
Abstract
The recent success of using methyltin(IV) cations in constructing multidimensional structures containing the Au–CN–Sn link with interesting physical properties will be surveyed. The methyltin(IV)-dicyanoaurates, Me3Sn[Au(CN)2] (1) and Me2Sn[Au(CN)2]2 (2) containing the Au–CN–Sn link can be easily prepared by aqueous reaction of Me3SnCl or Me2SnCl2 with stoichiometric amounts of an aqueous solution of K[Au(CN)2]. The room temperature solid-state emission spectrum of 1 excited at 254 nm shows two intense emission bands at 442 and 670 nm, and a shoulder at 390 nm. When excited at 320 nm, the crystalline sample shows two intense emission bands at 442 and 720 nm, and a shoulder at 380 nm. After 2 min of grinding, only the blue emission band at 442 nm is observed. In contrast, the emission spectrum of 2 shows only one emission maximum at 422 nm. The porosity of 1 and 2 was probed by gas sorption measurements performed at 77 K. 1 exhibited no detectable microporosity as revealed by the inspection of the N2, H2, as well as, O2 isotherms. The gas adsorption studies reveal that only a small amount of N2 and H2 (3.82 and 4.66 cm3 g−1, respectively) is adsorbed by the framework of 2 at 77 K. However, a CO uptake of 11.20 cm3 g−1 can be reached at 1 atm. The framework of 2 can take up significant amounts of O2 (23.27 cm3 g−1). In addition to intriguing photoluminescence and gas sorption behavior, these complexes also exhibit ion exchange properties in the presence of bivalent transition metal cations, such as cobalt(II), nickel(II), copper(II), and zinc(II).
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The rational design and synthesis of cyanide-bridged bimetallic supramolecules containing cyanometallate building blocks are the focus of widespread research interest because of their interesting topological structures and diverse properties [1–3]. Among many metal cyanide anions, the dicyanoaurate [Au(CN)2]− building block, due to its affinity to bridge transition metal centers has been used in the construction of 2D and 3D cyano-bridged bimetallic Au–CN–M coordination polymers [4–7]. The unique ability of linear [Au(CN)2]− anion to form Au–Au aurophilic interaction plays a key role in controlling the dimensionality and topology of these dicyanoaurate-based heterometallic polymers [4–7]. The cyanoaurate-based heterometallic polymers may exhibit unusual structural motifs and physical properties, such as luminescence [8–11], vapochromism [12, 13], birefringence [14–18], colossal thermal expansion [19, 20], magnetism [21–25] or ion exchange [26]. This dicyanoaurate anion also has significant importance in industrial and medical applications. It is used in gold electroplating [27], and it is considered as a pharmacologically active human metabolite of several antirheumatic gold(I) complexes of sulfur-containing ligands [28, 29]. In this light, it seems surprising that literature describing cyanoaurate-based coordination polymers of main-group metals is unusually sparse. A series of lead(II) coordination polymers containing the dicyanoaurate(I) bridging ligands were prepared and structurally characterized [14–18]. Some of them are highly birefringent materials [14–18]. We have previously reported the construction and structural characterization of multidimensional structures containing the Au–CN–Sn link generated by the reaction of the hard Lewis acidic organotin RnSn(4–n)+ cation and the soft Lewis basic dicyanoaurate [Au(CN)2]− anion [26]. Single crystals of both compounds can be obtained by slow interdiffusion of aqueous solutions of Me3SnCl or Me2SnCl2 and K[Au(CN)2]. As observed under polarized light microscopy, the crystals of both 1 and 2 display striking pleochroism (Fig. 1). Pleochroism is the optical property of a crystal, whereby certain wavelengths of polarized light are absorbed in different amounts in different crystallographic directions. This may produce different colors when the crystal is viewed in polarized light [30].

The crystals of both 1 and 2 display pleochroism as observed under polarized light microscopy
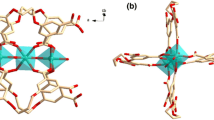
The X-ray structural analysis of 1 showed that each Me3Sn unit is linked to two others by two Au(CN)2 units and form infinite cyanide-bridged chains [26], as illustrated in Fig. 2. These cyanide-bridged chains are further crosslinked by Au–Au interactions of 3.12(1) Å into a 2D grid. The void space of these 2D grids is filled by the arrays of zigzag chains joined by weak Au–Au interactions of 3.42(1) Å.

