
Abstract
Au, NiO, and NiO/Au clusters of 2.5–16 nm, supported on Al2O3, ZrO2, TiO2, and ZnO, were studied in the purification of ethene feedstock from ethyne by hydrogenation at 357 K. The Au, NiO, and NiO/Au catalysts possessed 100 % selectivity to ethene. As the size of NiO clusters decreased from 7 to 3 nm, the turnover frequency (TOF) decreased from 812–1,023 to 276 h−1. In contrast with NiO, Au activity increased with decreasing particle size. NiO/Au catalysts possessed higher stability and activity in comparison with Au and NiO catalysts. The synergistic gain on NiO/Au clusters (SG) calculated as TOFNiO/Au – TOFAu – TOFNiO was 1,466; 1,147; 563; and 569 h−1 for NiO/Au/Al2O3, NiO/Au/TiO2, NiO/Au/ZnO, and NiO/Au/ZrO2, respectively. The reasons of the observed catalytic trends and the origin of the most active and selective sites are discussed.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Ethene feedstock for polymerization coming from the stage of hydrocarbons cracking contains 0.5–3 % of ethyne compounds [1, 2]. Even such a small content of ethyne impurities quickly poisons Ziegler–Natta polymerization catalysts [2, 3]. Thus, preliminary removal of alkynes via hydrogenation is necessary [1–4]. Traditionally, the Ag/Pd catalysts HO-21 (BASF), G-58, and G-83 (Süd-Chemie) are employed for the hydrogenation of С≡С bond to C=C bond. However, turnover frequency (TOF) and stability of Ag/Pd catalysts are often low. Moreover, undesirable “green-oil” and ethane by-products are usually formed on palladium surface [3, 4]. Nowadays, the studies directed to improving of catalytic properties of palladium-based catalysts are becoming less popular, giving way to the intensive searching for alternative and high-performance palladium-free catalysts for the ethyne hydrogenation [1–5].
Nano-sized gold is an effective catalyst for oxidation [6], isomerization [7], cyclization [8], hydrodechlorination [9], and other reactions [10, 11]. Composites based on bare gold clusters demonstrate high activity and/or selectivity in hydrogenation of 1,3-butadiene [2], 1-propyne [12], and ethynylbenzene [13] under the mild conditions. Bimetallic gold-nickel catalysts also may possess the enhanced catalytic properties in conversion of different hydrocarbons: n-butane [14], allylbenzene [5, 7], and 2,4-dichloropfenol [9]. Thus, the application of gold and gold-nickel clusters in selective hydrogenation is very promising.
This work is an extension of studies of the catalytic properties of Au and NiO/Au clusters in the hydrogenation of ethyne derivatives [13, 15]. It was reported that Au/Al2O3 catalysts possess high activity (up to 0.142 s−1) in the hydrogenation of ethynylbenzene into styrene at 423 K. A synergistic effect was revealed for NiO/Au/Al2O3 in the hydrogenation of ethyne into ethene [15]: The conversion of ethyne on the NiO/Au catalysts was higher than the sum of conversions on Au and Ni catalysts. It was found that synergistic gain on NiO/Au clusters was maximized at 357 K, and Au/Ni ratio equaled 1:1 [15]. According to the analysis of oxidation state of metals in gold-contained catalysts [7], it was proposed that synergistic effect could be caused by the formation of new Auδ+ catalytic sites. The goals of the present research were: (1) to study the regularities of catalytic action of Au and NiO/Au clusters (Au/Ni=1:1), supported on Al2O3, TiO2, ZrO2, and ZnO, in the hydrogenation of ethyne–ethene mixture at 357 K; (2) to study the impact of clusters shape and size to the catalysis by gold, and (3) to determine the origin of the active gold sites by density functional theory (DFT).
Experimental
Al2O3 (“Katalizator,” АОК-63-11) with a 160 m2/g surface area and an isoelectric point (IEP) equal to 7.0, TiO2 (“Degussa,” Aerolyst®7710, 50 m2/g, 6.0 IEP), ZrO2 (Degussa, Aerolyst®6100, 30 m2/g, 4.5 IEP), and ZnO (Degussa, Aerolyst®9000, 36 m2/g, 10.0 IEP) were used as the supports for metal clusters. Au/supports and Pd/Al2O3 were produced by deposition–precipitation [15, 16]. In typical synthesis, 50 ml of the aqueous solution of HAuCl4 (or PdCl2) with 5.8 × 10−6 g/ml metal content was adjusted to pH equal to the IEP of the support by adding the aqueous solution of NaOH (0.1 M). Then 1 g of the support was dispersed in the solution with stirring for 1 h at 307 K. The precursor obtained was washed to remove NaCl. The degree of NaCl removing was controlled via AgNO3 test. Then the sample was dried for 24 h at 298 K and calcined for 3 h in air at 623 K.
Monometallic NiO samples were prepared by impregnation of the calcined support (3 h, air, 623 K) with the aqueous solution of Ni(NO3)2, corresponding to the pore volume of the support, followed by drying for 24 h at 298 K and calcining for 3 h in air at 623 K [15]. Bimetallic NiO/Au/supports and Ag/Pd/Al2O3 were produced by pore volume impregnation of fresh Au/supports and Pd/Al2O3 with the aqueous solution of Ni(NO3)2 and AgNO3, respectively. The precursors obtained were dried for 24 h at 298 K, and calcined for 3 h in air at 623 K.
