
Abstract
Background
Colorectal cancer (CRC) is a cumulative term applied to a clinically and genetically heterogeneous group of neoplasms that occur in the bowel. Based on twin studies, up to 45 % of the CRC cases may involve a heritable component. Yet, only in 5–10 % of these cases high-penetrant germline mutations are found (e.g. mutations in APC and DNA mismatch repair genes) that result in a familial aggregation and/or an early onset of the disease. Genome-wide association studies have revealed that another ~5 % of the CRC cases may be explained by a cumulative effect of low-penetrant risk factors. Recent attempts to identify novel genetic factors using whole exome and whole genome sequencing has proven to be difficult since the remaining, yet to be discovered, high penetrant CRC predisposing genes appear to be rare. In addition, most of the moderately penetrant candidate genes identified so far have not been confirmed in independent cohorts. Based on literature examples, we here discuss how careful patient and cohort selection, candidate gene and variant selection, and corroborative evidence may be employed to facilitate the discovery of novel CRC predisposing genes.
Conclusions
The picture emerges that the genetic predisposition to CRC is heterogeneous, involving complex interplays between common and rare (inter)genic variants with different penetrances. It is anticipated, however, that the use of large clinically well-defined patient and control datasets, together with improved functional and technical possibilities, will yield enough power to unravel this complex interplay and to generate accurate individualized estimates for the risk to develop CRC.
Similar content being viewed by others


1 Colorectal cancer
Colorectal cancer (CRC) is the third most commonly diagnosed cancer in males and the second in females, with worldwide over 1.3 million new cancer cases and over 650,000 deaths reported each year [1]. The incidence of CRC is strongly age-dependent, increasing from the age of 40 years and reaching a median age of 70 years in the general population [2]. CRC is not a single disease, but rather a heterogeneous group of malignancies originating from precursor cells within the gastrointestinal tract between the cecum and the anus [3]. Clinically, these malignancies can differ in (i) localization (e.g. proximal or distal), (ii) pathology/histology (e.g. adenocarcinoma/serrated adenocarcinoma) and (iii) invasiveness/metastatic behaviour (e.g. loco-regional or distant organ site) [3]. About 90 % of CRCs present as adenocarcinomas originating from the epithelial lining of the colon [4]. Other types of CRC include neuroendocrine, adenosquamous, signet ring cell, squamous cell, spindle cell and undifferentiated carcinomas [4]. Although every CRC is unique, it is generally believed that tumours with similar clinicopathologic characteristics arise and behave in a similar way [5]. Along with the increase in our understanding of the pathology and etiology of CRCs in recent decades, improved classification systems have been developed [5, 6]. An example of this progress is the development of the pathological Dukes classification system [7] to the TNM staging system that grades tumour growth and dissemination to lymph nodes and other organ sites [8]. Recently, high-resolution molecular profiling of somatic mutations has resulted in new insights in CRC sub-phenotyping [9]. While the complete mutation profiles are, as yet, complex and not well enough understood to be used in routine clinical practice, classification systems have been introduced that primarily rely on specific mutation spectra [6].
2 Etiologic and pathologic hallmarks of CRC
A milestone in the classification of CRCs was achieved when it was first postulated in the late 1980’s that CRCs develop via a multistep process that is accompanied by the sequential accumulation of genetic mutations, often occurring over many years [10]. A more detailed comprehension of this multistep processes may provide a basis for understanding how various developmental and pathological characteristics of CRCs contribute to the apparent heterogeneous manifestation of this disease.
The first formulation of the multistep carcinogenic process was postulated by Fearon and Vogelstein, who described the timing of genetic mutations that induce chromosomal instability (CIN) [11]. The CIN pathway includes molecular events that occur during the development from hyperproliferative colon epithelium to adenomatous lesions and, finally, invasive adenocarcinoma. It has been put forward that mutations in so-called ‘gatekeeper genes’ are the major drivers of tumorigenesis [12]. Based on a genetic study of early adenomatous lesions it was found that the majority of the earliest lesions carry a mutated, inactive, APC gene [13]. Concurrently, it was found that the frequency of inactivating APC mutations remained constant as tumours progressed from benign to malignant stages [13]. Based on these findings, inactivation of APC was proposed to represent an initiating event of the CIN pathway. Another genetic event conferring neoplastic properties to colonic cells was found to occur in the KRAS proto-oncogene in intermediate adenomas [10, 11]. Currently, it is thought that additional mutations that further propagate or ‘drive’ tumour development target genes and/or pathways that lead to various developmental and pathological hallmarks of cancer. These hallmarks include, for instance, the evasion from growth control (TGF-β signalling [14]), the acquisition of chromosomal/mitotic instability (spindle-assembly checkpoint [15, 16]) and the loss of cell cycle control and apoptosis (p53 pathway [17]). As a consequence, mutations that are required for CRC development can affect any of the pathways mentioned, which hence may lead to molecular and genetic heterogeneity [18]. In addition to the CIN pathway, at least two alternative pathways have been proposed, i.e., the CpG island methylator phenotype (CIMP) pathway and the microsatellite instability (MSI) pathway.
The CIMP and MSI pathways are characterized by different mutational mechanisms, but they also lead to the acquisition of cancer-specific features reminiscent to those seen in the CIN pathway [18]. The CIMP phenotype is related to widespread promoter CpG island methylation [5, 6]. Tumours with a CIMP ‘high’ phenotype are thought to arise from sessile serrated adenomas and these tumours carry specific mutations in the BRAF gene reminiscent to those encountered in the KRAS gene in CIN tumours [18]. It has been suggested that the CpG island methylation observed may silence specific tumour-related genes, including the mismatch repair gene MLH1, which may give rise to microsatellite instable tumours [5, 6]. MSI-positive tumours tend to have stable karyotypes (CIN-negative). Instead, their genomes feature instability at e.g. mono- or dinucleotide repeats such as (A)n or (CA)n, referred to as microsatellites [19]. The MSI phenotype can also manifest itself independent of CIMP. It has been shown that biallelic somatic mutations in the MLH1 or MSH2 genes can explain a sizeable fraction of tumours that are characterized by microsatellite instability [20–22]. These tumours tend to feature KRAS mutations more frequently than CIMP tumours [5, 6]. It should be noted, however, that the CIN, CIMP and MSI pathways are non-exclusive, and that alternative pathways and clinical entities are still being discovered as well [23–29]. In addition to somatic mutations, also, germline mutations may affect the ultimate tumour characteristics.
3 CRC predisposition syndromes
Concurrent with the formulation of the adenoma-carcinoma sequence in sporadic tumours, fine mapping of genomic lesions in the germline of patients with familial adenomatous polyposis (FAP) syndrome has pointed at the same APC gene [30–32]. It is assumed that in these patients germline inactivation of one APC allele markedly increases the chance of adenoma formation and that somatic inactivation of the second APC allele is a critical (rate-limiting) event in adenoma formation [18]. Presumably, once adenomas in FAP have developed, they progress at a rate akin to that seen in sporadic adenomas because of the need to acquire additional rate-limiting mutations for carcinoma development [18].
Subsequently, the genes underlying the development of a second distinct group of familial tumours, now known as Lynch Syndrome, were identified. Patients with this syndrome differ from FAP patients in that they generally show a non-polyposis phenotype, with CRCs at a relatively young age and an excess of extra-colonic tumours [33]. The genes underlying Lynch Syndrome that were consecutively identified include MSH2 [34, 35], MLH1 [36, 37], PMS2 [38] and MSH6 [39]. In addition, our group found that 3′ EPCAM deletions underlie a heritable form of epigenetic silencing of the MSH2 gene [40]. It has been proposed that Lynch tumours result from major increases in mutation rates in the adenomatous lesions itself, thus leading to an accelerated progression towards carcinomas [18].
To facilitate the identification of individuals at risk for CRC development, clinical guidelines have been issued [41], including (i) a strong family history of colorectal cancers or polyps, (ii) multiple primary cancers in a patient with CRC, (iii) the occurrence of other cancers within a kindred consistent with a known CRC causing syndrome, and (iv) a relatively young age at the initial time of diagnosis. For a suspicion of specific syndromes further criteria may be added. For instance, FAP will generally be suspected in patients when at least 100 colonic adenomas are identified [42], and the presence of microsatellite instability (MSI) in the tumour in combination with a positive family history may serve as hallmarks of Lynch syndrome [43]. Besides its specific application to identify patients for example in FAP and Lynch syndrome, such clinical guidelines have also been used to distinguish additional CRC predisposition syndromes from sporadic forms of CRC.
4 Additional CRC predisposition syndromes
In addition to the most prevalent FAP and Lynch syndromes, germline mutations and aberrations in pathways relevant to cancer development have been identified in more rare highly penetrant Mendelian CRC predisposition syndromes (Table 1). Many of these genes were identified through targeted investigation of candidate genes. Among the first syndromes to be discovered this way were the hamartomatous polyposis syndromes Peutz-Jegher’s syndrome (PJS) and juvenile polyposis syndrome (JPS). These syndromes are characterized by mutations in the chromatin remodelling gene LBK1/STK11 [44, 45] and the TGF-β signalling genes SMAD4 and BMPR1A [46, 47], respectively. In later studies autosomal recessively inherited mutations in the DNA repair gene MUTYH were found to give rise to a specific subtype of polyposis, referred to as MUTYH-associated polyposis (MAP) [23]. Recently, genome-wide screening efforts led to the identification of mutations affecting the expression of GREM1 as the cause of hereditary mixed polyposis syndrome (HMPS) [48] and mutations in the POLD1 and POLE genes as the cause of polymerase proofreading-associated polyposis (PPAP) syndrome [27]. In addition, our group very recently identified mutations in the NTHL1 gene as the cause of NTHL1-associated polyposis (NAP) syndrome [29].
Besides the above-mentioned Mendelian CRC syndromes, CRC predisposition has also been observed in patients affected by various developmental syndromes (Table 1), including Bloom syndrome, which is caused by mutations in the BLM gene [49]. Also oligodontia-colorectal cancer syndrome, which is caused by defects in the AXIN2 gene, is associated with CRC predisposition [50]. In addition, an increased CRC risk has been reported in cases of Cowden syndrome [51] and Li-Fraumeni syndrome [52], which are caused by defects in the AKT signalling regulator PTEN and the cell cycle regulator TP53, respectively.
5 Missing heritability of CRC
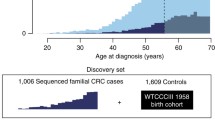
Together, the above-mentioned Mendelian cancer syndromes account for ~5–10 % of the total burden of CRC [33] (Fig. 1). Based on twin studies from the Swedish, Danish and Finnish twin registries, the heritability of CRC was suggested to be on average ~ 30 % (range: 15–45 %) [55–57]. The apparent discrepancy between the fraction accounted for by Mendelian cancer syndromes and the aforementioned estimates have led to the notion of ‘missing heritability’ [58–60]. It is thought that this missing heritability may be multifactorial in nature, i.e., may be associated with moderately to lowly penetrant genetic variants that, possibly in conjunction with environmental factors, may fail to give rise to clear-cut dominant or recessive inheritance patterns [61]. The more common heritable forms may still include some rare high-risk Mendelian non-syndromic forms of CRC, but likely exist predominantly of intermediate familial risk forms of CRC and relatively low-risk forms of CRC that depend on interactions between genetic and environmental factors [61, 62] (Fig. 1).

Distribution of CRC cases by familial background. Colorectal cancers can be divided into isolated cases, familial cases and hereditary cases. The latter result from known predisposition syndromes, like Lynch syndrome and familial adenomatous polyposis (FAP) syndrome. Additional, less frequent syndromes include polymerase proofreading-associated polyposis (PPAP), MUTYH-associated-polyposis (MAP), Peutz-Jegher’s syndrome (PJS), Juvenile Polyposis syndrome (JPS), NTHL1-associated polyposis (NAP), and some other colorectal cancer-associated syndromes as described in the main text. Currently, these forms of CRC predisposition account for 5–10 % of all cases. The black dots represent additional early-onset cases below the age of 50. About 10 % of CRC cases belong to this group. These cases can thus be familial or sporadic in origin and may result from variability in penetrance of the underlying risk alleles as described in the text. Modified from [53, 54].
In the search for this missing heritability, two contrasting hypotheses have been put forward: the common disease-common variant (CDCV) hypothesis and the common disease-rare variant (CDRV) hypothesis [63]. The CDCV hypothesis postulates that low-risk susceptibility variants occurring in >1 % of the population may contribute to the development of CRC [63]. In contrast, the CDRV hypothesis postulates that many rare variants, with frequencies <1 % in the population, may contribute to the development of CRC [61, 63]. Most of the initial candidate gene studies lacked the power and genomic resolution to address this latter hypothesis [64] and high-throughput identification of variants found in more than 1 % of the population only became feasible after the introduction of massively parallel genotyping techniques enabling genome-wide association studies (GWAS).