Structure of complex 1 showing the arrays of interpenetrating cyanide-bridged {Me3Sn–NC–Au–CN} n chains. Hydrogen atoms have been omitted. Color scheme: Au, yellow; Sn, green; N, blue; C, gray
The X-ray structural analysis of 2 showed that each Me2Sn unit is connected to four others by four Au(CN)2 bridges [26], so that an infinite set of self-penetrating layers are formed (Fig. 3). These cyanide-bridged networks are also joined by aurophilic interactions of 3.29(1) and 3.45(2) Å. By treating the Me2Sn units as nodes and connecting them according to the connectivity defined by the Au(CN)2 linkers, there are a set of cyanide-bridged uninodal four-connected 3D networks with 65·8 topology. This topology corresponds to the CdSO4 prototype [26].

Structure of complex 2 showing the arrays of fourfold-interpenetrated cyanide-bridged networks. Hydrogen atoms have been omitted. Color scheme: Au, yellow; Sn, green; N, blue; C, gray
Recently, we have shown that the solvent-free mechanochemical method (grinding stoichiometric amounts of K[Au(CN)2] and metal chlorides in a mortar with a pestle) is a fast, simple, and efficient route to the synthesis of cyanoaurate-based heterometallic coordination polymers [31]. This mechanochemical method was successfully applied also to main group metals to obtain Ph3Sn[Au(CN)2] and 2 [31].
In this paper, we report the infrared characterization, as well as the luminescence and gas sorption properties of these multidimensional structures 1 and 2, as well as, the ion exchange properties of 2.
Results and discussion
The IR spectra of both organotin(IV)-dicyanoaurates 1 and 2 are consistent with their solid-state structures. Thus, the presence of a sharp υCN band at 2,169 cm−1 shows that all of the bridging cyanide groups are in an identical coordination environment in both compounds. This band is shifted toward higher energy with respect to the 2,141 cm−1 υCN stretching vibration of K[Au(CN)2] [32]. As observed for other organotin(IV)-cyanometalates, the IR spectra of 1 and 2 exhibit υSnC stretching at 554 and 596 cm−1 [33, 34]. The υAuC stretching band is found at 455 cm−1 in 1 and at 459 cm−1 in 2. This band is also shifted toward higher energy with respect to the corresponding 427 cm−1 υAuC stretching of K[Au(CN)2] [32]. In the absence of low-frequency Raman spectra, we can tentatively assign bands at 62 (1) and 64 cm−1 (2) to the υAuAu stretching mode [35, 36].
The importance of the aurophilic interactions in influencing the luminescence properties of gold(I) compound is now well recognized [37]. The Au–Au bond distance in complexes showing aurophilic interactions ranges from 2.70 to 3.50 Å [38–41], and owing to this interaction, the gold(I) complexes are potentially luminescent. In the crystal structure of 1, there are aurophilic interactions with lengths of 3.12 and 3.45 Å, and indeed, this complex is emissive when exposed to UV light at room temperature. Upon irradiation with 254 nm UV light, the crystals of 1 exhibit intense pink luminescence. The room temperature solid-state emission spectrum of 1 excited at 254 nm shows two intense emission bands at 442 and 670 nm, and a shoulder at 390 nm (Fig. 4). Thus, the visually observed pink-colored emission was generated by additive color mixing of the red and blue emissions. The excitation spectrum shows a maximum at 278 nm.

Solid-state emission (λ ex = 254 nm) and excitation spectra of 1 at room temperature
The emission spectra of K[Au(CN)2] consists of two bands at about 390 and 630 nm, when excited with 337 nm [42]. The separation between the gold atoms in K[Au(CN)2] is 3.64 Å [37, 42], which is longer than those found in 1. The emission peak at 390 nm in K[Au(CN)2] has been attributed to this weak aurophilic interaction between the dicyanoaurate units [42]. An emission peak at 436 nm is observed for [C5H10NH2][Au(CN)2] and attributed to a short aurophilic interaction (3.10 Å) between the dicyanoaurate units arranged into columns [43]. Thus, the peak at 442 nm observed for 1 could be tentatively attributed to aurophilic interaction. As in the case of K[Au(CN)2] and Tl[Au(CN)2], the low-energy band at 670 nm in 1 is probably due to luminescence traps caused by imperfections in the microcrystalline sample [10, 42]. The emission band at 670 nm (full width at half-maximum is 230 nm) is much broader than the peak at 442 nm (full width at half-maximum is 100 nm), which gives an indication of longer lifetime of the low-energy band.