The metal contents in catalysts were determined by atomic absorption on a Thermo iCE 3000 AA spectrometer. The metals were preliminarily removed from the support by washing with aqua regia (HCl/HNO3=4:1). The relative error of metal content determination was less than 1 %. X-ray diffraction (XRD) patterns were accumulated with a Rigaku D/MAX 2500 instrument using Cu K(a) radiation with a step size of 0.02° two-theta (2θ) over the range 35–70°. Transmission electron microscopy (TEM) and energy-dispersive X-ray (EDX) analysis of catalysts were carried out on a JEOL JEM 2100F/UHR microscope with 0.1 nm resolution and a JED-2300 X-ray spectrometer, respectively. The size of spherical (SPH) and distorted (DIS) particles was calculated as diameter and maximum linear size, respectively. For each catalyst, 300–380 particles were processed to determine the particle size distribution. The average particle size was determined as the average size of the most frequent particles. The concentration of SPH particles in the sample was calculated as C(SPH)=n(SPH)×N × 100 %, where n(SPH) is the number of SPH particles, N (300–380) is the number of processed particles. The concentration of DIST particles was calculated in the same manner.
The hydrogenation of ethyne–ethene mixture was carried out using a fixed-bed flow reactor. A catalyst (1 g) was placed in a quartz reactor and heated to 357 K for 1 h in a stream of H2. The gas mixture containing C2H2, H2, and C2H4 in a 1:2:20 ratio was passed through the catalyst at a flow rate of 720 h−1. The reactor effluents were analyzed with Tcvet-800 gas chromatograph equipped with a flame ionization detector and a 30-m PoropakT capillary column. The ethyne conversion was calculated as Δ(С2H2) = [χ 0(С2H2) − χ i(С2H2)] × [χ 0(С2H2)]−1×100 %, where χ 0(С2H2) is the molar concentration of С2H2 in the initial gas mixture, and χ i(С2H2) is the molar concentration of С2H2 in the products after time on stream (i). The selectivity of the ethene formation was determined as S(С2H4) = [(χ i(С2H4) − χ 0(С2H4)] × Δ(С2H2)−1, where χ 0(С2H4) and χ i(С2H4) are the molar concentrations of С2H4 in the initial gas mixture and in the products after reaction time (i), respectively; Δ(С2H2) is the ethyne conversion. The reproducibility of the catalytic measurements was ±2 % with respect to the absolute value of conversion and selectivity.
TOFs were calculated by equation TOF = A × B −1 × t −1 as described in Mohr et al. and Okumura et al. [17, 18], where [A] = [mole] is the amount of ethyne converted per the reaction time [t]=[h], [B] = [mole] is the amount of the surface atoms in deposited clusters. B was calculated as (B total(Au) + B total(Ni)) × D, where B total(Au) and B total(Ni) is the total moles of Au and Ni in the sample, respectively, [D] is the surface-to-volume ratio for clusters with different average sizes given in Mohr et al. and Okumura et al. [17–19].
The calculations of C2H2, C2H4, and H2 adsorption on Au12 were performed within DFT framework using Perdow–Burke–Ernzerhof functional [20]. Relativistic effects of gold were taken into account in scalar–relativistic approach using Dirac–Coloumb–Breit Hamiltonian modified by Dyall [21]. The basis sets as implemented in the Priroda 06 program [22] were used in the calculations (Au {30s29p20d14f}/[8s7p5d2f]; H {6s2p}/[2s1p]; С {10s7p3d}/[3s2p1d]). Au12 cluster was considered as a model. It has a dynamic structure [23] and co-exists in flat (two-dimensional (2D)) and three-dimensional (3D) structures that allowed us to study the dependence of adsorption properties on the structural features of gold particles.
The adsorption energy of RH molecule (E ad(RH), RH = C2H2, C2H4, H2) on Au12 was calculated as E ad(RH) = E(Au12) + E(RH) − E(Au12RH), where E(Au12) is the total energy of an isolate gold cluster, E(RH) is the total energy of isolate hydrocarbon, E(Au12RH) is the total energy of the hydrocarbon absorbed on Au12. All the total energies included zero-point vibration energies. The heats of adsorption (Q) were calculated at 298 K on the basis of the formulas of statistical thermodynamics in the approximation of rigid rotato-harmonic oscillator. The calculation of all the structures was performed in singlet ground state. The types of stationary points on the potential energy surface were determined from the analysis of Hess matrix; the second derivatives were calculated analytically [23].
Results and discussion
In the present work, some M/X catalysts (M=Au, NiO, NiO/Au; X = Al2O3, TiO2, ZnO, ZrO2) numbered from №1 to №12 and reference Ag/Pd/Al2O3 catalyst ([Pd]=0.021 wt.%; [Ag]=0.006 wt.%) were synthesized. The actual metal loadings in the samples №s 1–12 are presented in Table 1. The actual gold content in the samples prepared by deposition–precipitation was 0.018–0.024 wt.% that is lower than theoretical loading. The loss of gold is attributed to washing treatment during the preparation [10]. The actual concentration of Ni in the impregnated samples was 0.006–0.007 wt.% that is in agreement with theoretical weight percent. The molar ratio of Ni/Au in bimetallic samples was close to the desired 1:1 ratio.
The size and shape of supported NiO, Au, and NiO/Au particles
The structural features of supported particles in the samples №s 1–12 are presented in Figs. 1, 2, 3, and 4 and Table 1. The particle size distributions in Ni samples were monomodal (see Fig. 1 as representative). The sizes of detectable nickel particles varied from 2 to 17 nm (Table 1). The average particle size in NiO/Al2O3, NiO/TiO2, NiO/ZnO, and NiO/ZrO2 was 3, 4.5, 7, and 7 nm, respectively (Table 1). The increase in particle size of NiO during transition from Al2O3 to ZrO2 could be caused by the decrease of the surface area of a support, which favors segregation of clusters during the calcination [3–5]. The shape of the NiO particles, deposited on Al2O3, was nearly spherical (Fig. 1). The similar shape was observed for NiO particles immobilized on TiO2, ZnO, and ZrO2 (Table 1). The obtained results are in agreement with the shape of nickel particles immobilized by impregnation from the aqueous solution of Ni(NO3)2 [9].