6 Genome-wide association studies
Since 2007, several genome-wide association studies (GWAS) have been conducted under the premise of the CDCV hypothesis for CRC [65–84]. These studies aimed to use unbiased genome-wide screens to identify non-random associations of alleles at nearby loci (linkage disequilibrium), which then served as proxies, or tagSNPs, for other nearby risk alleles [85]. To adequately power the detection of risk variants with small effect sizes through GWAS, the sizes of the case-control cohorts in the discovery phase have steadily been growing from roughly 1000 cases and controls included in the initial studies [65–67] to (meta)analyses of several 10,000 samples in the more recent studies [80, 86, 87]. In addition to these large cohorts, recent studies have also started to address the involvement of multi-ethnic disease loci [88].
Together, GWAS have led to the identification of approximately 25 distinct CRC risk loci [60, 89]. Some of the variants associated with CRC risk are in or near genes implicated in relevant functional pathways, such as the DNA repair [75], TGF-ß and MYC signalling pathways [65, 68–70, 80, 87]. As yet, however, a major fraction of the associated variants cannot be placed within any functional context [90]. Due to the small effect-sizes of the loci identified in GWAS, the clinical utility of the outcome of these studies has so far been limited [60, 89]. It has been estimated that individuals carrying multiple low-penetrance risk loci may have a 1–2 fold increase in their lifetime CRC risk [89]. Together, these common variants, including those that remain to be discovered, may explain another 5–10 % of the heritability of CRC [89], suggesting that a substantial fraction of its heritability may still be missing.
7 Common disease - rare variant hypothesis
The remainder of the CRC risk may be due to rare variants causing common non-Mendelian forms of CRC (CDRV paradigm) [61]. Features reminiscent to those of Mendelian syndromes characterize these CRCs in that they may occur at relatively young ages and may be found as familial aggregates. They are, however, distinct from the above-mentioned Mendelian forms in that the CRC patients usually do not exhibit a clearly recognizable clinical phenotype. Examples of the latter are the identification of germline mutations in the EPHB2 and GALNT12 genes through targeted screens of familial CRC patients [91–93]. Additionally, our group has previously implicated PTPRJ [94], BUB1 and BUB3 [95] variants in familial and early-onset CRC cases without polyposis and/or loss of mismatch repair capacity, which is again indicative of a contribution of rare variants to CRC predisposition.
Based on current knowledge of CRC susceptibility, a model has been suggested and expectations about the remaining genetic risk factors have been formulated (depicted in Fig. 2). According to this model, one side of the risk spectrum represents the highly penetrant variants underlying e.g. Mendelian cancer syndromes, and the other side of the risk spectrum represents the common low-penetrance variants identified by GWAS [62, 96]. It has been suggested that the majority of the missing heritability may involve variants with low minor allele frequencies (MAF), defined here as 0.5 % < MAF < 5 %, or rare variants (MAF < 0.5 %) [96]. Such variants are not frequent enough to be captured by current GWAS, nor do they exhibit sufficiently large effect sizes to be detected by classical linkage analyses in families [96]. Indeed, many of the above-mentioned examples of rare variants fall into this category [96]. Recently, the possibility has arisen to study more rare genomic variants through unbiased genome-wide screens using massively parallel sequencing (MPS) [97, 98] (Fig. 2). Indeed, recent reports implicating variants in RPS20 [28] and FAN1 [99] in familial CRC predisposition support the potential of MPS to identify rare variants with a moderate penetrance.

Model explaining the penetrance of genetic risk factors for CRC. In the top panel, the genetic risk factors are shown. The risk factors that, according to current knowledge, do contribute most to CRC are shown in the grey shaded bar. Highly penetrant mutations, like those found in FAP syndrome, Lynch syndrome and other hereditary/familial syndromes, are at the top left side of this plot. Usually, these syndromes are caused by very rare mutations. At the lower right side common low-penetrance risk factors are depicted. In between these two extremes, rare moderately penetrant risk factors may exist that still await discovery. The bottom panel marks the technologies that have led to the discovery of the various genetic risk factors, i.e., the light grey bar marks highly penetrant risk factors that have been identified by linkage analysis, the black bar marks risk factors that have been identified by GWAS. In addition, massively parallel sequencing (MPS) may allow the identification of additional moderately penetrant risk factors between these two extremes (dark grey bar)
8 Massively parallel sequencing and cancer predisposition
While it took almost 20 years for the first draft of the human genome to be completed [100, 101], the recent application of MPS, and in particular whole-exome and whole-genome sequencing (WES and WGS, respectively), allows the generation of comparable data within a few days at highly reduced costs [97, 102, 103]. Particularly for Mendelian traits, MPS has allowed the identification of numerous disease genes (reviewed in [104–107]) including a number of genes underlying rare highly penetrant well-phenotyped cancer predisposition syndromes. For CRC, this approach has resulted in the identification of distinct syndromes, such as PPAP [27] and NAP [29], which are caused by rare variants with a presumably high penetrance. Currently, it is unclear how many CRC cases can be explained by these syndromes, as the exact frequencies of the underlying variants still have to be established.
Next to these highly penetrant risk factors, the identification of low- to moderately-penetrant CRC risk factors remains of major interest [108]. Their identification, however, remains complex. Studies that focused on the identification of such risk factors were mainly based on (i) familial CRC cases [99, 109–111], (ii) sporadic early-onset CRC cases [112, 113] or (iii) combinations of such categories [57, 114, 115]. Unfortunately, the cohort sizes in these studies have thus far been relatively small and the identified candidates have not always been confirmed in independent studies. A good example of the latter are pathogenic mutations in the BLM gene. Recently, we identified two carriers of a pathogenic BLM mutation by exome sequencing in a cohort of 55 early-onset CRC patients (≤ 45 years of age) [115], the relevance of which could be confirmed in an additional larger cohort of early-onset CRC patients [116]. Still, the interpretation of studies aimed at the detection of low- to moderately-penetrant CRC factors remains difficult.
Several factors may underlie the complexity and limited success of these studies. First, many large-scale sequencing projects have yielded the conclusion that the human genome harbours a plethora of non-causative, possibly detrimental, rare variants [117–122]. As a result, it has become a major interpretation challenge to point out the causative variants and genes within a vast number of candidates. Secondly, these variants may not lead to obvious inheritance patterns per se, since their ultimate effect on the phenotype may rely on an interplay with other genetic and/or environmental factors [62]. Consequently, not all referral criteria used for the identification of high risk syndromes as outlined above may apply. Conversely, the distinction from individuals with sporadic non-inherited tumours is not clear-cut, making it impossible to distinguish between cases with or without predisposition on a case-by-case basis. Thirdly, since corroborative evidence is still missing for many novel candidates, and many variants and genes remain to be discovered, there is a lack of knowledge about the biological specifications of such variants.
9 Guidelines for gene discovery
9.1 Lessons learned from Mendelian cancer syndromes
In 2009, PALB2 was reported as the first candidate cancer predisposition gene discovered by MPS [123]. Since then, MPS has been spearheading identification strategies for novel cancer predisposition genes. Especially the discovery of novel Mendelian forms of cancer predisposition from well-selected cohorts of patients by WES and WGS appeared to be successful. This notion is exemplified by studies that have led to the identification of various novel candidate cancer predisposing genes, including GATA2, MAX, PALB2, SMARCE1, BAP1 and ERCC4, which predispose to the clinically well-defined entities Emberger-syndrome [124], pheochromocytoma [125], multiple spinal meningioma [126], melanocytic tumours [127] and Fanconi anaemia [128], respectively.
Beyond the successful identification of novel candidate genes for the rare cancer syndromes mentioned above, the methodologies and guidelines applied to these syndromes have benefited largely from those used in syndromes with Mendelian inheritance patterns [106, 107, 129]. As such, the success rates appear to be similar to the ~0.5 genes identified per disease studied with a Mendelian inheritance pattern [106]. The methodologies and criteria used to study these diseases encompass a couple of distinct entities, i.e., (1) patient and cohort selection, (2) candidate gene and variant selection and (3) corroborative evidence to support causality, including co-segregation, recurrence, functional and somatic evidence (Fig. 3).

Strategy for (colorectal) cancer predisposition gene identification. Commonalities of recently published successful MPS studies to identify cancer predisposition genes in hereditary CRC and potential improvements as described in the main text
9.1.1 Patient and control cohort selection
A well-designed study starts with a carefully selected cohort of patients and/or families, which will increase the chance to discover rare disease-causing candidate genes and/or variants (Fig. 3). Unfortunately, however, it is not always possible to select well-phenotyped cases. For some cancer syndromes, which are genetically homogeneous, this may not be a problem since candidate variants will be implicated through frequency if the number of cases included is large enough. This has recently been demonstrated e.g. by the finding of germline CTR9 mutations in 3 out of 35 patients with Wilms tumour [130]. However, for conditions such as CRC, the current cohorts of early-onset and familial cases appear to be too heterogeneous to use such strategies. It may, therefore, be necessary to apply more stringent definitions by e.g. performing endophenotyping (see below) of tumours or by focusing on rare phenotypes such as the occurrence of childhood CRC. In this way, germline mutations in the RPS20 gene could be identified using only a single four-generation family, which is probably unique since similar mutations have so far not been reported in other families [28].
Another strategy may be a search for founder mutations. The recruitment of families with similar phenotypes from local populations may be of help for such strategies. This approach has e.g. led to the identification of POT1 mutations in cutaneous melanoma [131] and an NTHL1 variant in patients with hereditary polyposis by our group [29].
Studies of Mendelian disorders suggest that the patient selection step may be crucial for increasing the power of the studies to reveal rare alleles with a high to moderate penetrance. This is especially so since in a cohort of resembling patients it is more likely that a single gene or a few genes with similar function will underlie a specific phenotype. This, in turn, would be more obvious in a plethora of non-causative rare variants [117–122] than would be the identification of a candidate gene by mere prioritization on an ad hoc hypothesis basis with the exclusion of common variants. Possibly, some CRC risk factors may be too rare (e.g. RPS20) or their penetrance may be too low to give rise to a distinctly recognizable phenotype. Nevertheless, the advantages of unravelling Mendelian or near-Mendelian CRC conditions are two-fold. First, they may reveal novel pathways involved in CRC predisposition that can be used in further candidate gene identification approaches. Second, the removal of resolved cases from cohorts will increase the specificity of novel searches for the underlying genes and/or pathways (Fig. 3).
Another step to improve the power of studies is to assemble a carefully selected set of normal controls for comparison. As many Mendelian disorders are rare and highly penetrant, it is common to compare the frequency of variants to population-matched controls from public databases, as exemplified by studies that identified the cancer predisposition genes MAX [126] and POLD1/POLE [27]. Given the fact that CRC is a common phenotype, selecting age- and population-matched controls may not be trivial, but unrelated gender- and age-matched family members may serve as proper and easily accessible controls.
9.1.2 Candidate gene and variant selection
The bottleneck in the MPS study design is rapidly moving away from the performance of genome-wide screens towards the development of bioinformatic tools for the selection of variants and the introduction of comprehensive pipelines for sequence analysis (see for detailed reviews [132, 133]). Particularly, the variant and gene selection step by which the vast number of identified variants is reduced is important (Fig. 3). The approach chosen for prioritization depends on prior knowledge of the patient selection criteria (as described above), the damaging effect of variants encountered in the genes, the inheritance model and the function of the genes affected [129]. Such approaches are not trivial as they are based on selecting one or two causative variants from several million variants that usually result from genome-wide sequencing efforts [117–122]. Overlap, linkage, trio-based sequencing (i.e., patient and parents) and shared homozygosity can be used to augment the prioritization of variants and genes in the exomes of patients with Mendelian conditions [105, 106]. The application of such strategies is not limited to WES but can also be used for WGS [107, 134].
For oligogenic disorders, detailed knowledge of gene and variant function remains important. A thorough understanding of the underlying biology can be of help to specify pathways and gene functions. Gala and colleagues focused in a recent sessile serrated polyposis study, with an increased CRC risk, on truncating variants in oncogene-induced senescence pathways. In doing so, they found several variants in five genes that were enriched in 20 patient-derived exomes as compared to 4500 exomes from healthy individuals deposited in a public database [135]. Although at least one of these variants (p.E49* in PIF1) appears to be more common in the healthy population than originally assumed and, therefore, is less likely to be causative (Weren and Herwaarden, personal communication), the study illustrates how insight into the genetic and functional interplay between different genes can assist in variant prioritization. Obviously, such a selection may also introduce biases since it is limited to known pathways. Therefore, further corroborative evidence is required to establish the causality of particular genes and variants.
9.1.3 Corroborative evidence to support causality
In order to establish a causal relationship between a variant and its predisposition to cancer, additional evidence is usually required (Fig. 3). This evidence may include (1) recurrence in cases versus controls, (2) co-segregation in families, (3) second-hit mutations in tumours and (4) modelling functional effects.
Recurrence in cases versus controls One of the best ways to proof that a rare variant, or a rarely mutated gene, plays a role in a Mendelian disease is by independent replication in a validation cohort. Such a replication is considered to be a strong indication that a candidate variant is not the product of an ascertainment bias [129]. Most recent studies that utilized MPS to identify the cause of Mendelian cancer predisposition syndromes aimed at addressing this point [27, 123, 125–128, 130, 136, 137]. Also, for moderately penetrant variants such a replication is considered to be one of the key requirements to demonstrate that a specific or comparable variant recurrently occurs in other patients. To really proof an association, and to assess the variation in the normal population, screening of a large control cohort is needed, the size of which depends on the size of the effect to be investigated. As discussed below, however, further considerations to lend adequate statistical power to such screens will become increasingly important. In cases where replication turns out to be difficult beyond the initial discovery cohort, additional functional evidence or familial co-segregation may be employed to underscore pathogenicity.