The room temperature solid-state emission spectrum of crystalline sample of 1 excited at 320 nm shows two intense emission bands at 442 and 720 nm, and a shoulder at 380 nm (Fig. 5). Interestingly, after 2 min of grinding in a mortar with a pestle, this pink-emitting solid was converted into a solid exhibiting intense blue emission (maximum at 444 nm). As shown in Fig. 5, gently scrapping and pressing of 1 with a spatula progressively decreases the intensity of the 720 and 380 nm bands. After total conversion, only the blue emission band at 442 nm is observed.

Solid-state emission (λ ex = 320 nm) spectra of 1 in various states (crystalline and solid samples scrapped and pressed with spatula for 5 to 20 s)
In contrast, the emission spectrum of 2 shows only one emission maximum at 422 nm (Fig. 6). The excitation spectrum of 2 shows two strong bands at 298 and 367 nm. The room temperature luminescence behavior of 2 is very similar to that of β- and γ-polymorphs of Zn[Au(CN)2]2, which have only one emission maximum at 450 and 440 nm, respectively. In crystal structures of these polymorphs, the network interpenetration is supported by Au–Au interactions with lengths of 3.15–3.29 Å [12].

Solid-state emission (λ ex = 300 nm) and excitation spectra of 2 at room temperature
Recently, Long and others published a series of paper using Prussian blue analogues for hydrogen storage at 77 K [44, 45]. In addition to the promising H2 storage materials, the cyano-bridged frameworks may be useful for CO2 storage and separation applications [46, 47]. Accordingly, we studied the gas sorption properties of these organotin(IV)-dicyanoaurates. To our knowledge, these are the first gas sorption measurements reported for organotin(IV)-dicyanoaurates. Thus, the porosity of 1 and 2 was probed by gas sorption measurements performed at 77 K. 1 exhibited no detectable microporosity as revealed by the inspection of the N2, H2, as well as, O2 isotherms. The gas adsorption studies reveal that only a small amount of N2 and H2 (3.82 and 4.66 cm3 g−1, respectively) is adsorbed by the framework of 2 at 77 K (Fig. 7). However, a CO uptake of 11.20 cm3 g−1 can be reached at 1 atm. In contrast, as seen in Fig. 7, the framework of 2 can take up significant amounts of O2 (23.27 cm3 g−1). This complex adsorbs higher amounts (more than six times) of O2 than N2 at 77 K.

Gas adsorption isotherms of 2 for N2 (green), CO (black), H2 (red), and O2 (blue) at 77 K
Some cyanometallates also show high selectivity for oxygen over nitrogen [48]. This could be due to the fact that the unsaturated metal centers of the framework interact with O2 more strongly than with N2 [49]. Other cyano-bridged coordination solids containing coordinatively unsaturated metal centers have been shown to interact with H2 at high loading [44, 45]. The maximum O2 uptake is 3.39 wt.%, which corresponds to approximately 0.67 O2 molecules per formula unit of 2. To our knowledge, there are only few cyano-bridged frameworks that adsorb O2 at 77 K [48, 50]. The oxygen plot represents a type I isotherm characteristic for microporous materials. The surface area, which was determined from the O2 adsorption data by applying Langmuir equation, was 67.8 m2/g. This result is comparable to the 49 m2/g estimated for dehydrated Prussian blue [51]. The pore volume of 0.029 cm3/g calculated from O2 isotherm is in good agreement with the pore volume (0.027 cm3/g) estimated from the crystal structure of 2.
In contrast to porous metal-organic frameworks and other zeolite-like materials, which upon standard ion exchange reactions preserve their original crystal structure, 1 shows unusual ion exchange behavior [26]. As we previously reported, the Me3Sn+ cations are removed completely from cyanide-bridged {Me3Sn–NC–Au–CN} n chains and replaced by Me2Sn2+ or bivalent transition metal cations [26]. In a concentrated aqueous solution of Me3SnCl, the dimethyltin(IV)-, cobalt(II)-, and nickel(II)-dicyanoaurates can be easily converted back into the starting compound 1. However, the metathesis of zinc(II)-dicyanoaurate with Me3SnCl afforded partial reaction, but for copper(II)-dicyanoaurate, no reaction occurs [26].