TEM image of the NiO/Al2O3 (a) and particle size distribution (b)

TEM image of the Au/Al2O3 (a) and particle size distribution (b)

TEM images of the NiO/Au/Al2O3 (a) and particle size distribution (b)

TEM image of the NiO/Au/Al2O3 (a); EDX elemental maps of the Au Mα (b) and Ni Kα (c) and typical TEM images of the NiO/Au particles
The particle size distributions in Au/Al2O3 (Fig. 2, №1) and monometallic gold samples №s 4, 7, and 10 were monomodal and varied from 2 to 23 nm (Table 1). The average particle size of Au in Au/Al2O3, Au/TiO2, Au/ZnO, and Au/ZrO2 was 2.5, 8, 10, and 9 nm, respectively. The shapes of the supported Au particles were spherical (Table 1) that is in agreement with the shape of Au particles on the same supports immobilized by deposition–precipitation from HAuCl4 × aq [10, 24].
The deposition of NiO particles on the Au catalysts resulted in a broadening of particle size distribution (see Figs. 1, 2, and 3 and Table 1). The shift of the average particle size to higher values was detected in bimetallic samples in comparison with corresponding monometallic ones (Table 1). The observed trend could be caused by the formation of decorated M1/M2 clusters, whose sizes are larger than M1 and M2 [3–5, 14]. The presence of NiO/Au particles in the bimetallic samples was confirmed by the TEM-EDX analysis. The TEM micrograph of the gold–nickel catalyst and the EDX elemental maps of Au and Ni are presented in Fig. 4. The elemental maps of bimetallic sample contained individual particles of Au and NiO (Au Mα and Ni Kα signals from these clusters do not overlap) along with mixed particles of NiO/Au (overlay Au Mα and Ni Kα signals from the same particle).
The fraction of NiO/Au particles in the bimetallic catalysts №s 3, 6, 9, and 12 ranged from 40 % to 19 % from the total number of supported particles. The size of NiO/Au particles varied from 4 to 20 nm, and the average size was always larger in comparison with Au and NiO. An important feature of NiO/Au clusters was their distortion (deviation from spherical shape) (see Fig. 4), which may be caused by the inclusion of NiO clusters into the surface of Au nanoparticles during the stage of catalyst preparation.
The catalytic properties of NiO, Au, and NiO/Au nanocomposites
The conversion, selectivity, and durability of catalysts №s 1–12 tested in hydrogenation of ethyne–ethene mixture at 357 K are presented in Figs. 5, 6, and 7, the calculated TOFs are summarized in Table 1. After hydrogen treatment, NiO particles with size of 3–4.5 nm supported on Al2O3 and TiO2 showed 18–22 % ethyne conversion (Fig. 5). At these conversions, the selectivity to ethene on NiO catalysts was high—95–99 % that is in agreement with the results obtained in Zhang et al. [25]. The initial TOF of NiO/Al2O3 and NiO/TiO2 was 497 and 608 h−1, respectively (Table 1). The TOFs of 3 nm NiO particles are lower by one to two orders of magnitude in comparison with C≡C bond hydrogenation activity of 3–10 nm clusters of Pt [26] and Ag/Pd [25] supported on Al2O3.

Hydrogenation of ethyne-ethene mixture at 357 K over NiO catalysts № 2, 5, 8, 11. The metal loading of Ni in samples is 0.006 wt.%

Hydrogenation of ethyne-ethene mixture at 357 K over Au catalysts № 1, 3, 4, 6, 9, 12 and Ag/Pd catalyst. The metal loadings of Pd and Ag are 0.021 and 0.006 wt.%, respectively. The metal loadings of Au and Ni are 0.018–0.02 wt.% and 0.006–0.007 wt.%, respectively

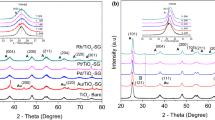
Powder X-ray diffraction patterns for a 0.21 % Au/Al2O3, b 0.15%Ni/Al2O3, and c 0.21%Au + 0.21%Ni/Al2O3. Planes (111), (200) are attributed to gold clusters. Planes (311), (222), (400) are attributed to alumina support
The 7 nm NiO particles supported on ZnO and ZrO2 showed better activity in comparison with 3–4.5 nm NiO particles supported on Al2O3 and TiO2 (Table 1). The negative-size effect of activity is the known feature of Pd, Pt, and Ni catalysts attributed to irreversible adsorption of unsaturated compounds on small clusters [3, 4, 10]. According to data presented in del Angel et al. [26], the TOF of Pt and Pd clusters decreases from 780 to 480 and from 9,910 to 8,640 h−1 as the cluster size decreases from 12 to 3 and from 10 to 3 nm, respectively. The reason of the better activity of larger NiO particles can also be influenced by their reducibility. The rate-determining step in hydrogenation is usually a dissociative adsorption of H2 that takes place at Ni0 sites not at Ni2+ [4]. Keane and Medina showed that large NiO clusters are reduced much easier to Ni0 in comparison with small NiO clusters that strongly interact with surface groups of the support [9, 27]. Therefore, the 7 nm NiO of the sample №s 8 and 11 possess higher activity in hydrogenation in comparison with the 3 and 4.5 nm NiO of the sample № 3 and № 5 (Table 1).
The stability of supported NiO clusters was low (Fig. 5). After 735 min of the reaction, the color of NiO/Al2O3 and NiO/TiO2 became darker, and TOFs decreased by approximately 55 % and 49 %, respectively. The same trends were observed for NiO/ZnO and NiO/ZrO2 (Fig. 5). The most probable mechanism of nickel deactivation is due to green-oil formation as described in Borodziński et al. and Nikolaev et al. [3, 4].
