
Abstract
In this paper, gold nanorods’ (GNRs) interaction with different proteins (i.e. carbonic anhydrase, lysozyme, ovalbumin and bovine serum albumin (BSA)) at physiological pH is investigated using localized surface plasmon resonance (LSPR) spectroscopy. We observe that the incubation of these proteins at different concentrations with cetyltrimethylammonium bromide-capped GNRs of three aspect ratios induces dramatic changes in the extinction spectra of the nanoparticles. In particular, we correlate the position and shape of the longitudinal LSPR peaks to the ability of the proteins to specifically interact with GNRs’ surface. The different types of behaviour observed are explained by the exposed molecular surface area of the proteins’ cysteine residues as modelled on the basis of their respective X-ray crystallographic data structures. Cysteine is the only amino acid that exhibits an SH group that is well known to have a strong affinity to gold. The presence and the accessibility of such a residue may explain the protein binding to GNRs. The isoelectric point of the proteins is also an important characteristic to take into account, as the electrostatic strength between GNRs and protein explains some of the cases where aggregates are formed.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
During the last decades when nanosciences have made their revolution, gold nanoparticles (GNPs) have attracted particular interests for many reasons. Among them, two have drawn our attention in the present study. First, their genuine optical properties make them of prime choice for many applications especially in medicine and biology [1, 2]. Second, they are highly stable in many liquid or gaseous environments, which makes them very versatile. GNPs provide high contrast in cellular and tissue imaging using confocal reflectance microscopy [3] or dark-field imaging [4]. They have been used in photothermal therapy of cancer [5]. Their optical relevance is mainly due to the localized surface plasmon resonance (LSPR) emerging in the visible spectral range for GNPs with size between ~2 to ~200 nm. Moreover, LSPR can be finely tuned by changing the GNPs size, environment or shape. The literature is rich in articles describing protocols to synthesize a wide range of GNPs of different shapes and geometries such as spherical particles, nanorods, nanoshells, nanostars and nanocages [6, 7]. Interestingly, a non-spherical nanoparticle will present several resonance characteristics of its shape [8, 9]. Gold nanorods (GNRs) are in this aspect, highly studied because their LSPR can be tuned on a wider wavelength scale. The aspect ratio of GNRs is also easily deduced from their absorption spectra. The LSPR sensitivity to the nanoparticle close environment makes GNPs good candidates for sensor use.
GNRs are more and more often used in biology and for biomedical applications. Reports are found on their use in biosensing, bio-imaging, photothermal therapy or even in theranostic [10–13]. The common point of all these works is the necessity to attach the appropriate biomolecules to the GNRs, in order for it to accomplish the task it was designed for. For this, several routs can be considered, namely ligand exchange, electrostatic interaction, biofunctional linkage and surface coating. All these techniques are described in detail in [14, 15]. Giving the increasing interest of scientist from all disciplines for working with these tremendous nanostructures in a bio-environment, it is of crucial importance to understand how they directly interact with biomolecules.
In the present study, cetyltrimethylammonium bromide (CTAB)-capped GNRs were synthesized using the seed-mediated growth method in order to study their direct interaction with four different proteins (i.e. carbonic anhydrase, lysozyme, ovalbumin and bovine serum albumin (BSA)). GNRs represent a unique class of metallic nanostructures with two well-defined LSPR bands in the visible/near infrared spectral range corresponding to electronic oscillations along the short and long axis of nanorods. In a previous work [16], we already showed that the position of these two LSPR bands is not dependent on the molecular weight of the proteins interacting with the GNRs surface. In this context, we are interested in investigating the interaction of the above mentioned proteins at physiological pH with GNRs as function of their concentrations using LSPR spectroscopy.
Experimental
Reagents
Tetrachloroauric(III) acid (HAuCl4), CTAB and ascorbic acid were purchased from Aldrich. Sodium borohydride (NaBH4, 99 %) and silver nitrate (AgNO3) were obtained from Merck. Lysozyme from chicken egg white (ref. 62970), carbonic anhydrase from bovine erythrocytes (86.5 %, ref. c3934) and albumin from bovine serum (BSA ref. a8806, 96 %) were purchased from Sigma-Aldrich. Ovalbumin (ref. W377206) has been provided by GE Healthcare. All proteins were dissolved in distilled water at concentrations ranging from 10−10 to 10−4 M. Table 1 gives the main characteristics of the proteins used.