Now, we performed subsequent metal exchange experiments to study the ion exchange behavior of 2 containing Me2Sn2+ framework ions. In this regard, 2 was soaked in concentrated aqueous solutions of bivalent transition-metal Co2+, Ni2+, Cu2+, and Zn2+ cations (ten times excess). After 2 days, the microcrystalline white powder displayed obvious color changes in the case of Co2+, Ni2+, and Cu2+ (Fig. 8). As was confirmed by IR spectroscopy, the Me2Sn2+ cations are exchanged with bivalent transition metal cations, and 2 converted into corresponding Co(H2O)2[Au(CN)2]2 [52, 53], Ni(H2O)2[Au(CN)2]2 [54], Zn[Au(CN)2]2 [12], and Cu(H2O)2[Au(CN)2]2 [13, 54] transition-metal dicyanoaurates. Consequently, these transition-metal dicyanoaurates were soaked into the concentrated aqueous solution of Me2SnCl2 (ten times excess) for 40 days. In striking contrast to 1, these transition-metal dicyanoaurates cannot be converted back into the starting compound 2.

Transition metal dicyanoaurates M(H2O)2[Au(CN)2]2 (M = Co2+, Ni2+ and Cu2+) and Zn[Au(CN)2]2 obtained in metathesis reactions of 2 with corresponding MCl2 salts
Experimental
General procedures and materials
All chemicals and solvents used for the syntheses were of reagent grade. The solvents for synthesis were used without further purification. All reactions were carried out at room temperature. The elemental analysis has been carried out with an Elementar Vario EL III apparatus at the Laboratory of Organic Analysis, Institute of Organic Chemistry, Research Centre for Natural Sciences, Hungarian Academy of Sciences. Infrared spectra were recorded in the 500 to 4,000 cm−1 spectral range on a Bio-Rad (Digilab Division) FTS-60A FTIR spectrometer equipped with UMA-500 infrared microscope with Ge plate and in the 50 to 500 cm−1 spectral range on a PIKE GladiATR spectrometer with diamond plate. Steady-state luminescence spectra were recorded on an Edinburgh Instrument FS920 spectrofluorometer. Spectral corrections were applied using excitation and emission correction functions of the instrument. Powder samples were placed on a Quartz Suprasil plate in a front-face sample holder. Longpass filters were used to exclude the scattered excitation light. The adsorption isotherms were measured by static volumetric method using fully automated Autosorb 1C (Quantachrome) equipment. Prior to analysis, the sample was heated and kept at 25°C under vacuum for 24 h to remove all the previously adsorbed gases from the surface and the pores. All the measurements were carried out at the boiling temperature of liquid nitrogen (77.3 K).
Synthesis of 1 and 2: The syntheses of both complexes have been previously reported [26]. Complex 1: Anal. Calcd for C5H9N2AuSn: C 14.55; H 2.20; N 6.79. Found: C 14.83; H 1.92; N 6.71; m. p. > 300°C; IR data: 2,169 (s), 791 (b, m), 554 (m), 453 (m), 200–80 (lattice vibration), 62 (s). Complex 2: Anal. Calcd for C6H6N4Au2Sn: C 11.14; H 0.94; N 8.66. Found: C 11.44; H 0.65; N 8.58; m. p. > 300°C; IR data: 2,167 (s), 809 (b, m), 596 (w), 459 (m), 216 (w), 200–80 (lattice vibration), 142 (w), 64 (s).
Metathesis of 2 with transition metal halides
The microcrystalline powder of 2 was immersed in concentrated aqueous solution of transition-metal MCl2 (M = Co, Ni, Zn, Cu) salts. After 2 days, the microcrystalline white powder displayed obvious color changes in the case of Co(II), Ni(II), and Cu(II). Upon decanting the metal chloride solution, the products were washed with water. The ion exchanged products were characterized by IR spectroscopy.
Co(H2O)2[Au(CN)2]2: starting from 2 (90 mg, 0.139 mmol), the metathesis yielded 39.5 mg (48%) product; IR (per centimeter): 2,999 (b, m), 2,206 (m), 2,194 (s), 2,172 (s), 2,161 (s), 1,535 (m), 889 (b, m), 748 (b, m). These characteristic vibrations in the IR spectrum indicate the formation of the Co(H2O)2[Au(CN)2]2 complex [26, 52, 53]
Ni(H2O)2[Au(CN)2]2: starting from 2 (90 mg, 0.139 mmol), the metathesis yielded 64.3 mg (78%) product; IR (per centimeter): 3,002 (b, m), 2,215 (m), 2,204 (sh), 2,172 (s), 2,165 (s), 1,540 (m), 907 (b, m), 757 (b, m). These characteristic vibrations in the IR spectrum indicate the formation of the Ni(H2O)2[Au(CN)2]2 complex [26, 54].