A 1.5 % conversion of ethyne with 100 % selectivity to ethene was detected on 2.5 nm Au deposited on Al2O3 (Fig. 6). The high selectivity of Au/Al2O3 is in agreement with results of Jia [28] and Gluhoi [29] reported that ethyne was hydrogenated to ethene on Au/Al2O3 with 100 % selectivity at 313–523 K. The initial TOF for 2.5 nm Au was 36 h−1 that is in agreement with gold hydrogenation activity [1, 13, 15, 18] and lower in comparison with the activity of either NiO (Table 1) or Pt and Pd clusters of the same size deposited on alumina [26, 30].
The positive-size effect of activity was found for gold catalysts. As the size of gold decreases from 8 to 2.5 nm, the initial TOF increases from 28 to 36 h−1 (Table 1). The better hydrogenation activity of 2.5 nm gold particles can be explained if the chemisorption of H2 is dependent on gold particle size. Our recent research on the TOF of Au particles of different sizes (2–30 nm) in hydrogenation of ethynylbenzene [13] and studies by Bus [30, 31], Serna [32], Boronat [33], and Jia [28] have proved the positive influence of small gold clusters on hydrogenation rate. It was concluded that H2 is dissociatively adsorbed only at the corners and edges of the supported gold particles [34, 35]. Gold atoms at the corner and edge sites have a low coordination number compared with face atoms and thus have a more reactive d-band, resulting in these atoms to be able to interact more easily with H2 [30] and facilitate the H2 dissociation.
It is interesting to note that almost identical 8–10 nm Au clusters supported on ZrO2, ZnO, and TiO2 possess different hydrogenation properties. Whereas Au/ZrO2 and Au/ZnO are inactive, Au/TiO2 converts 0.5 % of ethyne into ethene. It seems that the higher hydrogenation activity of Au/TiO2 is due to specific metal-support interaction. Fujitani [36] find no HD formation between H2 and D2 other than on Au/TiO2. No HD formation was observed at any single-crystal surface (Au(111), Au(311), and TiO2(110)). This suggests that the bulk gold and TiO2(110) surface did not make H2 dissociate, whereas the Au/TiO2 surface did. Recent DFT study [37] proved this phenomenon. Yong revealed the favored heterolytic dissociation of H2 with E a = 0.37 eV at the perimeter sites of Au/TiO2, where Au atom and a nearby surface O 2−(s) were involved. As a result of this process, oxygen atoms near the perimeter were passivated. Further H2 dissociation occurred on pure gold atoms via a homolytic mode with E a = 0.64 eV [37].
Some contribution to the observed decreasing of TOFs in the row Au/Al2O3 > Au/TiO2 > Au/ZnO > Au/ZrO2 (Table 1) could be provided by OH groups and metal cations of the supports. Thus, Zhang reported that dehydroxylated Au/ZrO2 was inactive in semi-hydrogenation of 1,3-butadiene but became active after regeneration of the surface –M–O–M– groups by water treatment [38]. Hydroxyl groups on ZrO2 were proposed to supply protons for hydrogenation and according to H/D exchange reactions between D 2 and the –OH groups occurred at above 323 K [38]. The specific surface of supports decreases in the row Al2O3 > TiO2 > ZnO > ZrO2, thus the concentration of hydroxyl groups in Au catalysts decreases during the transition from Al2O3 to ZrO2. As a result, the positive contribution of hydrogen from hydroxyl groups to the hydrogenation decreases.
The metal cations of the supports are of importance in the adsorption and activation of reactants [39, 40]. It was suggested that cooperation of coordinatively unsaturated Au atoms and the acid–base pair site (Alδ+–Oδ−) play an important role as in H2 dissociation step and preferential adsorption of hydrogenated group [39]. Alumina is the oxide with strong Lewis acid sites that able to activate the C–H bond of ethyne directly on the Mδ+–Oδ− pairs [40]. A decrease in the Lewis coordinative activity was found for the metal cation of basic oxides (CaO, ZnO, and MgO). The weakening of Lewis acid sites decreases the ability to form the acetylide species (ZrO2) up to the full inactivity (SiO2).
The durability of active Au/TiO2 and Au/Al2O3 catalysts was higher in comparison with nickel samples: No decrease in conversion occurred during 735 min (Figs. 5 and 6). The high durability of gold catalysts in hydrogenation of ethyne at 357 K is in good agreement with results of Jia [28], Zhang [38], and Azizi [41]. For example, Azizi and co-workers investigated the hydrogenation of ethyne and the formation of carbonaceous deposits using Au/CeO2. The catalysts were prepared by direct anionic exchange, which produced catalysts with an average gold particle size of 2 nm. H2/C2H2 ratios between 3 and 60 were tested in the temperature range 300–673 K. An ethene selectivity of 100 % was observed for all H2/C2H2 ratios up to 573 K. Above 573 K, the deactivation occurred swiftly and was accompanied by the decreased selectivity.
The decoration of Au by NiO led to a sharp increase in ethyne conversion up to 90 % while the selectivity still remains 100 %, and the time of stable operation remains at least 735 min (Fig. 6). Moreover, the activity and stability of NiO/Au catalysts at 357 K were found to be higher than the activity and stability of reference Ag/Pd catalyst for selective hydrogenation prepared by the same method as NiO/Au catalyst (Fig. 6). The conversion of ethyne on NiO/Au catalysts was higher than the sum of conversions on NiO and Au catalysts (Fig. 6) that points out the synergistic activity of NiO/Au catalysts. The synergistic gain on NiO/Au clusters (SG) after 735 min of time on stream was 1,466; 1,147; 563; and 569 h−1 for NiO/Au/Al2O3, NiO/Au/TiO2, NiO/Au/ZnO, and NiO/Au/ZrO2, respectively (Table 1).