Synthesis of GNRs
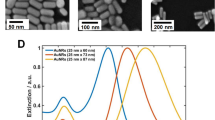
GNRs were synthesized utilizing the seed-mediated growth method described in details in [16]. The UV–visible spectra of each set of GNRs are presented on Fig. 1. The shift of the spectra is explained by the fact that the three solutions contain GNRs with different aspect ratios.

Normalized LSPR spectra of the three types of synthesized GNRs
Preparation of proteins/GNRs conjugates
All protein solutions were prepared in distilled water at pH = 7 ± 0.5. Six concentrations of protein solutions were prepared (100 pM, 1 nM, 10 nM, 100 nM, 1 μM, 10 μM and 100 μM) except for BSA for which just 1 nM, 100 nM and 10 μM solutions were prepared. Then, 250 μl of pure GNR solutions were mixed with 400 μl of the protein solution at different concentration values. The samples were incubated at room temperature for 20 min before the measurement.
Instrumentation
The interaction between CTAB-capped GNRs and the different proteins was characterized by extinction spectroscopy. Spectra were recorded using a Kontron Uvikon 941 spectrophotometer in the range of 400 to 800 nm with 1 nm step and 1 nm resolution. The extinction spectra were fitted using the “multipeak fit” tool on Origin® with a Lorentzian type.
The GNRs morphology was analysed with conventional transmission electron microscopy (TEM) using a JEOL 100 U type TEM microscope operated at 100 kV accelerating voltage. The ζ potential of the colloidal solutions was measured using a particle analyser (Nano ZS90 Zetasizer, Malvern Instruments) equipped with a He–Ne laser (633 nm, 5 mW) and a measurement angle of 90°. Each sample was measured three times, and the mean value was reported.
The solvent molecular surface area (MSA) of the proteins was estimated using a solvent probe of 1.4 Å; numerical results were obtained using GetArea [17] and graphically confirmed using Jmol (http://jmol.sourceforge.net).
Results
Figure 1 illustrates the normalized extinction spectra of the as-synthesized GNRs. The spectral position of transversal LSPR band and the longitudinal LSPR bands at 639, 662 and 686 nm correspond to GNRs with aspect ratios of 2.1 (average length/width, 46/22), 2.52 (average length/width, 43/17 nm) and 2.8 (average length/width, 42/15 nm), respectively, as given by TEM images (see inset in Fig. 1). In the following, data will be presented showing the interaction of proteins with one set of the GNR. All the experiences were repeated at least once with another set of GNR, and the results were reproducible. Herein, we will mainly focus on the study of the longitudinal LSPR (i.e. width and position) to explain the mechanism of GNRs/proteins interaction.
Figure 2 presents the modifications of the longitudinal LSPR peak of GNRs in the presence of different concentrations of proteins. Upon incubation of GNRs with carbonic anhydrase (Fig. 2a) and lysozyme (Fig. 2b) protein solutions, the positions of the longitudinal LSPR peaks vary as function of protein concentration. Moreover, we observe that the width of the bands is constant, indicating a good dispersion of the nanoparticles in the solution. On the opposite, dramatic changes of the longitudinal LSPR bands were observed after incubation of the colloidal solution with ovalbumin (Fig. 2c) and BSA (Fig. 2d) solutions. In the case of ovalbumin (Fig. 2c), the protein seems to interact with GNRs’ surface in the same manner as carbonic anhydrase or lysozyme except at 100 nM and 1 μM. At these concentrations, the intensity of plasmonic band suddenly decreases together with its broadening, clearly suggesting the aggregation of nanoparticles. Similarly, this phenomenon was also induced by BSA protein at the concentrations of 1 and 100 nM (Fig. 2d). Taking into account that the spectral position of the LSPR band is strongly dependent on the dielectric function of GNRs’ local environment, the modification of its position together with its broadening indicate the extent of individual or aggregated nanoparticles in colloidal solution.