Co-segregation in families To further assess the penetrance of a candidate risk allele and to check whether the anticipated inheritance model is correct, an assessment of the presence or absence of the candidate risk allele in respectively affected and unaffected family members should be performed. As most Mendelian cancer predisposition syndromes by definition feature either a dominant or a recessive pattern, this step can be very informative. Co-segregation analyses have, for instance, been used to substantiate the implication of POLD1, POLE and NTHL1 in CRC [27, 29]. Although there may be considerable ascertainment biases in the selection of families, and cancer syndromes may not exhibit a complete penetrance due to a multifactorial etiology, a familial risk is expected to contribute to our understanding of CRC development.
Translating this approach to moderately penetrant risk variants will, therefore, be important for an accurate risk assessment. A mutation may be significantly associated with early-onset CRC, but may not per se lead to a strong family history of CRC. This notion may be explained by the ‘genetic background’ in such families. Current study designs are not suited to detect polygenic inheritance in cancer. Indeed, as of today, only a few reports have been published that describe interactions between known predisposition genes, including MUTYH and OGG1 [138], MSH6 and APC [139], and MSH2 and APC [140].
Second-hit mutations in tumours One of the earliest forms of evidence used to support the causality of mutations was based on Knudson’s two-hit hypothesis [141]. Indeed, many of the well-established cancer predisposition genes, such as those underlying FAP and Lynch syndrome follow this scenario, as described above. Also, some of the recently discovered cancer predisposing genes appear to act in accordance with this scenario, such as for instance the MAX [125], SMARCE1 [126] and BAP1 [127] genes. Subsequent elaborate work on this scenario has revealed that (partial) deregulation of tumour suppressor genes and/or proto-oncogenes may add to the risk of tumour formation [142]. Therefore, this scenario should not be regarded as absolute. Perhaps, moderately penetrant risk factors may not exhibit second hits, as their effects are less prominently associated with gene dosage.
Instead of limiting mutation analyses to hits in the second allele of a candidate gene, more insight may be gained from genome-wide profiling through MPS. It is well-established now that specific cancer syndromes may follow distinct mutational pathways as has, for instance, been observed in Lynch syndrome (see above), and MUTYH-associated polyposis (MAP) [23]. Also, recent studies on CRC predisposition syndromes such as PPAP [27] and NAP [29] have revealed mutation profiles that are specifically associated with defects in DNA repair pathways. Given that MPS continuously improves the ease at which mutations can be detected, the generation of mutation profiles may become a cost-effective way to phenotype heritable tumours [9, 143, 144]. This concept, also known as endophenotyping, may make a clear difference in the study of CRC predisposition.
Modelling functional effects As novel genes and variants continue to be implicated in cancer predisposition, it is becoming increasingly imperative to unravel the functional consequences of such genes and variants. Well-established models, such as mouse models, are available for at least some of the currently known cancer predisposition syndromes. Existing knockout models of the POLE and NTHL1 genes have for instance been used to support their implication in CRC predisposition [27, 29]. In the absence of such in vivo models, in silico models can be used to make an educated guess of the functional consequences of specific variants. Ultimately, proof can be delivered by in vitro modelling. For variants in e.g. DNA repair genes this can be done through their exogenous expression in mammalian or yeast cells and assessment whether the sensitivity to DNA damaging agents has increased, as has been shown for ERCC4 [128] and POLE [27]. Also, the effects of variants on downstream signalling can be investigated. This has e.g. been done to show the effect of TGFBR1 variants on TGF-β signalling through SMAD2/3 [136]. Another example represents the influence of RHBDF2 variants on EGFR signalling [145]. In the case of SASH1 mutations, wound-healing assays on control and patient-derived fibroblasts were performed to reveal alterations in pathways governing cell migration [146]. It is anticipated that functional evidence will play an increasingly important role in CRC research. Indeed, in the recent literature, functional analyses have been used to implicate genes in gastrointestinal predisposition syndromes when other criteria such as replication could not be met due to the rarity of pathogenic mutations in the underlying genes. The implication of RPS20 in a single family with CRC (see above) was for instance supported by showing a defect in pre-rRNA maturation in patients with mutations in this gene [28]. Also, germline mutations in the IPMK gene were found to increase the resistance to apoptosis in cell lines derived from a single family with small intestinal carcinoids [147].
9.2 Beyond Mendelian disease gene identification
As alluded to above, novel insights gained from MPS approaches for Mendelian conditions may be limited in what they can contribute to our understanding of CRC predisposition. There are three possible reasons for this: (i) the underlying risk models may not be applicable to CRC, (ii) a significant part of the heritability may result from non-coding variants and (iii) not all familial CRC cases may result from genetic defects.
Firstly, few moderately penetrant genetic risk variants have been studied so far, and their contribution may deviate from currently known inheritance models. Most of our knowledge of moderately penetrant CRC risk variants stems from candidate gene approaches, of which only a few have significantly contributed to novel insights [64]. Interestingly, risk variants may also act as modifiers, which affect the penetrance, dominance, expressivity and pleiotropy of inherited traits [148]. An early example of a role of modifiers in CRC came from a FAP model in mice: APC min [149]. These mice have an average lifespan of 120 days in which they develop multiple polyps and CRC [149]. Through crossing with another mouse strain, a novel strain was obtained of which the mice lived disease-free for almost 300 days, suggesting that the APC min locus was influenced by a modifier [149]. Subsequently, the genetic locus for this modifier was mapped and named modifier of min (mom) [150]. In recent years several additional modifiers have been identified and mapped, indicating that modification is a recurring theme, particularly in the WNT signalling cascade [151–153]. Similar observations have been made in mouse models for DNA repair genes. For instance, in a model reminiscent of heterozygous BLM Ash carriers, in which CRC predisposition is only found in an APC min background [154], the overall predisposition effect depends both on BLM ablation and on the mouse strain used [155–157]. Such effects are generally not accounted for in Mendelian disease gene and GWAS studies. As a consequence, care should be taken when applying Mendelian prioritization guidelines to detect moderately penetrant CRC risk variants.
Secondly, the criteria used for Mendelian disease genetics are mostly based on the protein-coding regions of the genome (i.e., the exome). While the human exome itself still seems to be ill-defined [158], another significant part of the heritability may reside outside these regions of the genome. Many of the variants identified by GWAS are in fact located outside coding regions [90]. Also, variants implicated in Mendelian cancer syndromes may not necessarily be coding, examples of which include mutations in the MLH1 5’UTR region associated with CRC [159, 160] and presumed enhancers that may affect the expression of EPCAM-MSH2 read-through transcripts associated with CRC in EPCAM deletion carriers [161]. Likewise, a 40-kb duplication upstream of GREM1 has been linked to hereditary mixed polyposis syndrome by effecting an increased and ectopic expression of the BMP antagonist GREM1 [48]. Also primary epimutations, that is aberrations in gene expression due to an altered epigenotype not linked to genomic variation, have been suggested to play a role in cancer predisposition [162]. Examples include imprinting of the H19 locus in Beckwith–Wiedemann syndrome [163] and the de novo methylation of MLH1 associated with sporadic Lynch-syndrome [164–166]. It is reasonable to assume that many additional, also moderately penetrant, risk factors reside in non-coding regions. Therefore, care should be taken to not overrate the utility of exomes.
Thirdly, not all CRC cases with a suspected heritability may be caused by genetic variants. Recent work of Tomasetti and Vogelstein suggest that only a few mutations are required for CRC to become manifest [167, 168]. Their proposition raises some concern about how much of the CRC heritability still remains to be discovered. Furthermore, mere chance together with the above-mentioned modifying effects have not been taken into account in current heritability estimates [167, 169, 170]. Therefore, it is difficult to provide an accurate estimate of the remaining heritability of CRC and how much of it may be attributed to which class of variants.
Although the above limitations may appear challenging, some promising strategies and developments have been outlined in the current literature, i.e., (1) rare variant association studies, (2) whole-genome sequencing, (3) high-throughput functional genomics and (4) somatic driver mutation and germline mutation overlap (Fig. 3).
9.2.1 Rare variant association studies to power discoveries
Most of the studies presented in the survey above have sought to prioritize variants under the premise that they would be similar to Mendelian conditions. As already pointed out, the insights gained by these approaches have been limited. A particular shortcoming is the strong signal-to-noise ratios in WES and WGS experiments [171]. One way to filter out causative variants from ‘noise’, such as large numbers of neutral variants, may be to enhance the power of associations in large groups of cases and controls. Simulations have indicated that studies in which several thousands of samples are included may be adequately powered to detect loci explaining ~1 % of the phenotypic variance underlying a common dichotomous trait in WES screens [172]. It has been proposed that the power of such approaches could be enhanced by focusing on isolated populations, de novo mutations and/or specific genomic regions [171]. For WGS, probably much more cases and controls are needed, demanding cohort sizes similar or even larger than those used in recent GWAS. As GWAS consortia have already collected such cohorts, these may also be used for WGS. This notion may be a basis for future patient recruitment (Fig. 3).
9.2.2 WGS and the elucidation of non-coding and complex variation
From studies comparing WES with WGS, it has been concluded that WGS is much more sensitive than WES in calling single-nucleotide variants and short insertions/deletions (indels) at a comparable coverage [173, 174]. Moreover, it was found that WGS avoids the biases intrinsic to enrichment, thereby delivering improved uniformity of read coverage and reduced bias of allele ratios across the entire genome [173. This also translates into a greater ease in the detection of copy number variation across the genome by WGS [173]. It is considered likely that with further improvements in the technology towards applications of ‘third generation sequencing’, such as the Nanopore [175, 176] or PacBio platforms [177], increasing read lengths will allow the delineation of complex structural rearrangements and the phasing of variants by a single WGS experiment (Fig. 3). Currently, however, our understanding of non-coding variation is still limited. Therefore, a further development of conceptualized data interpretation approaches [178–180], as well as functional genomics, will be required.
9.2.3 High-throughput functional genomics
Recently, high-throughput functional technologies have become available to study specific gene functions and to develop human disease models through the application of genomic engineering via nuclease-induced genome modifications. These technologies allow forward as well as reverse genetic screens [181]. In particular, the CRISPR/Cas9 system has become a popular option based on its simplicity and versatility [182]. Findlay and colleagues already used the CRISPR/Cas9 system to e.g. mutagenize a region of 6 nucleotides in exon 18 of the BRCA1 gene with all possible hexamers and compared their impact on nonsense-mediated decay and exonic splicing [183].
Besides the recapitulation of specific variants, functional genetic analyses have become feasible in a much broader context in both in vitro and in vivo models. Promising models include organoids, which represent in vitro culture systems to grow cells in a three-dimensional (3D) organ-like fashion, such as intestinal epithelial structures derived from Lgr5-positive colonic crypt stem cells [184]. Importantly, these organoids are amenable to high-throughput genetic manipulation. Recently, the utility of these organoids was demonstrated by introducing mutations in the key driver tumour suppressor genes APC, SMAD4 and TP53, and the oncogenes KRAS and PIK3CA, in colonic organoids via a knock-out/knock-in approach to mimic tumorigenesis according to the classical adenoma-carcinoma sequence model [185, 186]. When comparing the tumorigenic and metastatic properties of these organoids, it was found that ‘driver’ pathway mutations are sufficient for clonal expansion and features of invasive carcinomas, but that additional molecular lesions are required for invasive behaviour [185, 186]. These findings underscore the notion that knowledge on genetic background and environmental variation may be important for our understanding of CRC development. As such, recent efforts to build organoid biobanks from CRC patients may hold promise for the elucidation of individual genetic and environmental risk factors [187] in a high-throughput setting. Last but not least, the findings may be translated into animal models and, ultimately, humans.
9.2.4 Using somatic driver mutations to identify novel predisposition genes
An interesting feature of many of the CRC predisposing variants is the apparent overlap between the corresponding pathways that drive cancer development somatically and those that predispose genetically. This notion is not new, as the very first models of cancer development such as the Knudson’s two-hit hypothesis discussed above originated from these parallels. Recent large-scale profiling efforts of cancer genomes have revealed genes frequently affected by somatic mutations in CRC [9, 143, 144]. These efforts were motivated by the aim to identify driver mutations. In a recent review, a comparison was made between drivers in the COSMIC (Catalogue of Somatic Mutations in Cancer) database featuring 468 genes that are somatically mutated in cancers, and known germline predisposition genes [188]. The result was that ~10 % of somatically mutated cancer genes also confer susceptibility to cancer when present as mutant variants in the germline, but that 40 % of the germline-mutated predisposition genes can also be mutated somatically in tumours [188]. The author suggested that this discrepancy may at least in part represent a bias due to the scarcity of studies on germline-associated mutations in tumours [188]. Indeed, holistic studies of somatic and germline variants in tumour material to reveal oncogenic drivers are getting increasingly popular [189]. Most of the current studies are limited because of their small numbers and the resulting lack of power. Therefore, more systematic screens of tumour and matched germline material using large patient cohorts will be needed. Since the screening for somatic mutations in cancer cells has been among the first fields to harness the power of MPS, and especially WGS [190], the methods used to identify driver mutations are better established than those to identify germline variants. Consequently, although missing driver variants have been reported, the identification of driver variants in (non-)coding regions [191] may lead to the discovery and improved prioritization of germline variants (Fig. 3).