The synergistic effects of activity resulted from the interaction between M1 and M2 metals can be attributed to the formation of alloy [3, 5, 8, 14, 42]. Besenbacher observed the formation of Au–Ni alloy in Au–Ni/MgAl2O4 catalyst with extremely high metal loading (Au + Ni = 17 wt.%) after reduction at 823 K [14]. The obtained catalyst possessed the enhanced catalytic properties in n-butane steam-reforming. Zhang prepared Au7Ni3 intermetallic alloy by melting the mixture of gold and nickel at 1,333 K [42]. This alloy exhibited the synergistic activity in electro-oxidation of formic acid. It was reported that, compared with the standard spectrum of pure Au, the diffraction angles of Au–Ni sample were shifted to the higher positions, reflecting Au–Ni alloy with lattice contraction due to the partial substitution of Au by Ni [42]. The attempts to obtain the XRD reflexes from supported metals of the sample №s 1–12 were failed due to the low concentration of gold (0.018–0.024 wt.%) and/or nickel (0.006–0.007 wt.%). The XRD patterns of specially prepared samples with relatively high metal contents (0.15–0.42 wt.%) are presented in Fig. 7. The XRD pattern of NiO/Au sample (Au = Ni = 0.21 wt.%) was described by the sum of XRD patterns of the gold and nickel catalysts (Fig. 7). No new reflexes or shifts of Au reflexes toward larger values pointed at alloy formation, as described in Lijuan et al. [42], were detected. The obtained XRD results allowed us to exclude the formation of new Au–Ni compounds in our samples with a high degree of certainty. Moreover, the synthesis of Au–Ni alloy [14, 42] was carried out at 823–1333 K and high metal contents (17–100 wt.%). Thus, alloy formation is not possible at the temperatures and metal loadings employed in our study.
The synergism in M1–M2 activity also can be attributed to the formation of the new structural sites. For example, Keane linked the increase of the rate of 2,4-dichlorophenol hydrodeclorination on Au/Ni/Al2O3 with decoration of nickel clusters with gold ones [9]. It was proposed that the Au component served to adsorb and activate the C–Cl bond whereas the H2 dissociated at the Ni0. The addition of tin to the Pt/SiO2 catalyst produced a remarkable positive shift in both the product distribution and TOF in the liquid-phase hydrogenation of unsaturated aldehyde to corresponding alcohols [43, 44]. The simultaneous increase in both activity and selectivity indicates the formation of a new type of mixed (Mx+)/(Pt)0 clusters, different in nature when compared with the original Pt and M ones. The promotion effect was proposed due to a (Mx+) activating the C=O bond, which becomes easily hydrogenated. The building of mixed (M1)/(M2) clusters is very similar to what is occurring in the well-known strong metal support interaction between reducible oxide support and deposited metal clusters [1–5, 10].
Our investigations of model Au, Ni/Au, and Ni catalysts by XPS, DRIFT, and XAS techniques showed that nickel in both Ni/Au and Ni catalysts exists as NiO, gold in the Au catalyst exists as Au0 nanoclusters, whereas the Au0 nanoclusters co-exist with Au3+ cations in the NiO/Au samples [7]. Then, the first probable explanation (I) of synergistic activity between NiO and Au in ethyne hydrogenation (Table 1) could be due to the electron transfer from the electron-rich Au0 particles to the electron-deficient NiO that give new Auδ+ catalytic sites.
According to the TEM data of the present research, there is a new significant fraction of distorted Au structures in NiO/Au catalysts (Table 1). Moreover, the concentration of distorted particles is correlated with SG values. It is known that the deviation from spherical shape of the particle led to increase in surface corners and edges [5]. Thus, the second explanation (II) of synergistic activity can be linked with the formation of new Au structural sites with a low coordination number in NiO/Au clusters.
The nature of Au activation in NiO/Au clusters
The DFT calculation of H2, C2H2 and C2H4 on flat Au12 (2D) and Au12 (3D) containing flat, corner and edge atoms, were performed to test the reasonability of the explanations (I) and (II). The optimized structures of the most stable (2D) and (3D) Au12 isomers are shown in Fig. 8. The calculated heats of adsorption of substrates are listed in Table 2.

The optimized structures of flat (2D) and three-dimensional (3D) Au12 clusters and the most stable complexes of Au12 with hydrogen, ethene, and ethyne molecules
The calculated heat (Q) of dissociative adsorption of H2 on the flat Au 012 (2D) was equal to 40 kJ/mol (Table 2). During transition to uncharged Au 012 (3D) structures that contain flat elements (planes) and structural defects (corners and edges), the Q(H2) increases up to 78 kJ/mol. The obtained results confirmed the existing hypotheses on the positive impact of cluster defects to the activation of H2 [13, 30, 32–35] and are in agreement with explanation of NiO/Au synergy (II).