Zn[Au(CN)2]2: starting from 2 (90 mg, 0.139 mmol), the metathesis yielded 68.8 mg (88%) product; IR (per centimeter): 2,198 (s), 2,160 (w). These characteristic vibrations in the IR spectrum indicate the formation of the Zn[Au(CN)2]2 complex [12, 26].
Cu(H2O)2[Au(CN)2]2: starting from 2 (90 mg, 0.139 mmol), the metathesis yielded 77.6 mg (93%) product; IR (per centimeter): 3,144 (b, s), 2,218 (s), 2,173 (s), 1,483 (b, m) 710 (b, m). These characteristic vibrations in the IR spectrum indicate the formation of the Cu(H2O)2[Au(CN)2]2 complex [13, 26, 54].
Metathesis of transition metal dicyanoaurates with Me2SnCl2
The microcrystalline powders of Co(H2O)2[Au(CN)2]2, Ni(H2O)2[Au(CN)2]2, Cu(H2O)2[Au(CN)2]2, and Zn[Au(CN)2]2, respectively, were immersed in concentrated aqueous solution of Me2SnCl2 and left undisturbed at room temperature. After 40 days, the powders retained their original colors, indicating the preservation of original transition metal dicyanoaurates. Upon decanting the solution, the powders were washed with successive aliquots of water (3 × 10 mL), and were characterized by IR spectroscopy. The IR spectroscopy data showed that the bivalent transition-metal Co2+, Ni2+, Cu2+, and Zn2+ cations are not exchanged with Me2Sn2+ cations. Thus, these transition-metal dicyanoaurates cannot be converted back into the starting compound 2.
References
Achim C, Shatruk M, Dragulescu-Andrasi A, Chambers KE, Stoian SA, Bominaar EL, Dunbar KR (2007) Properties of Prussian blue materials manifested in molecular complexes: observation of cyanide linkage isomerism and spin-crossover behavior in pentanuclear cyanide clusters. J Am Chem Soc 129(19):6104–6116. doi:10.1021/Ja066273x
Goodwin AL, Calleja M, Conterio MJ, Dove MT, Evans JSO, Keen DA, Peters L, Tucker MG (2008) Colossal positive and negative thermal expansion in the framework material Ag3[Co(CN)6]. Science 319(5864):794–797. doi:10.1126/science.1151442
Kepert CJ, Phillips AE, Goodwin AL, Halder GJ, Southon PD (2008) Nanoporosity and exceptional negative thermal expansion in single-network cadmium cyanide. Angew Chem Int Edit 47(8):1396–1399. doi:10.1002/anie.200704421
Balch AL, Pham DM, Rios D, Olmstead MM (2005) Assisted self-association of dicyanoaurate, [Au(CN)2]–, and dicyanoargentate, [Ag(CN)2]–, through hydrogen bonding to metal ammonia complexes. Inorg Chim Acta 358(14):4261–4269. doi:10.1016/j.ica.2005.06.033
Balch AL, Stork JR, Rios D, Pham D, Bicocca V, Olmstead MM (2005) Metal–metal interactions in platinum(II)/gold(I) or platinum(II)/silver(I) salts containing planar cations and linear anions. Inorg Chem 44(10):3466–3472. doi:10.1021/Ic048333a
Leznoff DB, Katz MJ, Sakai K (2008) The use of aurophilic and other metal–metal interactions as crystal engineering design elements to increase structural dimensionality. Chem Soc Rev 37(9):1884–1895. doi:10.1039/B709061g
Leznoff DB, Lefebvre J (2005) Coordination polymers with cyanoaurate building blocks: potential new industrial applications for gold. Gold Bull 38(2):47–54
Patterson HH, Rawashdeh-Omary MA, Omary MA, Fackler JP (2001) Excited-state interactions for [Au(CN) –2 ] n and [Ag(CN) –2 ] n oligomers in solution. Formation of luminescent gold–gold bonded excimers and exciplexes. J Am Chem Soc 123(45):11237–11247. doi:10.1021/Ja011176j
Balch AL, Stender M, White-Morris RL, Olmstead MM (2003) New structural features of unsupported chains of metal ions in luminescent [(NH3)4Pt][Au(CN)2]2·1.5(H2O) and related salts. Inorg Chem 42(15):4504–4506. doi:10.1021/Ic034383o