10 Conclusions
With the advent of MPS, expectations have been high that this technology may be able to reveal much of the missing heritability of CRC. Next to identifying a number of strong candidate genes affected by clearly interpretable rare risk variants, the application of MPS to CRC has thus far led to a plethora of potential candidate genes whose role still remains to be established. In this review, we have outlined how further adjustments of experimental approaches and strategies may be of help to obtain final evidence on true CRC risk factors as also on their clinical relevance. Knowledge on functional networks of known (highly penetrant) genetic risk factors may be rewarding in this respect, as they may reveal novel pathways involved in cancer predisposition. In the end, it is anticipated that the availability of large multi-center datasets from clinically well-defined patients and controls will yield enough power to perform independent comprehensive meta-analyses, which may unravel the complex interplay between common and rare variants within genes or intergenic regions, and to make an accurate individualized estimate for the risk to develop CRC (and other clinical entities).
References
L. A. Torre, F. Bray, R. L. Siegel, J. Ferlay, J. Lortet-Tieulent, A. Jemal, Global cancer statistics, 2012. CA-Cancer J. Clin. 65, 87–108 (2015)
F. A. Haggar, R. P. Boushey, Colorectal cancer epidemiology: incidence, mortality, survival, and risk factors. Clin. Colon Rectal Surg. 22, 191–197 (2009)
C.A.R. S.R. Hamilton, B. Vogelstein, L.H. Sobin, S. Kudo, F. Fogt, E. Riboli, S.J. Winawer, S. Nakamura, D.E. Goldgar, P. Hainaut, J.R. Jass, in Pathology and Genetics of Tumours of the Digestive System ed. By S.R. Hamilton,. L.A. Aaltonen (IARC, Lyon, France, 2000), p. 103–142
M. Fleming, S. Ravula, S. F. Tatishchev, H. L. Wang, Colorectal carcinoma: pathologic aspects. J. Gastrointest. Oncol. 3, 153–173 (2012)
S. Ogino, A. Goel, Molecular classification and correlates in colorectal cancer. J. Mol. Diagn. 10, 13–27 (2008)
J. R. Jass, Classification of colorectal cancer based on correlation of clinical, morphological and molecular features. Histopathology 50, 113–130 (2007)
C. E. Dukes, The classification of cancer of the rectum. J. Pathol. 35, 323–332 (1932)
L. Sobin, M. Gospodarowicz and C. Wittekind (TNM Classification of Malignant Tumours, Wiley-Blackwell, 2009)
N. Cancer, Genome atlas. Comprehensive molecular characterization of human colon and rectal cancer Nature 487, 330–337 (2012)
B. Vogelstein, E. R. Fearon, S. R. Hamilton, S. E. Kern, A. C. Preisinger, M. Leppert, Y. Nakamura, R. White, A. M. Smits, J. L. Bos, Genetic alterations during colorectal-tumor development. N. Engl. J. Med. 319, 525–532 (1988)
E. R. Fearon, B. Vogelstein, A genetic model for colorectal tumorigenesis. Cell 61, 759–767 (1990)
B. Vogelstein, K. W. Kinzler, Cancer genes and the pathways they control. Nat. Med. 10, 789–799 (2004)
S. M. Powell, N. Zilz, Y. Beazer-Barclay, T. M. Bryan, S. R. Hamilton, S. N. Thibodeau, B. Vogelstein, K. W. Kinzler, APC mutations occur early during colorectal tumorigenesis. Nature 359, 235–237 (1992)
S. Markowitz, J. Wang, L. Myeroff, R. Parsons, L. Sun, J. Lutterbaugh, R. S. Fan, E. Zborowska, K. W. Kinzler, B. Vogelstein, M. Brattain, J. Willson, Inactivation of the type II TGF-beta receptor in colon cancer cells with microsatellite instability. Science 268, 1336–1338 (1995)
D. P. Cahill, C. Lengauer, J. Yu, G. J. Riggins, J. K. Willson, S. D. Markowitz, K. W. Kinzler, B. Vogelstein, Mutations of mitotic checkpoint genes in human cancers. Nature 392, 300–303 (1998)
R. Fodde, R. Smits, H. Clevers, APC, signal transduction and genetic instability in colorectal cancer. Nat. Rev. Cancer 1, 55–67 (2001)
S. J. Baker, E. R. Fearon, J. M. Nigro, S. R. Hamilton, A. C. Preisinger, J. M. Jessup, P. vanTuinen, D. H. Ledbetter, D. F. Barker, Y. Nakamura, R. White, B. Vogelstein, Chromosome 17 deletions and p53 gene mutations in colorectal carcinomas. Science 244, 217–221 (1989)
E. R. Fearon, Molecular genetics of colorectal cancer. Annu. Rev. Pathol. 6, 479–507 (2011)
E. Vilar, S. B. Gruber, Microsatellite instability in colorectal cancer-the stable evidence. Nat. Rev. Clin. Oncol. 7, 153–162 (2010)
J. M. Carethers, Differentiating lynch-like from lynch syndrome. Gastroenterology 146, 602–604 (2014)
A. R. Mensenkamp, I. P. Vogelaar, W. A. van Zelst-Stams, M. Goossens, H. Ouchene, S. J. Hendriks-Cornelissen, M. P. Kwint, N. Hoogerbrugge, I. D. Nagtegaal, M. J. Ligtenberg, Somatic mutations in MLH1 and MSH2 are a frequent cause of mismatch-repair deficiency in Lynch syndrome-like tumors. Gastroenterology 146, 643–646 e648 (2014)
W. R. Geurts-Giele, C. H. Leenen, H. J. Dubbink, I. C. Meijssen, E. Post, H. F. Sleddens, E. J. Kuipers, A. Goverde, A. M. van den Ouweland, M. G. van Lier, E. W. Steyerberg, M. E. van Leerdam, A. Wagner, W. N. Dinjens, Somatic aberrations of mismatch repair genes as a cause of microsatellite-unstable cancers. J. Pathol. 234, 548–559 (2014)
N. Al-Tassan, N. H. Chmiel, J. Maynard, N. Fleming, A. L. Livingston, G. T. Williams, A. K. Hodges, D. R. Davies, S. S. David, J. R. Sampson, J. P. Cheadle, Inherited variants of MYH associated with somatic G:C– > T:A mutations in colorectal tumors. Nat. Genet. 30, 227–232 (2002)
S. Jones, P. Emmerson, J. Maynard, J. M. Best, S. Jordan, G. T. Williams, J. R. Sampson, J. P. Cheadle, Biallelic germline mutations in MYH predispose to multiple colorectal adenoma and somatic G:C– > T:A mutations. Hum. Mol. Genet. 11, 2961–2967 (2002)
L. Lipton, S. E. Halford, V. Johnson, M. R. Novelli, A. Jones, C. Cummings, E. Barclay, O. Sieber, A. Sadat, M. L. Bisgaard, S. V. Hodgson, L. A. Aaltonen, H. J. Thomas, I. P. Tomlinson, Carcinogenesis in MYH-associated polyposis follows a distinct genetic pathway. Cancer Res. 63, 7595–7599 (2003)
E. M. F. De Sousa, X. Wang, M. Jansen, E. Fessler, A. Trinh, L. P. de Rooij, J. H. de Jong, O. J. de Boer, R. van Leersum, M. F. Bijlsma, H. Rodermond, M. van der Heijden, C. J. van Noesel, J. B. Tuynman, E. Dekker, F. Markowetz, J. P. Medema, L. Vermeulen, Poor-prognosis colon cancer is defined by a molecularly distinct subtype and develops from serrated precursor lesions. Nat. Med. 19, 614–618 (2013)
C. Palles, J. B. Cazier, K. M. Howarth, E. Domingo, A. M. Jones, P. Broderick, Z. Kemp, S. L. Spain, E. Guarino, I. Salguero, A. Sherborne, D. Chubb, L. G. Carvajal-Carmona, Y. Ma, K. Kaur, S. Dobbins, E. Barclay, M. Gorman, L. Martin, M. B. KovPalles, J. B. Cazier, K. M. Howarth, E. Domingoac, S. Humphray, C. Consortium, W. G. S. Consortium, A. Lucassen, C. C. Holmes, D. Bentley, P. Donnelly, J. Taylor, C. Petridis, R. Roylance, E. J. Sawyer, D. J. Kerr, S. Clark, J. Grimes, S. E. Kearsey, H. J. Thomas, G. McVean, R. S. Houlston, I. Tomlinson, Germline mutations affecting the proofreading domains of POLE and POLD1 predispose to colorectal adenomas and carcinomas. Nat. Genet. 45, 136–144 (2013)
T. T. Nieminen, M. F. O’Donohue, Y. Wu, H. Lohi, S. W. Scherer, A. D. Paterson, P. Ellonen, W. M. Abdel-Rahman, S. Valo, J. P. Mecklin, H. J. Jarvinen, P. E. Gleizes, P. Peltomaki, Germline mutation of RPS20, encoding a ribosomal protein, causes predisposition to hereditary nonpolyposis colorectal carcinoma without DNA mismatch repair deficiency. Gastroenterology 147, 595–598 e595 (2014)
R. D. Weren, M. J. Ligtenberg, C. M. Kets, R. M. de Voer, E. T. Verwiel, L. Spruijt, W. A. van Zelst-Stams, M. C. Jongmans, C. Gilissen, J. Y. Hehir-Kwa, A. Hoischen, J. Shendure, E. A. Boyle, E. J. Kamping, I. D. Nagtegaal, B. B. Tops, F. M. Nagengast, A. Geurts van Kessel, J. H. van Krieken, R. P. Kuiper, N. Hoogerbrugge, A germline homozygous mutation in the base-excision repair gene NTHL1 causes adenomatous polyposis and colorectal cancer. Nat. Genet. 47, 668–671 (2015)
J. Groden, A. Thliveris, W. Samowitz, M. Carlson, L. Gelbert, H. Albertsen, G. Joslyn, J. Stevens, L. Spirio, M. Robertson, L. Sargeant, K. Krapcho, E. Wolff, R. Burt, J. P. Hughes, J. Warrington, J. McPherson, J. Wasmuth, D. Le Paslier, H. Abderrahim, D. Cohen, M. Leppert, R. White, Identification and characterization of the familial adenomatous polyposis coli gene. Cell 66, 589–600 (1991)
K. W. Kinzler, M. C. Nilbert, L. K. Su, B. Vogelstein, T. M. Bryan, D. B. Levy, K. J. Smith, A. C. Preisinger, P. Hedge, D. McKechnie, R. Finniear, A. Markham, J. Groffen, M. Boguski, S. Altschul, A. Horii, H. Ando, Y. Miyoshi, Y. Miki, I. Nishisho, Y. Nakamura, Identification of FAP locus genes from chromosome 5q21. Science 253, 661–665 (1991)
I. Nishisho, Y. Nakamura, Y. Miyoshi, Y. Miki, H. Ando, A. Horii, K. Koyama, J. Utsunomiya, S. Baba, P. Hedge, Mutations of chromosome 5q21 genes in FAP and colorectal cancer patients. Science 253, 665–669 (1991)
H. T. Lynch, A. de la Chapelle, Hereditary colorectal cancer. N. Engl. J. Med. 348, 919–932 (2003)
R. Fishel, M. K. Lescoe, M. R. Rao, N. G. Copeland, N. A. Jenkins, J. Garber, M. Kane, R. Kolodner, The human mutator gene homolog MSH2 and its association with hereditary nonpolyposis colon cancer. Cell 75, 1027–1038 (1993)