The presence of a positive charge on the Au12 (2D) and Au12 (3D) clusters led to an increase in the Q(H2) by 23 and 2 kJ/mol, respectively (Table 2). These results are in good agreement with the results of Zhang et al. [45, 46]. This group used Au/ZrO2 to investigate the effects of Au3+/Au0 ratio on 1,3-butadiene hydrogenation. It was reported that metallic Au0 atoms at the surface of the gold particles were not the only active catalytic sites and that isolated Au3+ ions were the most active [45, 46]. The enhanced activation of H2 on charged gold could be explained by mechanism of Aun–H2 formation. Recently, Gao and Lyalin in their DFT calculation [47] showed that H2 dissociates at the low coordinated corner Au atom with formation of the slightly bent H–Au–H bond. The main feature of the formation of this hydride structure is the catalytic oxidation–reduction cycle Mn → Mn+2 → Mn. In the case of non-charged zero-valence gold nanoparticles, this cycle should look like (Aun+1)0 → (Aun)Au+2 → (Aun+1)0. But these valence changes seem to be improbable for gold, as the typical gold oxidation states are 0, +1, and +3 rather than +2. However, if supported gold nanoparticles may carry a positive charge +3 or +1, then a highly probable catalytic cycle with common gold valences will be obtained: (Aun)Au+1 → (Aun)Au+3 → (Aun)Au+1.
The adsorption of ethene on Au 012 (2D) was accompanied by the formation of π-complex with one of the atoms in the cluster, while the adsorption of ethyne can be realized through π- and di-σ bonding (Table 2). The adsorption of C2H4 on Au 012 (3D) occurred through the formation of π-complex, and Q(C2H4) was slightly higher than Q(C2H4) on Au 012 (2D) (Table 2). The increase in the heat of adsorption was also detected for ethyne π-complex during transition from Au 012 (2D) to Au 012 (3D). It was also revealed that ethyne on Au 012 (3D) can form a new type of coordination—perpendicular (┴) and parallel (║) bridges (Fig. 8), with the high heat of adsorption (Table 2). Our calculation trends are in good agreement with results obtained in Jia et al. and Segura [28, 48]. Jia et al. [28] have shown that, at 273 K, the amount of ethyne adsorbed on 3.8-nm gold particles immobilized on Al2O3 was 18 times greater than that of ethene. Moreover, in contrast to ethene, adsorption of ethyne was irreversible. Segura [48] has demonstrated that 4-nm gold nanoparticles supported on CeO2 are extremely selective in the hydrogenation of C≡C bond in ethyne–ethene mixtures. His DFT calculation shows that differences in binding energy E b(C2H2)–Eb(C2H4) on flat Au 019 (2D) surface and on Au 019 (3D) cluster are 0.08 and 0.66 eV, respectively. Segura attributed the high selectivity of gold to the preferential adsorption of C≡C bond compared with C=C bond at the edges of gold clusters. Thus, Au defects favor the selective ethyne adsorption and its further conversion.
Ethyne and ethene are Lewis bases sensitive to electron acceptors [4, 5, 40]. An increase in the surface positive charge on Au due to electron transfer [7] improves the electron acceptor properties of gold and thereby should lead to stronger adsorption of ethyne or ethene on Auδ+ than on Au0 and, therefore, increase the probability of the chemical reaction. Table 2 shows that positive charge on the flat as well as on the Au +12 (2D) and Au +12 (3D) cluster leads to an increase in the heat of adsorption of ethene in π-complex up to 132 and 147 kJ/mol, respectively. Surprisingly, the calculated heats of adsorption of ethyne (113 and 102 kJ/mol for π- and di-σ form, respectively) were smaller in comparison to the heat of adsorption of ethene on flat gold surface, but the positive impact to growth of heat of ethyne adsorption remained unchanged. Moreover, the heat of ethyne adsorption in the most stable (║) bridge form on Au +12 (3D) increased up to 172 kJ/mol, which is larger by 25 kJ/mol in comparison with the heat of ethene adsorption on Au +12 (3D). Thus, positive charge on 3D clusters of Au favors preferential ethyne adsorption and increases its further conversion.
Conclusion
Monometallic Au and NiO particles supported on Al2O3, TiO2, ZnO, and ZrO2 possess 95–100 % selectivity to ethene in hydrogenation of ethyne–ethene mixture at 357 K. The negative-size effect was revealed for NiO particles: As the size of NiO clusters decreases from 7 to 3 nm, the TOF decreases from 812–1,023 to 276 h−1. In contrast to NiO, the size effect of activity was found to be positive for Au. As the size of gold decreases from 8 to 2.5 nm, the TOF increases from 0–28 to 36 h−1. The observed size trends of TOF result from different abilities of gold and nickel to adsorb and activate the ethyne, ethene, and hydrogen.
The bimetallic NiO/Au catalysts possess 100 % selectivity to ethene and higher activity and stability in comparison with mono- and bimetallic catalysts based on Pd, Ag, and Ni. The decoration of Au by NiO led to increase in TOFs of NiO/Au catalysts up to 1,778 h−1. The synergistic gain on NiO/Au clusters calculated as TOFNiO/Au − TOFAu − TOFNiO was 1,466; 1,147; 563; and 569 h−1 for NiO/Au/Al2O3, NiO/Au/TiO2, NiO/Au/ZnO, and NiO/Au/ZrO2, respectively. The formation of Auδ+ in NiO/Au particles and deviation of NiO/Au clusters shape from the spherical were detected. Using the methods of quantum chemistry, it was shown that distorted 3D and/or positively charged gold structures favor a dissociative adsorption of H2 and preferential adsorption of ethyne from ethyne–ethene mixture.