Assefa Z, Destefano F, Garepapaghi MA, Lacasce JH, Ouellete S, Corson MR, Nagle JK, Patterson HH (1991) Photoluminescence and electronic-structure of Tl[Au(CN)2]—evidence for relativistic effects in thallium–gold and gold–gold interactions. Inorg Chem 30(14):2868–2876
Liao DZ, Dong W, Zhu LN, Sun YQ, Liang M, Liu ZQ, Jiang ZH, Yan SP, Cheng P (2003) 3D porous and 3D interpenetrating triple framework structures constructed by aurophilicity-coordination interplay in {Mn[Au(CN)2]2(H2O)2} n and {KFe[Au(CN)2]3} n . Chem Commun 20:2544–2545. doi:10.1039/B306026h
Leznoff DB, Katz MJ, Ramnial T, Yu HZ (2008) Polymorphism of Zn[Au(CN)2]2 and its luminescent sensory response to NH3 vapor. J Am Chem Soc 130(32):10662–10673. doi:10.1021/Ja801773p
Leznoff DB, Lefebvre J, Batchelor RJ (2004) Cu[Au(CN)2]2(DMSO)2: golden polymorphs that exhibit vapochromic behavior. J Am Chem Soc 126(49):16117–16125. doi:10.1021/Ja049069n
Leznoff DB, Katz MJ, Kaluarachchi H, Batchelor RJ, Bokov AA, Ye ZG (2007) Highly birefringent materials designed using coordination polymer synthetic methodology. Angew Chem Int Edit 46(46):8804–8807. doi:10.1002/anie.200702885
Kroeker S, Katz MJ, Michaelis VK, Aguiar PM, Yson R, Lu H, Kaluarachchi H, Batchelor RJ, Schreckenbach G, Patterson HH, Leznoff DB (2008) Structural and spectroscopic impact of tuning the stereochemical activity of the lone pair in lead(II) cyanoaurate coordination polymers via ancillary ligands. Inorg Chem 47(14):6353–6363. doi:10.1021/Ic800425f
Leznoff DB, Katz MJ (2009) Highly birefringent cyanoaurate coordination polymers: the effect of polarizable C–X bonds (X = Cl, Br). J Am Chem Soc 131(51):18435–18444. doi:10.1021/Ja907519c
Ye ZG, Katz MJ, Aguiar PM, Batchelor RJ, Bokov AA, Kroeker S, Leznoff DB (2006) Structure and multinuclear solid-state NMR of a highly birefringent lead-gold cyanide coordination polymer. J Am Chem Soc 128(11):3669–3676. doi:10.1021/Ja0566634
Kroeker S, Greer BJ, Michaelis VK, Katz MJ, Leznoff DB, Schreckenbach G (2011) Characterising lone-pair activity of lead(II) by 207Pb solid-state NMR spectroscopy: coordination polymers of [N(CN)2]− and [Au(CN)2]− with terpyridine ancillary ligands. Chem-Eur J 17(13):3609–3618. doi:10.1002/chem.201002913
Leznoff DB, Korcok JL, Katz MJ (2009) Impact of metallophilicity on “colossal” positive and negative thermal expansion in a series of isostructural dicyanometallate coordination polymers. J Am Chem Soc 131(13):4866–4871. doi:10.1021/Ja809631r
Kepert CJ, Goodwin AL, Kennedy BJ (2009) Thermal expansion matching via framework flexibility in zinc dicyanometallates. J Am Chem Soc 131(18):6334–6335. doi:10.1021/Ja901355b
xReal JA, Niel V, Thompson AL, Munoz MC, Galet A, Goeta ASE (2003) Crystalline-state reaction with allosteric effect in spin-crossover, interpenetrated networks with magnetic and optical bistability. Angew Chem Int Edit 42(32):3760–3763. doi:10.1002/anie.200351853
Real JA, Galet A, Munoz MC (2006) Coordination polymers undergoing spin crossover and reversible ligand exchange in the solid. Chem Commun 41:4321–4323. doi:10.1039/B606434e
Real JA, Galet A, Munoz MC, Martinez V (2004) Supramolecular isomerism in spin crossover networks with aurophilic interactions. Chem Commun 20:2268–2269. doi:10.1039/B409974e