F. S. Leach, N. C. Nicolaides, N. Papadopoulos, B. Liu, J. Jen, R. Parsons, P. Peltomaki, P. Sistonen, L. A. Aaltonen, M. Nystrom-Lahti, X. Guan, J. Zhang, P. Meltzer, J. Yu, K. Fa-Ten, D. Chen, K. Cerpsaletti, R. Fournier, S. Todd, T. Lewis, R. Leach, S. L. Naylor, J. Weissenbach, J. P. Mecklin, J. Heikki, G. M. Petersen, S. R. Hamilton, J. Green, J. Jass, P. Watson, H. T. Lynch, J. Trent, A. De la Chapelle, K. W. Kinzler, B. Vogelstein, Mutations of a mutS homolog in hereditary nonpolyposis colorectal cancer. Cell 75, 1215–1225 (1993)
C. E. Bronner, S. M. Baker, P. T. Morrison, G. Warren, L. G. Smith, M. K. Lescoe, M. Kane, C. Earabino, J. Lipford, A. Lindblom, P. Tannergård, R. J. Bollag, A. R. Godwin, D. C. Ward, M. Nordenskjold, R. Fishel, R. Kolodner, R. M. Liskay, Mutation in the DNA mismatch repair gene homologue hMLH1 is associated with hereditary non-polyposis colon cancer. Nature 368, 258–261 (1994)
N. Papadopoulos, N. C. Nicolaides, Y. F. Wei, S. M. Ruben, K. C. Carter, C. A. Rosen, W. A. Haseltine, R. D. Fleischmann, C. M. Fraser, M. D. Adams, J. C. Venter, S. R. Hamilton, G. M. Petersen, P. Watson, H. T. Lynch, P. Peltomaki, J. P. Mecklin, A. De la Chapelle, K. W. Kinzler, B. Vogelstein, Mutation of a mutL homolog in hereditary colon cancer. Science 263, 1625–1629 (1994)
N. C. Nicolaides, N. Papadopoulos, B. Liu, Y. F. Wei, K. C. Carter, S. M. Ruben, C. A. Rosen, W. A. Haseltine, R. D. Fleischmann, C. M. Fraser, M. D. Adams, J. C. Venter, M. G. Dunlop, S. R. Hamilton, G. M. Petersen, A. De la Chapelle, B. Vogelstein, K. W. Kinzler, Mutations of two PMS homologues in hereditary nonpolyposis colon cancer. Nature 371, 75–80 (1994)
M. Miyaki, M. Konishi, K. Tanaka, R. Kikuchi-Yanoshita, M. Muraoka, M. Yasuno, T. Igari, M. Koike, M. Chiba, T. Mori, Germline mutation of MSH6 as the cause of hereditary nonpolyposis colorectal cancer. Nat. Genet. 17, 271–272 (1997)
M. J. Ligtenberg, R. P. Kuiper, T. L. Chan, M. Goossens, K. M. Hebeda, M. Voorendt, T. Y. Lee, D. Bodmer, E. Hoenselaar, S. J. Hendriks-Cornelissen, W. Y. Tsui, C. K. Kong, H. G. Brunner, A. Geurts van Kessel, S. T. Yuen, J. H. van Krieken, S. Y. Leung, N. Hoogerbrugge, Heritable somatic methylation and inactivation of MSH2 in families with lynch syndrome due to deletion of the 3′ exons of TACSTD1. Nat. Genet. 41, 112–117 (2009)
E. M. Stoffel, C. R. Boland, Genetics and genetic testing in hereditary colorectal cancer. Gastroenterology 149, 1191–1203 e1192 (2015)
H. F. Vasen, I. Tomlinson, A. Castells, Clinical management of hereditary colorectal cancer syndromes. Nat. Rev. Gastroenterol. Hepatol. 12, 88–97 (2015)
A. Umar, C. R. Boland, J. P. Terdiman, S. Syngal, A. de la Chapelle, J. Ruschoff, R. Fishel, N. M. Lindor, L. J. Burgart, R. Hamelin, S. R. Hamilton, R. A. Hiatt, J. Jass, A. Lindblom, H. T. Lynch, P. Peltomaki, S. D. Ramsey, M. A. Rodriguez-Bigas, H. F. Vasen, E. T. Hawk, J. C. Barrett, A. N. Freedman, S. Srivastava, Revised Bethesda guidelines for hereditary nonpolyposis colorectal cancer (lynch syndrome) and microsatellite instability. J. Natl. Cancer Inst. 96, 261–268 (2004)
A. Hemminki, D. Markie, I. Tomlinson, E. Avizienyte, S. Roth, A. Loukola, G. Bignell, W. Warren, M. Aminoff, P. Hoglund, H. Jarvinen, P. Kristo, K. Pelin, M. Ridanpaa, R. Salovaara, T. Toro, W. Bodmer, S. Olschwang, A. S. Olsen, M. R. Stratton, A. de la Chapelle, L. A. Aaltonen, A serine/threonine kinase gene defective in Peutz-Jeghers syndrome. Nature 391, 184–187 (1998)
D. E. Jenne, H. Reimann, J. Nezu, W. Friedel, S. Loff, R. Jeschke, O. Muller, W. Back, M. Zimmer, Peutz-Jeghers syndrome is caused by mutations in a novel serine threonine kinase. Nat. Genet. 18, 38–43 (1998)
J. R. Howe, S. Roth, J. C. Ringold, R. W. Summers, H. J. Jarvinen, P. Sistonen, I. P. Tomlinson, R. S. Houlston, S. Bevan, F. A. Mitros, E. M. Stone, L. A. Aaltonen, Mutations in the SMAD4/DPC4 gene in juvenile polyposis. Science 280, 1086–1088 (1998)
J. R. Howe, J. L. Bair, M. G. Sayed, M. E. Anderson, F. A. Mitros, G. M. Petersen, V. E. Velculescu, G. Traverso, B. Vogelstein, Germline mutations of the gene encoding bone morphogenetic protein receptor 1 a in juvenile polyposis. Nat. Genet. 28, 184–187 (2001)
E. Jaeger, S. Leedham, A. Lewis, S. Segditsas, M. Becker, P. R. Cuadrado, H. Davis, K. Kaur, K. Heinimann, K. Howarth, J. East, J. Taylor, H. Thomas, I. Tomlinson, Hereditary mixed polyposis syndrome is caused by a 40-kb upstream duplication that leads to increased and ectopic expression of the BMP antagonist GREM1. Nat. Genet. 44, 699–703 (2012)
S. Balci, D. Aktas, Mucinous carcinoma of the colon in a 16-year-old Turkish boy with bloom syndrome: cytogenetic, histopathologic, TP53 gene and protein expression studies. Cancer Genet. Cytogenet. 111, 45–48 (1999)
L. Lammi, S. Arte, M. Somer, H. Jarvinen, P. Lahermo, I. Thesleff, S. Pirinen, P. Nieminen, Mutations in AXIN2 cause familial tooth agenesis and predispose to colorectal cancer. Am. J. Hum. Genet. 74, 1043–1050 (2004)
R. Pilarski, Cowden syndrome: a critical review of the clinical literature. J. Genet. Couns. 18, 13–27 (2009)
P. Wong, S. J. Verselis, J. E. Garber, K. Schneider, L. DiGianni, D. H. Stockwell, F. P. Li, S. Syngal, Prevalence of early onset colorectal cancer in 397 patients with classic Li-Fraumeni syndrome. Gastroenterology 130, 73–79 (2006)
R. W. Burt, Colon Cancer screening. Gastroenterology 119, 837–853 (2000)
H. T. Lynch, B. D. Riley, S. M. Weissman, S. M. Coronel, Y. Kinarsky, J. F. Lynch, T. G. Shaw, W. S. Rubinstein, Hereditary nonpolyposis colorectal carcinoma (HNPCC) and HNPCC-like families: problems in diagnosis, surveillance, and management. Cancer 100, 53–64 (2004)
P. Lichtenstein, N. V. Holm, P. K. Verkasalo, A. Iliadou, J. Kaprio, M. Koskenvuo, E. Pukkala, A. Skytthe, K. Hemminki, Environmental and heritable factors in the causation of cancer–analyses of cohorts of twins from Sweden, Denmark, and Finland. N. Engl. J. Med. 343, 78–85 (2000)
W. M. Grady, Genetic testing for high-risk colon cancer patients. Gastroenterology 124, 1574–1594 (2003)
L. A. Mucci, J. B. Hjelmborg, J. R. Harris, K. Czene, D. J. Havelick, T. Scheike, R. E. Graff, K. Holst, S. Moller, R. H. Unger, C. McIntosh, E. Nuttall, I. Brandt, K. L. Penney, M. Hartman, P. Kraft, G. Parmigiani, K. Christensen, M. Koskenvuo, N. V. Holm, K. Heikkila, E. Pukkala, A. Skytthe, H. O. Adami, J. Kaprio, Familial risk and heritability of cancer among twins in nordic countries. JAMA.- J. Am. Med. Assoc. 315, 68–76 (2016)
N. Risch, The genetic epidemiology of cancer: interpreting family and twin studies and their implications for molecular genetic approaches. Cancer Epidemiol. Biomark. Prev. 10, 733–741 (2001)
K. Czene, P. Lichtenstein, K. Hemminki, Environmental and heritable causes of cancer among 9.6 million individuals in the Swedish family-cancer database. Int. J. Cancer 99, 260–266 (2002)
S. Jiao, U. Peters, S. Berndt, H. Brenner, K. Butterbach, B. J. Caan, C. S. Carlson, A. T. Chan, J. Chang-Claude, S. Chanock, K. R. Curtis, D. Duggan, J. Gong, T. A. Harrison, R. B. Hayes, B. E. Henderson, M. Hoffmeister, L. N. Kolonel, L. L. Marchand, J. D. Potter, A. Rudolph, R. E. Schoen, D. Seminara, M. L. Slattery, E. White, L. Hsu, Estimating the heritability of colorectal cancer. Hum. Mol. Genet. 23, 3898–3905 (2014)
W. Bodmer, C. Bonilla, Common and rare variants in multifactorial susceptibility to common diseases. Nat. Genet. 40, 695–701 (2008)
A. de la Chapelle, Genetic predisposition to colorectal cancer. Nat. Rev. Cancer 4, 769–780 (2004)
G. Gibson, Rare and common variants: twenty arguments. Nat. Rev. Genet. 13, 135–145 (2011)
X. Ma, B. Zhang, W. Zheng, Genetic variants associated with colorectal cancer risk: comprehensive research synopsis, meta-analysis, and epidemiological evidence. Gut 63, 326–336 (2014)
P. Broderick, L. Carvajal-Carmona, A. M. Pittman, E. Webb, K. Howarth, A. Rowan, S. Lubbe, S. Spain, K. Sullivan, S. Fielding, E. Jaeger, J. Vijayakrishnan, Z. Kemp, M. Gorman, I. Chandler, E. Papaemmanuil, S. Penegar, W. Wood, G. Sellick, M. Qureshi, A. Teixeira, E. Domingo, E. Barclay, L. Martin, O. Sieber, C. Consortium, D. Kerr, R. Gray, J. Peto, J. B. Cazier, I. Tomlinson, R. S. Houlston, A genome-wide association study shows that common alleles of SMAD7 influence colorectal cancer risk. Nat. Genet. 39, 1315–1317 (2007)
I. Tomlinson, E. Webb, L. Carvajal-Carmona, P. Broderick, Z. Kemp, S. Spain, S. Penegar, I. Chandler, M. Gorman, W. Wood, E. Barclay, S. Lubbe, L. Martin, G. Sellick, E. Jaeger, R. Hubner, R. Wild, A. Rowan, S. Fielding, K. Howarth, C. Consortium, A. Silver, W. Atkin, K. Muir, R. Logan, D. Kerr, E. Johnstone, O. Sieber, R. Gray, H. Thomas, J. Peto, J. B. Cazier, R. Houlston, A genome-wide association scan of tag SNPs identifies a susceptibility variant for colorectal cancer at 8q24.21. Nat. Genet. 39, 984–988 (2007)
B. W. Zanke, C. M. Greenwood, J. Rangrej, R. Kustra, A. Tenesa, S. M. Farrington, J. Prendergast, S. Olschwang, T. Chiang, E. Crowdy, V. Ferretti, P. Laflamme, S. Sundararajan, S. Roumy, J. F. Olivier, F. Robidoux, R. Sladek, A. Montpetit, P. Campbell, S. Bezieau, A. M. O’Shea, G. Zogopoulos, M. Cotterchio, P. Newcomb, J. McLaughlin, B. Younghusband, R. Green, J. Green, M. E. Porteous, H. Campbell, H. Blanche, M. Sahbatou, E. Tubacher, C. Bonaiti-Pellie, B. Buecher, E. Riboli, S. Kury, S. J. Chanock, J. Potter, G. Thomas, S. Gallinger, T. J. Hudson, M. G. Dunlop, Genome-wide association scan identifies a colorectal cancer susceptibility locus on chromosome 8q24. Nat. Genet. 39, 989–994 (2007)