References
McEwan L, Julius M, Roberts S, Fletcher JCQ (2010) A review of the use of gold catalysts in selective hydrogenation reactions. Gold Bull 43:298–306. doi:10.1007/BF03214999
Hugon A, Delannoy L, Louis C (2008) Supported gold catalysts for selective hydrogenation of 1,3-butadiene in the presence of an excess of alkenes. Gold Bull 41:127–138. doi:10.1007/BF03216590
Borodziński A, Bond GC (2008) Selective hydrogenation of ethyne in ethene-rich streams on palladium catalysts, part 2: steady-state kinetics and effects of palladium particle size, carbon monoxide, and promoters. Catal Rev Sci Eng 50:379–469. doi:10.1080/01614940802142102
Nikolaev SA, Zanaveskin LN, Smirnov VV, Averyanov VA, Zanaveskin KL (2009) Catalytic hydrogenation of alkyne and alkadiene impurities in alkenes. Practical and theoretical aspects. Russ Chem Rev 78:231–247. doi:10.1070/RC2009v078n03ABEH003893
Rostovshchikova TN, Lokteva ES, Nikolaev SA, Golubina EV, Gurevich SA, Kozhevin VM, Yavsin DA, Lunin VV (2011) New approaches to design of nanostructured catalysts. In: Song M (ed) Catalysis: principles, types and applications. Nova Science Publishers, New York, pp 245–306
Grisel R, Weststrate K-J, Gluhoi A, Nieuwenhuys BE (2002) Catalysis by gold nanoparticles. Gold Bull 35:39–45. doi:10.1007/BF03214836
Tkachenko OP, Kustov LM, Nikolaev SA, Smirnov VV, Klementiev KV et al (2009) DRIFT, XPS and XAS investigation of Au-Ni/Al2O3 synergetic catalyst for allylbenzene isomerization. Topics in Catalysis 52:344–350. doi:10.1007/s11244-008-9167-z
Hashmi A, Stephen K, Hutchings GJ (2006) Gold catalysis. Angew Chem Int Ed 45:7896–7936. doi:10.1002/anie.200602454
Keane MA, Gómez-Quero S, Cárdenas-Lizana F (2009) Alumina-supported Ni–Au: surface synergistic effects in catalytic hydrodechlorination. Chem Cat Chem 1:270–278. doi:10.1002/cctc.200900070
Bond GC, Louis C, Thompson DT (2006) Catalysis by gold. Imperial College Press, London, 366 P
Simakova OA, Campo B, Murzin DY (2010) Gold on carbon catalysts. In: Chow PE (ed) Gold nanoparticles: preparation, characterization and fabrication. Nova Science Publishers, New York, pp 147–171
Lopez-Sanchez JA, Lennon D (2005) The use of titania- and iron oxide-supported gold catalysts for the hydrogenation of propyne. Appl Catal A 291:230–237. doi:10.1016/j.apcata.2005.01.048
Nikolaev SA, Smirnov VV (2009) Selective hydrogenation of phenylacetylene on gold nanoparticles. Gold Bull 42:182–189. doi:10.1007/BF03214932
Besenbacher F, Chorkendorff I, Clausen BS, Hammer B, Molenbroek AM, Nørskov JK, Stensgaard I (1998) Design of a surface alloy catalyst for steam reforming. Science 279:1913–1915. doi:10.1126/science.279.5358.1913
Nikolaev SA, Smirnov VV (2009) Synergistic and size effects in selective hydrogenation of alkynes on gold nanocomposites. Catal Today 147S:S336–S341. doi:10.1016/j.cattod.2009.07.032
Haruta M (2004) Nano particulate gold catalysts for low-temperature CO oxidation. J New Mat Elect Syst 7:163–172
Mohr C, Hofmeister H, Claus P (2003) The influence of real structure of gold catalysts in the partial hydrogenation of acrolein. J Catal 213:86–94. doi:10.1016/S0021-9517(02)00043-X
Okumura M, Akita T, Haruta M (2002) Hydrogenation of 1,3-butadiene and of crotonaldehyde over highly dispersed Au catalysts. Catal Today 74:265–269. doi:10.1016/S0920-5861(02)00034-2
Mohr C, Hofmeister H, Radnik J, Claus P (2003) Identification of active sites in gold-catalyzed hydrogenation of acrolein. J Am Chem Soc 125:1905–1911. doi:10.1021/ja027321q
Perdew JP, Burke K, Ernzerhof M (1996) Generalized gradient approximation made simple. Phys Rev Lett 77:3865–3868. doi:10.1103/PhysRevLett.77.3865
Visscher L (2002) Chapter 6. Post Dirac-Hartree-Fock methods—electron correlation. Theor and Comput Chem 11:291–331. doi:10.1016/S1380-7323(02)80032-2
Laikov DN (1997) Fast evaluation of density functional exchange-correlation terms using the expansion of the electron density in auxiliary basis sets. Chem Phys Lett 281:151–156. doi:10.1016/S0009-2614(97)01206-2
Mukhamedzyanova DF, Ratmanova NK, Pichugina DA, Kuz’menko NE (2012) A structural and stability evaluation of Au12 from an isolated cluster to the deposited material. J Phys Chem C 116:11507–11518. doi:10.1021/jp212367z
Claus P (2005) Heterogeneously catalysed hydrogenation using gold catalysts. Appl Catal A 291:222–229. doi:10.1016/j.apcata.2004.12.048
Zhang Q, Li J, Liu X, Zhu Q (2000) Synergetic effect of Pd and Ag dispersed on Al2O3 in the selective hydrogenation of acetylene. Appl Catal A 197:221–228. doi:10.1016/S0926-860X(99)00463-9
Del Angel G, Benitez JL (1993) Selective hydrogenation of phenylacetylene on Pd/Al203: effect of the addition of Pt and particle size. React Kinet Catal Lett 51:547–553. doi:10.1007/BF02069103
Medina F, Salagre P, Sueiras J-E, Fierro J-LG (1994) Characterization of several γ-alumina-supported nickel catalysts and activity for selective hydrogenation of hexanedinitrile. J Chem Soc Faraday Trans 90:1455–1459. doi:10.1039/FT9949001455