Real JA, Agusti G, Gaspar AB, Munoz MC (2007) Thermal- and pressure-induced cooperative spin transition in the 2D and 3D coordination polymers {Fe(5-Br-pmd)z[M(CN)x]y} (M = AgI, AuI, NiII, PdII, PtII). Inorg Chem 46(23):9646–9654. doi:10.1021/Ic700993s
Real JA, Agusti G, Munoz MC, Gaspar AB (2008) Spin-crossover behavior in cyanide-bridged iron(II)-gold(I) bimetallic 2D Hofmann-like metal-organic frameworks. Inorg Chem 47(7):2552–2561. doi:10.1021/Ic701865k
Deak A, Tunyogi T, Palinkas G (2009) Synthesis and structure of a cyanoaurate-based organotin polymer exhibiting unusual ion-exchange properties. J Am Chem Soc 131(8):2815–2817. doi:10.1021/Ja809067t
Eisenmann ET (1977) The precipitation of KCo[Au(CN)2]3 and similar cyanoaurate complexes. J Electrochem Soc 124:1957–1958
Eisler R (2003) Chrysotherapy: a synoptic review. Inflamm Res 52(12):487–501. doi:10.1007/s00011-003-1208-2
Graham GG, Whitehouse MW, Bushell GR (2008) Aurocyanide, dicyano-aurate (I), a pharmacologically active metabolite of medicinal gold complexes. Inflammopharmacology 16(3):126–132. doi:10.1007/s10787-007-0020-y
Burns RG (1993) Mineralogical applications of crystal field theory. Cambridge University Press, Great Britain
Jobbagy C, Tunyogi T, Palinkas G, Deak A (2011) A versatile solvent-free mechanochemical route to the synthesis of heterometallic dicyanoaurate-based coordination polymers. Inorg Chem 50(15):7301–7308. doi:10.1021/Ic200893n
Penneman RA, Staritzky E, Jones LH (1956) Preparation and some properties of HAu(CN)2. J Am Chem Soc 78:62
Soliman TM, Etaiw SEH, Fendesak G, Fischer RD (1991) Stabilization of the binuclear organotin(IV) cation [(μ-OH)(Me3Sn)2]+ within the planar, heterobimetallic macrocyclic anion—[((μ-OH)(Me3Sn)2)2((μ-NC)2Ni(CN)2)2]2–. J Organomet Chem 415(1):C1–C5
Siebel E, Fischer RD, Davies NA, Apperley DC, Harris RK (2000) The organometallic double metal cyanide [(Me2Sn)3{Co(CN)6}2·6H2O]. A three-dimensional framework of infinite, stapled ribbons. J Organomet Chem 604(1):34–42
Perreault D, Drouin M, Michel A, Miskowski VM, Schaefer WP, Harvey PD (1992) Silver and gold dimers—crystal and molecular-structures of Ag2(Dmpm)2Br2 and [Au2(Dmpm)2](PF6)2 and relation between metal metal force-constants and metal–metal separations. Inorg Chem 31(4):695–702
Clark RJH, Tocher JH, Fackler JP, Neira R, Murray HH, Knackel H (1986) A vibrational study by Raman-spectroscopy of some dinuclear gold ylide complexes. J Organomet Chem 303(3):437–442
Fackler JP, Assefa Z, Omary MA, McBurnett BG, Mohamed AA, Patterson HH, Staples RJ (2002) Syntheses, structure, and photoluminescence properties of the 1-dimensional chain compounds [(TPA)2Au][Au(CN)2] and (TPA)AuCl (TPA = 1,3,5-triaza-7-phosphaadamantane). Inorg Chem 41(24):6274–6280. doi:10.1021/Ic025784r
Schmidbaur H (2000) The aurophilicity phenomenon: a decade of experimental findings, theoretical concepts and emerging applications. Gold Bull 33(1):3–10
Schmidbaur H, Schier A (2008) A briefing on aurophilicity. Chem Soc Rev 37(9):1931–1951. doi:10.1039/B708845k
Schmidbaur H, Graf W, Muller G (1988) Weak intramolecular bonding relationships—the conformation-determining attractive interaction between gold(I) centers. Angew Chem Int Ed Engl 27(3):417–419
Scherbaum F, Grohmann A, Huber B, Kruger C, Schmidbaur H (1988) Aurophilicity as a consequence of relativistic effects—the hexakis(triphenylphosphaneaurio) methane dication [(Ph3PAu)6 C]2+. Angew Chem Int Ed Engl 27(11):1544–1546