R. S. Houlston, E. Webb, P. Broderick, A. M. Pittman, M. C. Di Bernardo, S. Lubbe, I. Chandler, J. Vijayakrishnan, K. Sullivan, S. Penegar, C. Colorectal Cancer Association Study, L. Carvajal-Carmona, K. Howarth, E. Jaeger, S. L. Spain, A. Walther, E. Barclay, L. Martin, M. Gorman, E. Domingo, A. S. Teixeira, R.G.I.C. Co, D. Kerr, J. B. Cazier, I. Niittymaki, S. Tuupanen, A. Karhu, L. A. Aaltonen, I. P. Tomlinson, S. M. Farrington, A. Tenesa, J. G. Prendergast, R. A. Barnetson, R. Cetnarskyj, M. E. Porteous, P. D. Pharoah, T. Koessler, J. Hampe, S. Buch, C. Schafmayer, J. Tepel, S. Schreiber, H. Volzke, J. Chang-Claude, M. Hoffmeister, H. Brenner, B. W. Zanke, A. Montpetit, T. J. Hudson, C. S. Gallinger, International Colorectal Cancer Genetic Association, H. Campbell and M.G. Dunlop, Meta-analysis of genome-wide association data identifies four new susceptibility loci for colorectal cancer. Nat. Genet. 40, 1426–1435 (2008)
A. Tenesa, S. M. Farrington, J. G. Prendergast, M. E. Porteous, M. Walker, N. Haq, R. A. Barnetson, E. Theodoratou, R. Cetnarskyj, N. Cartwright, C. Semple, A. J. Clark, F. J. Reid, L. A. Smith, K. Kavoussanakis, T. Koessler, P. D. Pharoah, S. Buch, C. Schafmayer, J. Tepel, S. Schreiber, H. Volzke, C. O. Schmidt, J. Hampe, J. Chang-Claude, M. Hoffmeister, H. Brenner, S. Wilkening, F. Canzian, G. Capella, V. Moreno, I. J. Deary, J. M. Starr, I. P. Tomlinson, Z. Kemp, K. Howarth, L. Carvajal-Carmona, E. Webb, P. Broderick, J. Vijayakrishnan, R. S. Houlston, G. Rennert, D. Ballinger, L. Rozek, S. B. Gruber, K. Matsuda, T. Kidokoro, Y. Nakamura, B. W. Zanke, C. M. Greenwood, J. Rangrej, R. Kustra, A. Montpetit, T. J. Hudson, S. Gallinger, H. Campbell, M. G. Dunlop, Genome-wide association scan identifies a colorectal cancer susceptibility locus on 11q23 and replicates risk loci at 8q24 and 18q21. Nat. Genet. 40, 631–637 (2008)
I. P. Tomlinson, E. Webb, L. Carvajal-Carmona, P. Broderick, K. Howarth, A. M. Pittman, S. Spain, S. Lubbe, A. Walther, K. Sullivan, E. Jaeger, S. Fielding, A. Rowan, J. Vijayakrishnan, E. Domingo, I. Chandler, Z. Kemp, M. Qureshi, S. M. Farrington, A. Tenesa, J. G. Prendergast, R. A. Barnetson, S. Penegar, E. Barclay, W. Wood, L. Martin, M. Gorman, H. Thomas, J. Peto, D. T. Bishop, R. Gray, E. R. Maher, A. Lucassen, D. Kerr, D. G. Evans, C. Consortium, C. Schafmayer, S. Buch, H. Volzke, J. Hampe, S. Schreiber, U. John, T. Koessler, P. Pharoah, T. van Wezel, H. Morreau, J. T. Wijnen, J. L. Hopper, M. C. Southey, G. G. Giles, G. Severi, S. Castellvi-Bel, C. Ruiz-Ponte, A. Carracedo, A. Castells, E. Consortium, A. Forsti, K. Hemminki, P. Vodicka, A. Naccarati, L. Lipton, J. W. Ho, K. K. Cheng, P. C. Sham, J. Luk, J. A. Agundez, J. M. Ladero, M. de la Hoya, T. Caldes, I. Niittymaki, S. Tuupanen, A. Karhu, L. Aaltonen, J. B. Cazier, H. Campbell, M. G. Dunlop, R. S. Houlston, A genome-wide association study identifies colorectal cancer susceptibility loci on chromosomes 10p14 and 8q23.3. Nat. Genet. 40, 623–630 (2008)
R. S. Houlston, J. Cheadle, S. E. Dobbins, A. Tenesa, A. M. Jones, K. Howarth, S. L. Spain, P. Broderick, E. Domingo, S. Farrington, J. G. Prendergast, A. M. Pittman, E. Theodoratou, C. G. Smith, B. Olver, A. Walther, R. A. Barnetson, M. Churchman, E. E. Jaeger, S. Penegar, E. Barclay, L. Martin, M. Gorman, R. Mager, E. Johnstone, R. Midgley, I. Niittymaki, S. Tuupanen, J. Colley, S. Idziaszczyk, C. Consortium, H. J. Thomas, A. M. Lucassen, D. G. Evans, E. R. Maher, C. Consortium, C. C. Group, C. C. Group, T. Maughan, A. Dimas, E. Dermitzakis, J. B. Cazier, L. A. Aaltonen, P. Pharoah, D. J. Kerr, L. G. Carvajal-Carmona, H. Campbell, M. G. Dunlop, I. P. Tomlinson, Meta-analysis of three genome-wide association studies identifies susceptibility loci for colorectal cancer at 1q41, 3q26.2, 12q13.13 and 20q13.33. Nat. Genet. 42, 973–977 (2010)
J. Lascorz, A. Forsti, B. Chen, S. Buch, V. Steinke, N. Rahner, E. Holinski-Feder, M. Morak, H.K. Schackert, H. Gorgens, K. Schulmann, T. Goecke, M. Kloor, C. Engel, R. Buttner, N. Kunkel, M. Weires, M. Hoffmeister, B. Pardini, A. Naccarati, L. Vodickova, J. Novotny, S. Schreiber, M. Krawczak, C.D. Broring, H. Volzke, C. Schafmayer, P. Vodicka, J. Chang-Claude, H. Brenner, B. Burwinkel, P. Propping, J. Hampe and K. Hemminki, Genome-wide association study for colorectal cancer identifies risk polymorphisms in German familial cases and implicates MAPK signalling pathways in disease susceptibility. Carcinogenesis 31, 1612–1619 (2010)
R. Cui, Y. Okada, S. G. Jang, J. L. Ku, J. G. Park, Y. Kamatani, N. Hosono, T. Tsunoda, V. Kumar, C. Tanikawa, N. Kamatani, R. Yamada, M. Kubo, Y. Nakamura, K. Matsuda, Common variant in 6q26-q27 is associated with distal colon cancer in an Asian population. Gut 60, 799–805 (2011)
J. C. Figueiredo, J. P. Lewinger, C. Song, P. T. Campbell, D. V. Conti, C. K. Edlund, D. J. Duggan, J. Rangrej, M. Lemire, T. Hudson, B. Zanke, M. Cotterchio, S. Gallinger, M. Jenkins, J. Hopper, R. Haile, P. Newcomb, J. Potter, J. A. Baron, L. Le Marchand, G. Casey, Genotype-environment interactions in microsatellite stable/microsatellite instability-low colorectal cancer: results from a genome-wide association study. Cancer Epidemiol. Biomark. Prev. 20, 758–766 (2011)
M. G. Dunlop, S. E. Dobbins, S. M. Farrington, A. M. Jones, C. Palles, N. Whiffin, A. Tenesa, S. Spain, P. Broderick, L. Y. Ooi, E. Domingo, C. Smillie, M. Henrion, M. Frampton, L. Martin, G. Grimes, M. Gorman, C. Semple, Y. P. Ma, E. Barclay, J. Prendergast, J. B. Cazier, B. Olver, S. Penegar, S. Lubbe, I. Chander, L. G. Carvajal-Carmona, S. Ballereau, A. Lloyd, J. Vijayakrishnan, L. Zgaga, I. Rudan, E. Theodoratou, C. Colorectal Tumour Gene Identification, J. M. Starr, I. Deary, I. Kirac, D. Kovacevic, L. A. Aaltonen, L. Renkonen-Sinisalo, J. P. Mecklin, K. Matsuda, Y. Nakamura, Y. Okada, S. Gallinger, D. J. Duggan, D. Conti, P. Newcomb, J. Hopper, M. A. Jenkins, F. Schumacher, G. Casey, D. Easton, M. Shah, P. Pharoah, A. Lindblom, T. Liu, G. Swedish Low-Risk Colorectal Cancer Study, C. G. Smith, H. West, J. P. Cheadle, C. C. Group, R. Midgley, D. J. Kerr, H. Campbell, I. P. Tomlinson, R. S. Houlston, Common variation near CDKN1A, POLD3 and SHROOM2 influences colorectal cancer risk. Nat. Genet. 44, 770–776 (2012)
S. Jiao, L. Hsu, S. Berndt, S. Bezieau, H. Brenner, D. Buchanan, B. J. Caan, P. T. Campbell, C. S. Carlson, G. Casey, A. T. Chan, J. Chang-Claude, S. Chanock, D. V. Conti, K. R. Curtis, D. Duggan, S. Gallinger, S. B. Gruber, T. A. Harrison, R. B. Hayes, B. E. Henderson, M. Hoffmeister, J. L. Hopper, T. J. Hudson, C. M. Hutter, R. D. Jackson, M. A. Jenkins, E. D. Kantor, L. N. Kolonel, S. Kury, L. Le Marchand, M. Lemire, P. A. Newcomb, J. D. Potter, C. Qu, S. A. Rosse, R. E. Schoen, F. R. Schumacher, D. Seminara, M. L. Slattery, C. M. Ulrich, B. W. Zanke, U. Peters, Genome-wide search for gene-gene interactions in colorectal cancer. PLoS ONE 7, e52535 (2012)
C. Fernandez-Rozadilla, J. B. Cazier, I. P. Tomlinson, L. G. Carvajal-Carmona, C. Palles, M. J. Lamas, M. Baiget, L. A. Lopez-Fernandez, A. Brea-Fernandez, A. Abuli, L. Bujanda, J. Clofent, D. Gonzalez, R. Xicola, M. Andreu, X. Bessa, R. Jover, X. Llor, V. Moreno, A. Castells, A. Carracedo, S. Castellvi-Bel, C. Ruiz-Ponte, A colorectal cancer genome-wide association study in a Spanish cohort identifies two variants associated with colorectal cancer risk at 1p33 and 8p12. BMC Genomics 14, 55 (2013)
S. N. Hong, C. H. Park, J. I. Kim, D. H. Kim, H. C. Kim, D. K. Chang, P. L. Rhee, J. J. Kim, J. C. Rhee, H. J. Son, Y. H. Kim, Colorectal cancer-susceptibility single nucleotide polymorphisms in Korean population. J. Gastroenterol. Hepatol. 30, 849–857 (2013)
W. H. Jia, B. Zhang, K. Matsuo, A. Shin, Y. B. Xiang, S. H. Jee, D. H. Kim, Z. Ren, Q. Cai, J. Long, J. Shi, W. Wen, G. Yang, R. J. Delahanty, B. T. Ji, Z. Z. Pan, F. Matsuda, Y. T. Gao, J. H. Oh, Y. O. Ahn, E. J. Park, H. L. Li, J. W. Park, J. Jo, J. Y. Jeong, S. Hosono, G. Casey, U. Peters, X. O. Shu, Y. X. Zeng, W. Zheng, Genome-wide association analyses in east Asians identify new susceptibility loci for colorectal cancer. Nat. Genet. 45, 191–196 (2013)
U. Peters, S. Jiao, F. R. Schumacher, C. M. Hutter, A. K. Aragaki, J. A. Baron, S. I. Berndt, S. Bezieau, H. Brenner, K. Butterbach, B. J. Caan, P. T. Campbell, C. S. Carlson, G. Casey, A. T. Chan, J. Chang-Claude, S. J. Chanock, L. S. Chen, G. A. Coetzee, S. G. Coetzee, D. V. Conti, K. R. Curtis, D. Duggan, T. Edwards, C. S. Fuchs, S. Gallinger, E. L. Giovannucci, S. M. Gogarten, S. B. Gruber, R. W. Haile, T. A. Harrison, R. B. Hayes, B. E. Henderson, M. Hoffmeister, J. L. Hopper, T. J. Hudson, D. J. Hunter, R. D. Jackson, S. H. Jee, M. A. Jenkins, W. H. Jia, L. N. Kolonel, C. Kooperberg, S. Kury, A. Z. Lacroix, C. C. Laurie, C. A. Laurie, L. Le Marchand, M. Lemire, D. Levine, N. M. Lindor, Y. Liu, J. Ma, K. W. Makar, K. Matsuo, P. A. Newcomb, J. D. Potter, R. L. Prentice, C. Qu, T. Rohan, S. A. Rosse, R. E. Schoen, D. Seminara, M. Shrubsole, X. O. Shu, M. L. Slattery, D. Taverna, S. N. Thibodeau, C. M. Ulrich, E. White, Y. Xiang, B. W. Zanke, Y. X. Zeng, B. Zhang, W. Zheng, L. Hsu, R. Colon cancer family registry and the genetics and epidemiology of colorectal cancer consortium, Identification of genetic susceptibility loci for colorectal tumors in a genome-wide meta-analysis. Gastroenterology 144, 799–807 e724 (2013)
