, 111 patients treated with risankizumab 150 mg q12w for 88 weeks displayed sustained clinical efficacy and only 4 (3.6%) patients discontinued because of AEs [19]. Finally, in the predecessor to the current trial, only one patient in each of the 18 mg (single-dose) and 90 mg (q12w) risankizumab groups (2.3 and 2.4%, respectively), and no patients in the 180 mg q12w group, discontinued treatment because of AEs through 48 weeks of treatment [21].
The major strength of this study was the long duration (48 weeks), resulting in up to 96 weeks of observation when considering the predecessor study. However, the results are limited by the open-label design, the lack of a control group for comparisons, and the relatively small sample size. Additionally, patients in this study were not treated with risankizumab doses that are currently approved for clinical use.
Conclusion
In conclusion, the results from this 48-week OLE trial reveal that risankizumab provides sustained clinical efficacy during long-term treatment, with clinical outcomes that indicate sustained efficacy over time and a trend toward continued improvement over time. These results also demonstrate that risankizumab has good tolerability when used as a long-term therapy as no new safety signals were observed and no patients discontinued treatment because of AEs.
References
Parisi R, Symmons DP, Griffiths CE, et al. Identification and Management of Psoriasis and Associated ComorbidiTy (IMPACT) project team. Global epidemiology of psoriasis: a systematic review of incidence and prevalence. J Investig Dermatol. 2013;133(2):377–85.
Lebwohl MG, Bachelez H, Barker J, et al. Patient perspectives in the management of psoriasis: results from the population-based Multinational Assessment of Psoriasis and Psoriatic Arthritis Survey. J Am Acad Dermatol. 2014;70(5):871-81.e30.
Chandran V, Raychaudhuri SP. Geoepidemiology and environmental factors of psoriasis and psoriatic arthritis. J Autoimmun. 2010;34(3):J314–21.
Ni C, Chiu MW. Psoriasis and comorbidities: links and risks. Clin Cosmet Investig Dermatol. 2014;7:119–32.
Boehncke WH, Schon MP. Psoriasis. Lancet. 2015;386(9997):983–94.
Di Meglio P, Villanova F, Nestle FO. Psoriasis. Cold Spring Harb Perspect Med. 2014;4(8):a015354.
Bilal J, Berlinberg A, Bhattacharjee S, et al. A systematic review and meta-analysis of the efficacy and safety of the interleukin (IL)-12/23 and IL-17 inhibitors ustekinumab, secukinumab, ixekizumab, brodalumab, guselkumab and tildrakizumab for the treatment of moderate to severe plaque psoriasis. J Dermatolog Treat. 2018;29(6):569–78.
Blauvelt A, Chiricozzi A, Springer US. The immunologic role of IL-17 in psoriasis and psoriatic arthritis pathogenesis. Clin Rev Allergy Immunol. 2018;55(3):379–90.
Menter A, Feldman SR, Weinstein GD, et al. A randomized comparison of continuous vs. intermittent infliximab maintenance regimens over 1 year in the treatment of moderate-to-severe plaque psoriasis. J Am Acad Dermatol. 2007;56(1):31.e1-15.
Papp K, Menter A, Poulin Y, et al. Long-term outcomes of interruption and retreatment vs. continuous therapy with adalimumab for psoriasis: subanalysis of REVEAL and the open-label extension study. J Eur Acad Dermatol Venereol. 2013;27(5):634–42.
Tyring S, Gordon KB, Poulin Y, et al. Long-term safety and efficacy of 50 mg of etanercept twice weekly in patients with psoriasis. Arch Dermatol. 2007;143(6):719–26.
Gaffen SL, Jain R, Garg AV, et al. The IL-23-IL-17 immune axis: from mechanisms to therapeutic testing. Nat Rev Immunol. 2014;14(9):585–600.
Puig L. The role of IL 23 in the treatment of psoriasis. Expert Rev Clin Immunol. 2017;13(6):525–34.