Nagasundaram N, Roper G, Biscoe J, Chai JW, Patterson HH, Blom N, Ludi A (1986) Single-crystal luminescence study of the layered compound KAu(CN)2. Inorg Chem 25(17):2947–2951
Balch AL, Stender M, Olmstead MM, Rios D, Attar S (2003) Cation and hydrogen bonding effects on the self-association and luminescence of the dicyanoaurate ion, [Au(CN)2]–. Dalton Trans 22:4282–4287. doi:10.1039/B310085e
Long JR, Dinca M (2008) Hydrogen storage in microporous metal-organic frameworks with exposed metal sites. Angew Chem Int Ed 47(36):6766–6779. doi:10.1002/anie.200801163
Long JR, Kaye SS (2007) Hydrogen adsorption in dehydrated variants of the cyano-bridged framework compounds A2Zn3[Fe(CN)6]2·xH2O (A = H, Li, Na, K, Rb). Chem Commun 43:4486–4488. doi:10.1039/B709082j
Thallapally PK, Motkuri RK, Fernandez CA, McGrail BP, Behrooz GS (2010) Prussian blue analogues for CO2 and SO2 capture and separation applications. Inorg Chem 49(11):4909–4915. doi:10.1021/Ic902397w
Culp JT, Smith MR, Bittner E, Bockrath B (2008) Hysteresis in the physisorption of CO2 and N2 in a flexible pillared layer nickel cyanide. J Am Chem Soc 130(37):12427–12434. doi:10.1021/Ja802474b
Reguera E, Zamora B, Autie M, Contreras JL, Centeno M (2010) Separation of oxygen and nitrogen by porous cyanometallates. Sep Sci Technol 45(5):692–699. doi:10.1080/01496390903571101
Suh MP, Prasad TK, Hong DH (2010) High gas sorption and metal-ion exchange of microporous metal-organic frameworks with incorporated imide groups. Chem-Eur J 16(47):14043–14050. doi:10.1002/chem.201002135
Long JR, Kaye SS, Choi HJ (2008) Generation and O2 adsorption studies of the microporous magnets CsNi[Cr(CN)6] (T C = 75 K) and Cr3[Cr(CN)6]2·6H2O(T N = 219 K). J Am Chem Soc 130(50):16921–16925. doi:10.1021/Ja803926y
Kaye SS, Long JR (2005) Hydrogen storage in the dehydrated Prussian blue analogues M3[Co(CN)6]2 (M = Mn, Fe, Co, Ni, Cu, Zn). J Am Chem Soc 127(18):6506–6507. doi:10.1021/Ja051168t
Sonier JE, Lefebvre J, Tyagi P, Trudel S, Pacradouni V, Kaiser C, Leznoff DB (2009) Magnetic frustration and spin disorder in isostructural M(μ-OH2)2[Au(CN)2]2 (M = Mn, Fe, Co) coordination polymers containing double aqua-bridged chains: SQUID and mu SR studies. Inorg Chem 48(1):55–67. doi:10.1021/Ic801094m
Leznoff DB, Lefebvre J, Chartrand D (2007) Synthesis, structure and magnetic properties of 2-D and 3-D [cation] M[Au(CN)2]3 (M = Ni, Co) coordination polymers. Polyhedron 26(9–11):2189–2199. doi:10.1016/j.poly.2006.10.045
Leznoff DB, Lefebvre J, Callaghan F, Katz MJ, Sonier JE (2006) A new basic motif in cyanometallate coordination polymers: structure and magnetic behavior of M(μ-OH2)2[Au(CN)2]2 (M = Cu, Ni). Chem-Eur J 12(26):6748–6761. doi:10.1002/chem.200600303
Acknowledgments
We gratefully acknowledge the financial support of this work by the Hungarian Scientific Research Funds (OTKA) K68498 project and National Development Agency (Project ID: KMOP-1.1.2-07/1-2008-0002). We are grateful to Dr. Csaba Németh for his assistance in recording the FTIR spectra. We also thank Mrs. Márta Rockov for the EA measurements.
Open Access
This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Deák, A., Tunyogi, T., Jobbágy, C. et al. Cyanide-bridged bimetallic multidimensional structures derived from organotin(IV) and dicyanoaurate building blocks: ion exchange, luminescence, and gas sorption properties. Gold Bull 45, 35–41 (2012). https://doi.org/10.1007/s13404-012-0041-1
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13404-012-0041-1