S. Siegert, J. Hampe, C. Schafmayer, W. von Schonfels, J. H. Egberts, A. Forsti, B. Chen, J. Lascorz, K. Hemminki, A. Franke, M. Nothnagel, U. Nothlings, M. Krawczak, Genome-wide investigation of gene-environment interactions in colorectal cancer. Hum. Genet. 132, 219–231 (2013)
J. C. Figueiredo, L. Hsu, C. M. Hutter, Y. Lin, P. T. Campbell, J. A. Baron, S. I. Berndt, S. Jiao, G. Casey, B. Fortini, A. T. Chan, M. Cotterchio, M. Lemire, S. Gallinger, T. A. Harrison, L. Le Marchand, P. A. Newcomb, M. L. Slattery, B. J. Caan, C. S. Carlson, B. W. Zanke, S. A. Rosse, H. Brenner, E. L. Giovannucci, K. Wu, J. Chang-Claude, S. J. Chanock, K. R. Curtis, D. Duggan, J. Gong, R. W. Haile, R. B. Hayes, M. Hoffmeister, J. L. Hopper, M. A. Jenkins, L. N. Kolonel, C. Qu, A. Rudolph, R. E. Schoen, F. R. Schumacher, D. Seminara, D. L. Stelling, S. N. Thibodeau, M. Thornquist, G. S. Warnick, B. E. Henderson, C. M. Ulrich, W. J. Gauderman, J. D. Potter, E. White, U. Peters, Genome-wide diet-gene interaction analyses for risk of colorectal cancer. PLoS Genet. 10, e1004228 (2014)
N. Whiffin, F. Hosking, S. M. Farrington, C. Palles, S. Dobbins, L. Zgaga, A. Lloyd, B. Kinnersley, M. Gorman, A. Tenesa, P. Broderick, Y. Wang, E. Barclay, C. Hayward, L. Martin, D. D. Buchanan, A. K. Win, J. Hopper, M. Jenkins, N. M. Lindor, P. A. Newcomb, S. Gallinger, D. Conti, F. Schumacher, G. Casey, T. Liu, G. The Swedish Low-Risk Colorectal Cancer Study, H. Campbell, A. Lindblom, R. S. Houlston, I. Tomlinson, M. G. Dunlop, Identification of susceptibility loci for colorectal cancer in a genome-wide meta-analysis. Hum. Mol. Genet. 23, 4729–4737 (2014)
B. Zhang, W. H. Jia, K. Matsuo, A. Shin, Y. B. Xiang, K. Matsuda, S. H. Jee, D. H. Kim, P. Y. Cheah, Z. Ren, Q. Cai, J. Long, J. Shi, W. Wen, G. Yang, B. T. Ji, Z. Z. Pan, F. Matsuda, Y. T. Gao, J. H. Oh, Y. O. Ahn, M. Kubo, L. F. Thean, E. J. Park, H. L. Li, J. W. Park, J. Jo, J. Y. Jeong, S. Hosono, Y. Nakamura, X. O. Shu, Y. X. Zeng, W. Zheng, Genome-wide association study identifies a new SMAD7 risk variant associated with colorectal cancer risk in east Asians. Int. J. Cancer 135, 948–955 (2014)
Z. K. Stadler, P. Thom, M. E. Robson, J. N. Weitzel, N. D. Kauff, K. E. Hurley, V. Devlin, B. Gold, R. J. Klein, K. Offit, Genome-wide association studies of cancer. J. Clin. Oncol. 28, 4255–4267 (2010)
E. Theodoratou, Z. Montazeri, S. Hawken, G. C. Allum, J. Gong, V. Tait, I. Kirac, M. Tazari, S. M. Farrington, A. Demarsh, L. Zgaga, D. Landry, H. E. Benson, S. H. Read, I. Rudan, A. Tenesa, M. G. Dunlop, H. Campbell, J. Little, Systematic meta-analyses and field synopsis of genetic association studies in colorectal cancer. J. Natl. Cancer Inst. 104, 1433–1457 (2012)
U. Peters, C.M. Hutter, L. Hsu, F.R. Schumacher, D.V. Conti, C.S. Carlson, C.K. Edlund, R.W. Haile, S. Gallinger, B.W. Zanke, M. Lemire, J. Rangrej, R. Vijayaraghavan, A.T. Chan, A. Hazra, D.J. Hunter, J. Ma, C.S. Fuchs, E.L. Giovannucci, P. Kraft, Y. Liu, L. Chen, S. Jiao, K.W. Makar, D. Taverna, S.B. Gruber, G. Rennert, V. Moreno, C.M. Ulrich, M.O. Woods, R.C. Green, P.S. Parfrey, R.L. Prentice, C. Kooperberg, R.D. Jackson, A.Z. Lacroix, B.J. Caan, R.B. Hayes, S.I. Berndt, S.J. Chanock, R.E. Schoen, J. Chang-Claude, M. Hoffmeister, H. Brenner, B. Frank, S. Bezieau, S. Kury, M.L. Slattery, J.L. Hopper, M.A. Jenkins, L. Le Marchand, N.M. Lindor, P.A. Newcomb, D. Seminara, T.J. Hudson, D.J. Duggan, J.D. Potter and G. Casey , Meta-analysis of new genome-wide association studies of colorectal cancer risk. Hum. Genet. 131, 217–234 (2012)
S. L. Schmit, F. R. Schumacher, C. K. Edlund, D. V. Conti, L. Raskin, F. Lejbkowicz, M. Pinchev, H. S. Rennert, M. A. Jenkins, J. L. Hopper, D. D. Buchanan, N. M. Lindor, L. Le Marchand, S. Gallinger, R. W. Haile, P. A. Newcomb, S. C. Huang, G. Rennert, G. Casey, S. B. Gruber, A novel colorectal cancer risk locus at 4q32.2 identified from an international genome-wide association study. Carcinogenesis 35, 2512–2519 (2014)
M. G. Dunlop, A. Tenesa, S. M. Farrington, S. Ballereau, D. H. Brewster, T. Koessler, P. Pharoah, C. Schafmayer, J. Hampe, H. Volzke, J. Chang-Claude, M. Hoffmeister, H. Brenner, S. von Holst, S. Picelli, A. Lindblom, M. A. Jenkins, J. L. Hopper, G. Casey, D. Duggan, P. A. Newcomb, A. Abuli, X. Bessa, C. Ruiz-Ponte, S. Castellvi-Bel, I. Niittymaki, S. Tuupanen, A. Karhu, L. Aaltonen, B. Zanke, T. Hudson, S. Gallinger, E. Barclay, L. Martin, M. Gorman, L. Carvajal-Carmona, A. Walther, D. Kerr, S. Lubbe, P. Broderick, I. Chandler, A. Pittman, S. Penegar, H. Campbell, I. Tomlinson, R. S. Houlston, Cumulative impact of common genetic variants and other risk factors on colorectal cancer risk in 42,103 individuals. Gut 62, 871–881 (2013)
L. A. Hindorff, P. Sethupathy, H. A. Junkins, E. M. Ramos, J. P. Mehta, F. S. Collins, T. A. Manolio, Potential etiologic and functional implications of genome-wide association loci for human diseases and traits. Proc. Natl. Acad. Sci. U S A 106, 9362–9367 (2009)
G. Zogopoulos, C. Jorgensen, J. Bacani, A. Montpetit, P. Lepage, V. Ferretti, L. Chad, S. Selvarajah, B. Zanke, T. J. Hudson, T. Pawson, S. Gallinger, Germline EPHB2 receptor variants in familial colorectal cancer. PLoS One 3, e2885 (2008)
K. Guda, H. Moinova, J. He, O. Jamison, L. Ravi, L. Natale, J. Lutterbaugh, E. Lawrence, S. Lewis, J. K. Willson, J. B. Lowe, G. L. Wiesner, G. Parmigiani, J. Barnholtz-Sloan, D. W. Dawson, V. E. Velculescu, K. W. Kinzler, N. Papadopoulos, B. Vogelstein, J. Willis, T. A. Gerken, S. D. Markowitz, Inactivating germ-line and somatic mutations in polypeptide N-acetylgalactosaminyltransferase 12 in human colon cancers. Proc. Natl. Acad. Sci. U S A 106, 12921–12925 (2009)
E. Clarke, R. C. Green, J. S. Green, K. Mahoney, P. S. Parfrey, H. B. Younghusband, M. O. Woods, Inherited deleterious variants in GALNT12 are associated with CRC susceptibility. Hum. Mutat. 33, 1056–1058 (2012)
R. Venkatachalam, M. J. Ligtenberg, N. Hoogerbrugge, H. K. Schackert, H. Gorgens, M. M. Hahn, E. J. Kamping, L. Vreede, E. Hoenselaar, E. van der Looij, M. Goossens, M. Churchman, L. Carvajal-Carmona, I. P. Tomlinson, D. R. de Bruijn, A. Geurts van Kessel, R. P. Kuiper, Germline epigenetic silencing of the tumor suppressor gene PTPRJ in early-onset familial colorectal cancer. Gastroenterology 139, 2221–2224 (2010)
R. M. de Voer, A. Geurts van Kessel, R. D. Weren, M. J. Ligtenberg, D. Smeets, L. Fu, L. Vreede, E. J. Kamping, E. T. Verwiel, M. M. Hahn, M. Ariaans, L. Spruijt, T. van Essen, G. Houge, H. K. Schackert, J. Q. Sheng, H. Venselaar, C. M. van Ravenswaaij-Arts, J. H. van Krieken, N. Hoogerbrugge, R. P. Kuiper, Germline mutations in the spindle assembly checkpoint genes BUB1 and BUB3 are risk factors for colorectal cancer. Gastroenterology 145, 544–547 (2013)
T. A. Manolio, F. S. Collins, N. J. Cox, D. B. Goldstein, L. A. Hindorff, D. J. Hunter, M. I. McCarthy, E. M. Ramos, L. R. Cardon, A. Chakravarti, J. H. Cho, A. E. Guttmacher, A. Kong, L. Kruglyak, E. Mardis, C. N. Rotimi, M. Slatkin, D. Valle, A. S. Whittemore, M. Boehnke, A. G. Clark, E. E. Eichler, G. Gibson, J. L. Haines, T. F. Mackay, S. A. McCarroll, P. M. Visscher, Finding the missing heritability of complex diseases. Nature 461, 747–753 (2009)
M. L. Metzker, Sequencing technologies - the next generation. Nat. Rev. Genet. 11, 31–46 (2010)
E. R. Mardis, Next-generation sequencing platforms. Annu. Rev. Anal. Chem. 6, 287–303 (2013)
N. Segui, L. B. Mina, C. Lazaro, R. Sanz-Pamplona, T. Pons, M. Navarro, F. Bellido, A. Lopez-Doriga, R. Valdes-Mas, M. Pineda, E. Guino, A. Vidal, J. L. Soto, T. Caldes, M. Duran, M. Urioste, D. Rueda, J. Brunet, M. Balbin, P. Blay, S. Iglesias, P. Garre, E. Lastra, A. B. Sanchez-Heras, A. Valencia, V. Moreno, M. A. Pujana, A. Villanueva, I. Blanco, G. Capella, J. Surralles, X. S. Puente, L. Valle, Germline mutations in FAN1 cause hereditary colorectal cancer by impairing DNA repair. Gastroenterology 149, 563–566 (2015)