Longitudinal LSPR band of synthesized GNRs in the absence and presence of: a carbonic anhydrase, b lysozyme, c ovalbumin and d BSA of different concentrations
In order to study these spectral modifications, we have fitted the longitudinal LSP band with Lorentzian curves. As it reveals on Fig. 2c, d, the widest LSP were fitted with two Lorentzian when only one was needed for the unaltered LSP. The experimental recorded curve reflects a composite plasmonic resonances of (1) GNRs interacting with the protein as mentioned above (individual mode) and (2) protein-induced aggregation of GNRs (aggregation mode).
To understand the mechanism underlying these two modes, Fig. 3 was plotted. For this, the shift between the highest LSPR (aggregation mode if it exists, otherwise the normal mode) and the pure GNRs is measured and then plotted. Giving the resolution of the spectrometer, there is no observable shift for carbonic anhydrase. On the opposite, the GNRs’ individual mode in presence of lysozyme and ovalbumin proteins, respectively, displays a red shift for a high enough concentration (~10 nM), indicating an increase of the dielectric constant of the GNRs surrounding medium. In other words, a material with higher dielectric constant than CTAB has replaced or at least attached to the surfactant molecules. Obviously, this material can only be the proteins.

Longitudinal LSPR peak shift versus protein concentration. The longitudinal LSPR shift is calculated by comparing the position of the LSPR band of pure GNRs solution with the position of the corresponding band of conjugates
The extent of this red shift varies in a specific way. It is constant or slightly decreases as the protein concentrations increase until it reaches a minimum value at 10 nM for lysozyme and 1 nM for ovalbumin. For higher concentration, the extent of the shift increases again. We believe that this behaviour highlights some aspect of the GNRs/protein interactions as it is discussed in the next section.
Discussion
We explain the behaviour of the proteins toward GNRs by exploiting the available data on their electric charges and structures. The electric charge of the proteins at the working pH was obtained from their isoelectric point (Table 1). The electrostatic interaction between proteins and GNRs was studied, taking into account that the GNRs are capped with CTAB, which makes them positively charged (the ζ potential was measured to be +20 mV).
The structure of the proteins was gathered from the RCSB Protein Data Bank©. The structure of the proteins studied here was obtained from X-ray crystallographic data [18–21]. The cysteine (Cys) residue is the centre of our interest since it is the only amino acid which owns a thiol group. In chemistry, thiols are compounds which are very famous to demonstrate strong affinity to gold. In the presence of thiols, strong sulphur–gold bonds are formed, which are renowned to be strong covalent bonds. Previous studies [22, 23] on GNPs/protein interactions have shown that the binding of protein to a very small GNP depends firstly on the existence of a Cys inside the protein and secondly, on the accessibility of the thiol group of the Cys residue. Herein, the sulphur accessibility is estimated via the notion of MSA [24]. This is defined as the contact surface area of a solvent probe sphere that rolls along the surface of the protein. Using the software GetArea, we have calculated for each Cys residue, its MSA and the surface energy of the corresponding sulphur. The idea here is to measure the availability of the sulphur to bind to the GNRs. The same approach was followed using Jmol. Figure 4 shows the MSA (red dots) for a 1.4-Å sphere rolled on the protein surface.

CPK view of a carbonic anhydrase, b lysozyme, c ovalbumin and d BSA. The red dots materialized the MSA; the Cys residues are coloured in green and the sulphur atoms in yellow
In particular, carbonic anhydrase has only one Cys residue which is located inside its volume, making the sulphur non-accessible for binding with GNRs. Thus, the only existing interaction between carbonic anhydrase and GNRs is through electrostatic forces. As the carbonic anhydrase is negatively charged at the working pH of 7, it can only bind to positively charged structures, here, the CTAB-capped GNRs. We suppose that carbonic anhydrase can only weakly adsorb on the CTAB double layer and not on the GNRs’ surface. This explains that no shift is observed for carbonic anhydrase on Fig. 3a: the GNRs environment does not change and so its longitudinal LSPR. However, small deviations can occur, that are probably due to the imperfection of the CTAB capping. Especially, it is known that the CTAB double layer is sparse at the GNRs extremities.