Hawkes JE, Chan TC, Krueger JG. Psoriasis pathogenesis and the development of novel targeted immune therapies. J Allergy Clin Immunol. 2017;140(3):645–53.
Krueger JG, Ferris LK, Menter A, et al. Anti-IL-23A mAb BI 655066 for treatment of moderate-to-severe psoriasis: safety, efficacy, pharmacokinetics, and biomarker results of a single-rising-dose, randomized, double-blind, placebo-controlled trial. J Allergy Clin Immunol. 2015;136(1):116-24.e7.
Singh S, Kroe-Barrett RR, Canada KA, et al. Selective targeting of the IL23 pathway: generation and characterization of a novel high-affinity humanized anti-IL23A antibody. MAbs. 2015;7(4):778–91.
Gordon KB, Strober B, Lebwohl M, et al. Efficacy and safety of risankizumab in moderate-to-severe plaque psoriasis (UltIMMa-1 and UltIMMa-2): results from two double-blind, randomised, placebo-controlled and ustekinumab-controlled phase 3 trials. Lancet. 2018;392(10148):650–61.
Reich K, Gooderham M, Thaçi D, et al. Risankizumab compared with adalimumab in patients with moderate-to-severe plaque psoriasis (IMMvent): a randomised, double-blind, active-comparator-controlled phase 3 trial. Lancet. 2019;394(10198):576–86.
Blauvelt A, Leonardi CL, Gooderham M, et al. Efficacy and safety of continuous risankizumab therapy vs treatment withdrawal in patients with moderate to severe plaque psoriasis: a phase 3 randomized clinical trial. JAMA Dermatol. 2020;156(6):649–58.
McKeage K, Duggan S. Risankizumab: first global approval. Drugs. 2019;79(8):893–900.
Papp KA, Blauvelt A, Bukhalo M, et al. Risankizumab versus ustekinumab for moderate-to-severe plaque psoriasis. N Engl J Med. 2017;376(16):1551–60.
Strober BE. Why biologic therapies sometimes lose efficacy. Semin Cutan Med Surg. 2016;35(4 Suppl 4):S78–80.
Jabbar-Lopez ZK, Yiu ZZN, Ward V, et al. Quantitative evaluation of biologic therapy options for psoriasis: a systematic review and network meta-analysis. J Invest Dermatol. 2017;137(8):1646–54.
Megna M, Fabbrocini G, Ruggiero A, et al. Efficacy and safety of risankizumab in psoriasis patients who failed anti-IL-17, anti-12/23 and/or anti IL-23: preliminary data of a real-life 16-week retrospective study. Dermatol Ther. 2020;33:e14144.
Warren RB, Blauvelt A, Poulin Y, et al. Efficacy and safety of risankizumab vs. secukinumab in patients with moderate-to-severe plaque psoriasis (IMMerge): results from a phase III, randomized, open-label, efficacy-assessor-blinded clinical trial. Br J Dermatol. 2021;184(1):50–9. https://doi.org/10.1111/bjd.19341.
Ohtsuki M, Fujita H, Watanabe M, et al. Efficacy and safety of risankizumab in Japanese patients with moderate to severe plaque psoriasis: results from the SustaIMM phase 2/3 trial. J Dermatol. 2019;46(8):686–94.
Acknowledgements
AbbVie and the authors thank the patients who participated in this clinical trial and all the study investigators for their contributions.
Funding
Boehringer Ingelheim participated in the study design and analysis of data. AbbVie participated in the analysis and interpretation of data and writing, reviewing, and approval of this publication. All authors had access to the data and participated in the development, review, and approval of this manuscript and in the decision to submit this manuscript to Dermatology and Therapy for consideration. AbbVie and Boehringer Ingelheim funded the research for this study. The journal’s Rapid Service Fee was funded by AbbVie.
Medical Writing Assistance
Medical writing assistance, funded by AbbVie, was provided by Nate Connors, PhD, ISMPP CMPP™, and Jennifer C Jaworski, MS, of JB Ashtin.