Jia J, Haraki K, Kondo JN, Domen K, Tamaru K (2000) Selective hydrogenation of acetylene over Au/Al2O3 catalyst. J Phys Chem B 104:11153–11156. doi:10.1021/jp001213d
Gluhoi AC, Bakker JW, Nieuwenhuys BE (2010) Gold, still a surprising catalyst: selective hydrogenation of acetylene to ethylene over Au nanoparticles. Catal Today 154:13–20. doi:10.1016/j.cattod.2010.02.021
Bus E, Prins R, van Bokhoven JA (2007) Origin of the cluster-size effect in the hydrogenation of cinnamaldehyde over supported Au catalysts. Catal Comm 8:1397–1402. doi:10.1016/j.catcom.2006.11.040
Bus E, Miller JT, van Bokhoven JA (2005) Hydrogen chemisorption on Al2O3-supported gold catalysts. J Phys Chem B 109:14581–14587. doi:10.1021/jp051660z
Boronat M, Concepción P, Corma A, González S, Illas F, Serna P (2007) A molecular mechanism for the chemoselective hydrogenation of substituted nitroaromatics with nanoparticles of gold on TiO2 Catalysts: a cooperative effect between gold and the support. J Am Chem Soc 129:16230–16237. doi:10.1021/ja076721g
Boronat M, Lllas F, Corma A (2009) Active sites for H2 adsorption and activation in Au/TiO2 and the role of the support. J Phys Chem A 113:3750–3757. doi:10.1021/jp808271y
Boronat M, Concepción P, Corma A (2009) Unraveling the nature of gold surface sites by combining IR spectroscopy and DFT calculations. Implications in catalysis. J Phys Chem C 113:16772–16784. doi:10.1021/jp905157r
Kartusch C, van Bokhoven JA (2009) Hydrogenation over gold catalysts: the interaction of gold with hydrogen. Gold Bull 42:343–348. doi:10.1007/BF03214957
Fujitani T, Nakamura I, Akita T, Okumura M, Haruta M (2009) Hydrogen dissociation by gold clusters. Angew Chem Int Ed 48:9515–9518. doi:10.1002/anie.200905380
Yong B, Cao X-M, Gong X-Q, Hu P (2012) A density functional theory study of hydrogen dissociation and diffusion at the perimeter sites of Au/TiO2. Phys Chem Chem Phys 114:3741–3745. doi:10.1039/C2CP23755E
Zhang X, Shi H, Xu B-Q (2011) Vital roles of hydroxyl groups and gold oxidation states in Au/ZrO2 catalysts for 1,3-butadiene hydrogenation. J Catal 279:75–87. doi:10.1016/j.jcat.2011.01.002
Shimizu K-i, Yamamoto T, Tai Y, Satsuma A (2011) Selective hydrogenation of nitrocyclohexane to cyclohexanone oxime by alumina-supported gold cluster catalysts. J Mol Catal A 345:54–59. doi:10.1016/j.molcata.2011.05.018
Ivanov AV, Koklin AE, Uvarova EB, Kustov LM (2003) A DRIFT spectroscopic study of acetylene adsorbed on metal oxides. Phys Chem Chem Phys 5:4718–4723. doi:10.1039/B307138C
Azizi Y, Petit C, Pitchon V (2008) Formation of polymer-grade ethylene by selective hydrogenation of acetylene over Au/CeO2 catalyst. J Catal 256:338–344. doi:10.1016/j.jcat.2008.04.003
Lijuan Z, Tian Ruili HP, Yuru M, Dingguo X (2010) A gold-nickel alloy as anodic catalyst in a direct formic acid fuel cell. Rare Metal Mat Eng 39:945–948. doi:10.1016/S1875-5372(10)60102-6
Merlo AB, Machado BF, Vetere V, Faria JL, Casella ML (2010) PtSn/SiO2 catalysts prepared by surface controlled reactions for the selective hydrogenation of cinnamaldehyde. Appl Catal A 383:43–49. doi:10.1016/j.apcata.2010.05.020
Coq B, Figueras F (2001) Bimetallic palladium catalysts: influence of the co-metal on the catalyst performance. J Mol Catal A 173:117–134. doi:10.1016/S1381-1169(01)00148-0
Zhang X, Shi H, Xu B-Q (2005) Catalysis by gold: isolated surface Au3+ ions are active sites for selective hydrogenation of 1,3-butadiene over Au/ZrO2 catalysts. Angew Chem Int Ed 44:7132–7135. doi:10.1002/anie.200502101
Zhang X, Shi H, Xu B-Q (2007) Comparative study of Au/ZrO2 catalysts in CO oxidation and 1,3-butadiene hydrogenation. Catal Today 122:330–337. doi:10.1016/j.cattod.2007.02.016
Gao M, Lyalin A, Taketsugu T (2011) Role of the support effects on the catalytic activity of gold clusters: a density functional theory study. Catalysts 1:18–39. doi:10.3390/catal1010018
Segura Y, López N, Pérez-Ramírez J (2007) Origin of the superior hydrogenation selectivity of gold nanoparticles in alkyne + alkene mixtures: triple-versus double-bond activation. J Catal 247:383–386. doi:10.1016/j.jcat.2007.02.019
Acknowledgments
This work was supported by the Russian Foundation for Basic Research (Grant № 11-01-00280, № 10-03-00999, № 11-03-01011, and № 11-03-00403), the Russian Federation Ministry of Education and Science (State Contracts № 16.513.11.3137) and by the grants from the Russian Federation President (MK-107.2011.3, МК-2917.2012.3, and МК-1621.2012.3).
Open Access
This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 2.0 International License (https://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Nikolaev, S.А., Pichugina, D.А. & Mukhamedzyanova, D.F. Sites for the selective hydrogenation of ethyne to ethene on supported NiO/Au catalysts. Gold Bull 45, 221–231 (2012). https://doi.org/10.1007/s13404-012-0066-5
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13404-012-0066-5