As mentioned above, in the case of lysozyme and ovalbumin, the behaviour of individual mode is quite different. For both proteins, a red shift is observed for all the concentrations. If the proteins were just electrostatically adsorbed to the CTAB double layer, we would observe no shift just as for carbonic anhydrase. Thus, we can conclude that lysozyme and ovalbumin replace CTAB. We also observe for both proteins that the red shift extent first decreases until a specific concentration before it increases again. The CTAB bilayer surrounding the GNRs is most probably in competition with the protein. As shown in Table 2 and Fig. 4, lysozyme and ovalbumin have one accessible thiol. It is known that the functionalization of CTAB-capped GNR with thiolated molecules is trivially made by the sulphur–gold interaction that overcomes the weak binding of CTAB to gold [14]. The consequence of the addition of lysozyme (respectively ovalbumin) is the binding of the protein to gold GNR and the removal of CTAB. We observe here that a minimum value of the concentration has to be reached before proteins replace most of the CTAB (when the red shift extent increases).
If the first part of the LSPR, shift is similar for lysozyme and ovalbumin; the latter shows two main specificities. First of all, in the case of ovalbumin, the lowest red shift of the LSPR band occurs for a lower concentration than for lysozyme. Contrary to lysozyme, ovalbumin is negatively charged in the experimental conditions (see Table 1). As the GNRs are strongly positively charged, the electrostatic attraction forces are added to the thiol attraction. The result is an easier attachment of the proteins to the GNRs.
The second point to rise is the aggregation taking place at 100 nM and 1 μM (Fig. 3b). Once again, we explain this behaviour by the surface charge of ovalbumin. Electrostatic attraction forces act on several GNRs at the same time allowing the formation of aggregates. This is why the ovalbumin/GNRs ratio has to be very well controlled to avoid charge neutralization and hence aggregation. Many studies have already shown that a stabilized attachment of proteins to GNPs thanks to electrostatic interaction required an optimization of both the pH and the concentration of the solutions [14, 25, 26].
The case of BSA has in common with ovalbumin that it is negatively charge at pH 7 and thus is electrostatically attracted to the CTAB-capped GNRs. Nevertheless, BSA has also the characteristic of containing quite a lot of Cys residues (see Table 2). Among them, only five are solvent accessible, and among these five, two sulphur have enough surface energy to bind to gold surface. Not only BSA has twice more accessible sulphur than lysozyme or ovalbumin but these two sulphurs (in Cys 200 and Cys 559) are located on opposite sides of the protein (Fig. 4d). This way, one BSA can bind to two different GNRs. This is how very strong aggregates are formed.
The longitudinal LSPR is becoming closer to the same band of pure GNRs as the BSA concentration increases. The origin of this can be the fact that a strong excess of BSA in the presence of CTAB is known to form aggregates of proteins [27]. The concentration of BSA actually interacting with GNRs is thus much lower than the nominal concentration. Instead of increasing, the number of BSA interacting with the GNRs is decreasing, explaining the reduction of the longitudinal LSPR shift.
Conclusion
In this article, we have investigated the interaction of GNRs with carbonic anhydrase, lysozyme, ovalbumin, and BSA regarding the proteins concentration using LSPR spectroscopy. In a previous study [16], we have already demonstrated that the longitudinal LSPR shift is the most significant for the understanding of GNRs/protein interaction. In the same reference, we established the fact that the molecular weight of the proteins is not a relevant parameter to characterize this interaction. In the present letter, we have considered the protein concentration, isoelectric point and the MSA to explain the experimental results. The conclusion is that the number of accessible sulphurs is of crucial importance to explain the shape of the longitudinal LSPR shift evolution with the concentration. If there are too many, like in the case of BSA, strong aggregates are formed. If there is only one, the evolution of the longitudinal LSPR with the concentration presents two parts: the first one decreases and the second one increases. Then, the isoelectric point gives insight of the electric charge of the protein and explains some of the GNRs/proteins aggregation cases. Carbonic anhydrase which has no accessible sulphur and is negatively charged barely binds to GNRs. In comparison, ovalbumin has one accessible sulphur and is negatively charged; it binds to GNRs and in some conditions can even form electrostatic aggregates.