Authorship
All named authors meet the International Committee of Medical Journal Editors (ICMJE) criteria for authorship for this article, take responsibility for the integrity of the work as a whole, and have given their approval for this version to be published.
Authorship Contributions
All the authors critically contributed to data acquisition, reviewed this manuscript, and gave final approval for publication. KAP, MF, SKT contributed to the study design. KAP, SdV, SKT contributed to data acquisition. JZ contributed to statistical analysis.
Disclosures
Kim A. Papp is a(n) advisor/speaker/consultant/steering committee member/researcher for AbbVie, Akros, Allergan, Amgen, Anacor, Arcutis, Astellas, AstraZeneca, Bausch Health (Valeant), Baxalta, Baxter, Boehringer Ingelheim, Bristol-Myers Squibb, Can-Fite, Celgene, Coherus, Dermira, Dow, Forward Pharma, Galderma, Genentech, Gilead, GlaxoSmithKline, InflaRx, Janssen, Kyowa-Hakko Kirin, LEO Pharma, Lilly, MedImmune, Meiji Seika Pharma, Merck, Mitsubishi Pharma, Moberg Pharma, Novartis, Pfizer, Regeneron, Roche, Sanofi Genzyme, Takeda, and UCB. Saskia de Vente, Jiewei Zeng, Byron Padilla are full-time employees of AbbVie and may hold AbbVie stock or stock options. Mary Flack is a full-time employee of Boehringer Ingelheim. Stephen K. Tyring is an investigator for AbbVie, Bausch Health (Valeant), Boehringer Ingelheim, Celgene, Coherus, Dermira, Lilly, Janssen, LEO Pharma, Merck, Novartis, Ortho Dermatologics, Pfizer, Regeneron-Sanofi, and Sun Pharma; a speaker for AbbVie, Bausch Health (Valeant), Janssen, LEO Pharma, Lilly, Novartis, Pfizer, and Regeneron-Sanofi; and a consultant for AbbVie, Janssen, Novartis, and UCB.
Compliance with Ethics Guidelines
This study was conducted in accordance with the Good Clinical Practice Guideline as defined by the International Conference on Harmonisation, the Declaration of Helsinki, and/or all applicable federal and local regulations and the following institutional review boards: Institutional Review Board Services, Aurora, Ontario, Canada; Research Review Board Inc., Richmond Hill, Ontario, Canada; CPP- Ile de France IV, Hôpital Saint-Louis, Paris, France; Ethikkommission des Landes Berlin, Landesamt fur Gesundheit und Soziales, Berlin, Germany; Chesapeake IRB, Columbia, Maryland, USA. All patients provided informed consent and signed approved consent forms prior to any study procedures in accordance with Good Clinical Practice and local legislation.
Data Availability
AbbVie is committed to responsible data sharing regarding the clinical trials we sponsor. This includes access to anonymized, individual, and trial-level data (analysis data sets), as well as other information (e.g., protocols and Clinical Study Reports), as long as the trials are not part of an ongoing or planned regulatory submission. This includes requests for clinical trial data for unlicensed products and indications. This clinical trial data set can be requested by any qualified researchers who engage in rigorous, independent scientific research and will be provided following review and approval of a research proposal and Statistical Analysis Plan (SAP) and execution of a Data Sharing Agreement (DSA). Data requests can be submitted at any time and the data will be accessible for 12 months, with possible extensions considered. For more information on the process, or to submit a request, visit the following link: https://www.AbbVie.com/our-science/clinical-trials/clinical-trials-data-and-information-sharing/data-and-information-sharing-with-qualified-researchers.html.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Papp, K.A., de Vente, S., Zeng, J. et al. Long-Term Safety and Efficacy of Risankizumab in Patients with Moderate-to-Severe Chronic Plaque Psoriasis: Results from a Phase 2 Open-Label Extension Trial. Dermatol Ther (Heidelb) 11, 487–497 (2021). https://doi.org/10.1007/s13555-021-00490-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13555-021-00490-3