Liu et al. [28] conducted a similar study in 2012 using dynamic light scattering (DLS) instead of extinction spectroscopy. They come to the same conclusion as us, concerning the interaction between GNRs and BSA or human serum albumin, and they show that immunoglobulin (IgG and IgA) follow the same behaviour as ovalbumin. However, they fail to explain the difference of behaviour between these proteins. When looking at the available sulphur in IgG or IgA (Table 2) [29], we found that the three of them are in the same location (bottom of Fab and top of Fc). Steric interaction most probably cancels the duplicity of thiols, so they behave as there was only one accessible sulphur. This is in complete agreement with the explanation developed in our study.
References
El-Sayed IH, Huang X, El-Sayed M (2005) Surface plasmon resonance scattering and absorption of anti-EGFR antibody conjugated gold nanoparticles in cancer diagnostics: applications in oral cancer. Nano letters 5:829–834. doi:10.1021/nl050074e
Aslan K, Lakowicz JR, Geddes CD (2005) Plasmon light scattering in biology and medicine: new sensing approaches, visions and perspectives. Current opinion in chemical biology 9:538–544. doi:10.1016/j.cbpa.2005.08.021
Aaron J, Nitin N, Travis K et al (2007) Plasmon resonance coupling of metal nanoparticles for molecular imaging of carcinogenesis in vivo. Journal of biomedical optics 12:034007. doi:10.1117/1.2737351
Liu GL, Yin Y, Kunchakarra S et al (2006) A nanoplasmonic molecular ruler for measuring nuclease activity and DNA footprinting. Nature nanotechnology 1:47–52. doi:10.1038/nnano.2006.51
Li W, Cai X, Kim C et al (2011) Gold nanocages covered with thermally-responsive polymers for controlled release by high-intensity focused ultrasound. Nanoscale 3:1724–1730. doi:10.1039/c0nr00932f
Chen J, Saeki F, Wiley BJ et al (2005) Gold nanocages: bioconjugation and their potential use as optical imaging contrast agents. Nano letters 5:473–477. doi:10.1021/nl047950t
Geddes CD (ed) (2012) Reviews in Plasmonics 2010. Springer, New York. doi:10.1007/978-1-4614-0884-0
Hu M, Chen J, Li Z-Y et al (2006) Gold nanostructures: engineering their plasmonic properties for biomedical applications. Chemical Society reviews 35:1084–1094. doi:10.1039/b517615h
Sun Y, Xia Y (2003) Gold and silver nanoparticles: a class of chromophores with colors tunable in the range from 400 to 750 nm. Analyst 128:686. doi:10.1039/b212437h
Chen C-D, Cheng S-F, Chau L-K, Wang CRC (2007) Sensing capability of the localized surface plasmon resonance of gold nanorods. Biosensors & bioelectronics 22:926–932. doi:10.1016/j.bios.2006.03.021
Yong K-T, Swihart MT, Ding H, Prasad PN (2009) Preparation of gold nanoparticles and their applications in anisotropic nanoparticle synthesis and bioimaging. Plasmonics 4:79–93. doi:10.1007/s11468-009-9078-2
Huang X, El-Sayed IH, Qian W, El-Sayed MA (2006) Cancer cell imaging and photothermal therapy in the near-infrared region by using gold nanorods. Journal of the American Chemical Society 128:2115–2120. doi:10.1021/ja057254a
Young JK, Figueroa ER, Drezek RA (2012) Tunable nanostructures as photothermal theranostic agents. Annals of biomedical engineering 40:438–459. doi:10.1007/s10439-011-0472-5
Huang X, Neretina S, El-Sayed MA (2009) Gold nanorods: from synthesis and properties to biological and biomedical applications. Adv Mater 21:4880–4910. doi:10.1002/adma.200802789
Joshi PP, Yoon SJ, Hardin WG et al (2013) Conjugation of antibodies to gold nanorods through Fc. Synthesis and Molecular Specific Imaging. Bioconjugate chemistry, Portion. doi:10.1021/bc3004815
Cottat M, Thioune N, Gabudean A-M et al (2012) Localized surface plasmon resonance (lspr) biosensor for the protein detection. Plasmonics 8:699–704. doi:10.1007/s11468-012-9460-3
Fraczkiewicz R, Braun W (1998) Exact and efficient analytical calculation of the accessible surface areas and their gradients for macromolecules. J Comput Chem 19(3):319–333
Diamond R (1974) Real-space refinement of the structure of hen egg-white lysozyme. J Mol Biol 82:371–391
Eriksson AE, Jones TA, Liljas A (1988) Refined structure of human carbonic anhydrase II at 2.0 A resolution. Proteins 4:274–282. doi:10.1002/prot.340040406
Stein PE, Leslie AGW, Finch JT, Carrell RW (1991) Crystal structure of uncleaved ovalbumin at 1 · 95 Å resolution. J Mol Biol 221:941–959
Bhattacharya A, Grüne T, Curry S (2000) Crystallographic analysis reveals common modes of binding of medium and long-chain fatty acids to human serum albumin. Journal of molecular biology 303:721. doi:10.1006/jmbi.2000.4158
Lidgi-Guigui N, Leung C, Palmer RE (2008) Weak precursor state binding of protein molecules to size-selected gold nanoclusters on surfaces. Surf Sci 602:1006–1009. doi:10.1016/j.susc.2007.12.025
Prisco U, Leung C, Xirouchaki C et al (2005) Residue-specific immobilization of protein molecules by size-selected clusters. Journal of the Royal Society, Interface / the Royal Society 2:169–175. doi:10.1098/rsif.2005.0032
Richards FM (1977) Areas, volumes, packing and protein structure. Annual review of biophysics and bioengineering 6:151–176. doi:10.1146/annurev.bb.06.060177.001055
Pissuwan D, Valenzuela SM, Killingsworth MC et al (2007) Targeted destruction of murine macrophage cells with bioconjugated gold nanorods. J Nanoparticle Res 9:1109–1124. doi:10.1007/s11051-007-9212-z
Geoghegan WD, Ackerman GA (1977) Adsorption of horseradish peroxidase, ovomucoid and anti-immunoglobulin to colloidal gold for the indirect detection of concanavalin A, wheat germ agglutinin and goat anti-human immunoglobulin G on cell surfaces at the electron microscopic level: a new method, theory and application. Journal of Histochemistry & Cytochemistry 25:1187–1200. doi:10.1177/25.11.21217
Sharma A, Pasha JM, Deep S (2010) Effect of the sugar and polyol additives on the aggregation kinetics of BSA in the presence of N-cetyl-N, N, N-trimethyl ammonium bromide. Journal of colloid and interface science 350:240–248. doi:10.1016/j.jcis.2010.06.054
Liu H, Pierre-Pierre N, Huo Q (2012) Dynamic light scattering for gold nanorod size characterization and study of nanorod–protein interactions. Gold Bull 45:187–195. doi:10.1007/s13404-012-0067-4
Harris LJ, Larson SB, Hasel KW, Mcpherson A (1997) Refined structure of an intact IgG2a monoclonal antibody. Biochemistry 36(7):1581–97
Acknowledgments
This work was supported by the Programme Hubert Curien Brancusi, by the ANCS, project number PN II Capacitati/Brancusi, 489/2011 and by the Nanoantenna European project (FP7-HEALTH-F5-2009 241818)
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
About this article
Cite this article
Thioune, N., Lidgi-Guigui, N., Cottat, M. et al. Study of gold nanorods–protein interaction by localized surface plasmon resonance spectroscopy. Gold Bull 46, 275–281 (2013). https://doi.org/10.1007/s13404-013-0118-5
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13404-013-0118-5