E. S. Lander, L. M. Linton, B. Birren, C. Nusbaum, M. C. Zody, J. Baldwin, K. Devon, K. Dewar, M. Doyle, W. FitzHugh, R. Funke, D. Gage, K. Harris, A. Heaford, J. Howland, L. Kann, J. Lehoczky, R. LeVine, P. McEwan, K. McKernan, J. Meldrim, J. P. Mesirov, C. Miranda, W. Morris, J. Naylor, C. Raymond, M. Rosetti, R. Santos, A. Sheridan, C. Sougnez, N. Stange-Thomann, N. Stojanovic, A. Subramanian, D. Wyman, J. Rogers, J. Sulston, R. Ainscough, S. Beck, D. Bentley, J. Burton, C. Clee, N. Carter, A. Coulson, R. Deadman, P. Deloukas, A. Dunham, I. Dunham, R. Durbin, L. French, D. Grafham, S. Gregory, T. Hubbard, S. Humphray, A. Hunt, M. Jones, C. Lloyd, A. McMurray, L. Matthews, S. Mercer, S. Milne, J. C. Mullikin, A. Mungall, R. Plumb, M. Ross, R. Shownkeen, S. Sims, R. H. Waterston, R. K. Wilson, L. W. Hillier, J. D. McPherson, M. A. Marra, E. R. Mardis, L. A. Fulton, A. T. Chinwalla, K. H. Pepin, W. R. Gish, S. L. Chissoe, M. C. Wendl, K. D. Delehaunty, T. L. Miner, A. Delehaunty, J. B. Kramer, L. L. cook, R. S. Fulton, D. L. Johnson, P. J. Minx, S. W. Clifton, T. Hawkins, E. Branscomb, P. Predki, P. Richardson, S. Wenning, T. Slezak, N. Doggett, J. F. Cheng, A. Olsen, S. Lucas, C. Elkin, E. Uberbacher, M. Frazier, R. A. Gibbs, D. M. Muzny, S. E. Scherer, J. B. Bouck, E. J. Sodergren, K. C. Worley, C. M. Rives, J. H. Gorrell, M. L. Metzker, S. L. Naylor, R. S. Kucherlapati, D. L. Nelson, G. M. Weinstock, Y. Sakaki, A. Fujiyama, M. Hattori, T. Yada, A. Toyoda, T. Itoh, C. Kawagoe, H. Watanabe, Y. Totoki, T. Taylor, J. Weissenbach, R. Heilig, W. Saurin, F. Artiguenave, P. Brottier, T. Bruls, E. Pelletier, C. Robert, P. Wincker, D. R. Smith, L. Doucette-Stamm, M. Rubenfield, K. Weinstock, H. M. Lee, J. Dubois, A. Rosenthal, M. Platzer, G. Nyakatura, S. Taudien, A. Rump, H. Yang, J. Yu, J. Wang, G. Huang, J. Gu, L. Hood, L. Rowen, A. Madan, S. Qin, R. W. Davis, N. A. Federspiel, A. P. Abola, M. J. Proctor, R. M. Myers, J. Schmutz, M. Dickson, J. Grimwood, D. R. Cox, M. V. Olson, R. Kaul, C. Raymond, N. Shimizu, K. Kawasaki, S. Minoshima, G. A. Evans, M. Athanasiou, R. Schultz, B. A. Roe, F. Chen, H. Pan, J. Ramser, H. Lehrach, R. Reinhardt, W. R. McCombie, M. de la Bastide, N. Dedhia, H. Blocker, K. Hornischer, G. Nordsiek, R. Agarwala, L. Aravind, J. A. Bailey, A. Bateman, S. Batzoglou, E. Birney, P. Bork, D. G. Brown, C. B. Burge, L. Cerutti, H. C. Chen, D. Church, M. Clamp, R. R. Copley, T. Doerks, S. R. Eddy, E. E. Eichler, T. S. Furey, J. Galagan, J. G. Gilbert, C. Harmon, Y. Hayashizaki, D. Haussler, H. Hermjakob, K. Hokamp, W. Jang, L. S. Johnson, T. A. Jones, S. Kasif, A. Kaspryzk, S. Kennedy, W. J. Kent, P. Kitts, E. V. Koonin, I. Korf, D. Kulp, D. Lancet, T. M. Lowe, A. McLysaght, T. Mikkelsen, J. V. Moran, N. Mulder, V. J. Pollara, C. P. Ponting, G. Schuler, J. Schultz, G. Slater, A. F. Smit, E. Stupka, J. Szustakowski, D. Thierry-Mieg, J. Thierry-Mieg, L. Wagner, J. Wallis, R. Wheeler, A. Williams, Y. I. Wolf, K. H. Wolfe, S. P. Yang, R. F. Yeh, F. Collins, M. S. Guyer, J. Peterson, A. Felsenfeld, K. A. Wetterstrand, A. Patrinos, M. J. Morgan, P. de Jong, J. J. Catanese, K. Osoegawa, H. Shizuya, S. Choi, Y. J. Chen, C. International Human Genome Sequencing, Initial sequencing and analysis of the human genome. Nature 409, 860–921 (2001)
J. C. Venter, M. D. Adams, E. W. Myers, P. W. Li, R. J. Mural, G. G. Sutton, H. O. Smith, M. Yandell, C. A. Evans, R. A. Holt, J. D. Gocayne, P. Amanatides, R. M. Ballew, D. H. Huson, J. R. Wortman, Q. Zhang, C. D. Kodira, X. H. Zheng, L. Chen, M. Skupski, G. Subramanian, P. D. Thomas, J. Zhang, G. L. Gabor Miklos, C. Nelson, S. Broder, A. G. Clark, J. Nadeau, V. A. McKusick, N. Zinder, A. J. Levine, R. J. Roberts, M. Simon, C. Slayman, M. Hunkapiller, R. Bolanos, A. Delcher, I. Dew, D. Fasulo, M. Flanigan, L. Florea, A. Halpern, S. Hannenhalli, S. Kravitz, S. Levy, C. Mobarry, K. Reinert, K. Remington, J. Abu-Threideh, E. Beasley, K. Biddick, V. Bonazzi, R. Brandon, M. Cargill, I. Chandramouliswaran, R. Charlab, K. Chaturvedi, Z. Deng, V. Di Francesco, P. Dunn, K. Eilbeck, C. Evangelista, A. E. Gabrielian, W. Gan, W. Ge, F. Gong, Z. Gu, P. Guan, T. J. Heiman, M. E. Higgins, R. R. Ji, Z. Ke, K. A. Ketchum, Z. Lai, Y. Lei, Z. Li, J. Li, Y. Liang, X. Lin, F. Lu, G. V. Merkulov, N. Milshina, H. M. Moore, A. K. Naik, V. A. Narayan, B. Neelam, D. Nusskern, D. B. Rusch, S. Salzberg, W. Shao, B. Shue, J. Sun, Z. Wang, A. Wang, X. Wang, J. Wang, M. Wei, R. Wides, C. Xiao, C. Yan, A. Yao, J. Ye, M. Zhan, W. Zhang, H. Zhang, Q. Zhao, L. Zheng, F. Zhong, W. Zhong, S. Zhu, S. Zhao, D. Gilbert, S. Baumhueter, G. Spier, C. Carter, A. Cravchik, T. Woodage, F. Ali, H. An, A. Awe, D. Baldwin, H. Baden, M. Barnstead, I. Barrow, K. Beeson, D. Busam, A. Carver, A. Center, M. L. Cheng, L. Curry, S. Danaher, L. Davenport, R. Desilets, S. Dietz, K. Dodson, L. Doup, S. Ferriera, N. Garg, A. Gluecksmann, B. Hart, J. Haynes, C. Haynes, C. Heiner, S. Hladun, D. Hostin, J. Houck, T. Howland, C. Ibegwam, J. Johnson, F. Kalush, L. Kline, S. Koduru, A. Love, F. Mann, D. May, S. McCawley, T. McIntosh, I. McMullen, M. Moy, L. Moy, B. Murphy, K. Nelson, C. Pfannkoch, E. Pratts, V. Puri, H. Qureshi, M. Reardon, R. Rodriguez, Y. H. Rogers, D. Romblad, B. Ruhfel, R. Scott, C. Sitter, M. Smallwood, E. Stewart, R. Strong, E. Suh, R. Thomas, N. N. Tint, S. Tse, C. Vech, G. Wang, J. Wetter, S. Williams, M. Williams, S. Windsor, E. Winn-Deen, K. Wolfe, J. Zaveri, K. Zaveri, J. F. Abril, R. Guigo, M. J. Campbell, K. V. Sjolander, B. Karlak, A. Kejariwal, H. Mi, B. Lazareva, T. Hatton, A. Narechania, K. Diemer, A. Muruganujan, N. Guo, S. Sato, V. Bafna, S. Istrail, R. Lippert, R. Schwartz, B. Walenz, S. Yooseph, D. Allen, A. Basu, J. Baxendale, L. Blick, M. Caminha, J. Carnes-Stine, P. Caulk, Y. H. Chiang, M. Coyne, C. Dahlke, A. Mays, M. Dombroski, M. Donnelly, D. Ely, S. Esparham, C. Fosler, H. Gire, S. Glanowski, K. Glasser, A. Glodek, M. Gorokhov, K. Graham, B. Gropman, M. Harris, J. Heil, S. Henderson, J. Hoover, D. Jennings, C. Jordan, J. Jordan, J. Kasha, L. Kagan, C. Kraft, A. Levitsky, M. Lewis, X. Liu, J. Lopez, D. Ma, W. Majoros, J. McDaniel, S. Murphy, M. Newman, T. Nguyen, N. Nguyen, M. Nodell, S. Pan, J. Peck, M. Peterson, W. Rowe, R. Sanders, J. Scott, M. Simpson, T. Smith, A. Sprague, T. Stockwell, R. Turner, E. Venter, M. Wang, M. Wen, D. Wu, M. Wu, A. Xia, A. Zandieh, X. Zhu, The sequence of the human genome. Science 291, 1304–1351 (2001)
A. Geurts van Kessel, The cancer genome: from structure to function. Cell. Oncol. 37, 155–165 (2014)
D. Sie, P. J. Snijders, G. A. Meijer, M. W. Doeleman, M. I. van Moorsel, H. F. van Essen, P. P. Eijk, K. Grunberg, N. C. van Grieken, E. Thunnissen, H. M. Verheul, E. F. Smit, B. Ylstra, D. A. Heideman, Performance of amplicon-based next generation DNA sequencing for diagnostic gene mutation profiling in oncopathology. Cell. Oncol. 37, 353–361 (2014)
M. J. Bamshad, S. B. Ng, A. W. Bigham, H. K. Tabor, M. J. Emond, D. A. Nickerson, J. Shendure, Exome sequencing as a tool for Mendelian disease gene discovery. Nat. Rev. Genet. 12, 745–755 (2011)
C. Gilissen, A. Hoischen, H. G. Brunner, J. A. Veltman, Disease gene identification strategies for exome sequencing. Eur. J. Hum. Genet. 20, 490–497 (2012)
J. X. Chong, K. J. Buckingham, S. N. Jhangiani, C. Boehm, N. Sobreira, J. D. Smith, T. M. Harrell, M. J. McMillin, W. Wiszniewski, T. Gambin, Z. H. Coban Akdemir, K. Doheny, A. F. Scott, D. Avramopoulos, A. Chakravarti, J. Hoover-Fong, D. Mathews, P. D. Witmer, H. Ling, K. Hetrick, L. Watkins, K. E. Patterson, F. Reinier, E. Blue, D. Muzny, M. Kircher, K. Bilguvar, F. Lopez-Giraldez, V. R. Sutton, H. K. Tabor, S. M. Leal, M. Gunel, S. Mane, R. A. Gibbs, E. Boerwinkle, A. Hamosh, J. Shendure, J. R. Lupski, R. P. Lifton, D. Valle, D. A. Nickerson, M. J. Bamshad, The genetic basis of mendelian phenotypes: discoveries, challenges, and opportunities. Am. J. Hum. Genet. 97, 199–215 (2015)
J. C. Taylor, H. C. Martin, S. Lise, J. Broxholme, J. B. Cazier, A. Rimmer, A. Kanapin, G. Lunter, S. Fiddy, C. Allan, A. R. Aricescu, M. Attar, C. Babbs, J. Becq, D. Beeson, C. Bento, P. Bignell, E. Blair, V. J. Buckle, K. Bull, O. Cais, H. Cario, H. Chapel, R. R. Copley, R. Cornall, J. Craft, K. Dahan, E. E. Davenport, C. Dendrou, O. Devuyst, A. L. Fenwick, J. Flint, L. Fugger, R. D. Gilbert, A. Goriely, A. Green, I. H. Greger, R. Grocock, A. V. Gruszczyk, R. Hastings, E. Hatton, D. Higgs, A. Hill, C. Holmes, M. Howard, L. Hughes, P. Humburg, D. Johnson, F. Karpe, Z. Kingsbury, U. Kini, J. C. Knight, J. Krohn, S. Lamble, C. Langman, L. Lonie, J. Luck, D. McCarthy, S. J. McGowan, M. F. McMullin, K. A. Miller, L. Murray, A. H. Nemeth, M. A. Nesbit, D. Nutt, E. Ormondroyd, A. B. Oturai, A. Pagnamenta, S. Y. Patel, M. Percy, N. Petousi, P. Piazza, S. E. Piret, G. Polanco-Echeverry, N. Popitsch, F. Powrie, C. Pugh, L. Quek, P. A. Robbins, K. Robson, A. Russo, N. Sahgal, P. A. van Schouwenburg, A. Schuh, E. Silverman, A. Simmons, P. S. Sorensen, E. Sweeney, J. Taylor, R. V. Thakker, I. Tomlinson, A. Trebes, S. R. Twigg, H. H. Uhlig, P. Vyas, T. Vyse, S. A. Wall, H. Watkins, M. P. Whyte, L. Witty, B. Wright, C. Yau, D. Buck, S. Humphray, P. J. Ratcliffe, J. I. Bell, A. O. Wilkie, D. Bentley, P. Donnelly, G. McVean, Factors influencing success of clinical genome sequencing across a broad spectrum of disorders. Nat. Genet. 47, 717–726 (2015)
D. T. Chang, R. K. Pai, L. A. Rybicki, M. A. Dimaio, M. Limaye, P. Jayachandran, A. C. Koong, P. A. Kunz, G. A. Fisher, J. M. Ford, M. Welton, A. Shelton, L. Ma, D. A. Arber, R. K. Pai, Clinicopathologic and molecular features of sporadic early-onset colorectal adenocarcinoma: an adenocarcinoma with frequent signet ring cell differentiation, rectal and sigmoid involvement, and adverse morphologic features. Mod. Pathol. 25, 1128–1139 (2012)
M. S. DeRycke, S. R. Gunawardena, S. Middha, Y. W. Asmann, D. J. Schaid, S. K. McDonnell, S. M. Riska, B. W. Eckloff, J. M. Cunningham, B. L. Fridley, D. J. Serie, W. R. Bamlet, M. S. Cicek, M. A. Jenkins, D. J. Duggan, D. Buchanan, M. Clendenning, R. W. Haile, M. O. Woods, S. N. Gallinger, G. Casey, J. D. Potter, P. A. Newcomb, L. Le Marchand, N. M. Lindor, S. N. Thibodeau, E. L. Goode, Identification of novel variants in colorectal cancer families by high-throughput exome sequencing. Cancer Epidemiol. Biomarkers Prev. 22, 1239–1251 (2013)
A. E. Gylfe, R. Katainen, J. Kondelin, T. Tanskanen, T. Cajuso, U. Hanninen, J. Taipale, M. Taipale, L. Renkonen-Sinisalo, H. Jarvinen, J. P. Mecklin, O. Kilpivaara, E. Pitkanen, P. Vahteristo, S. Tuupanen, A. Karhu, L. A. Aaltonen, Eleven candidate susceptibility genes for common familial colorectal cancer. PLoS Genet. 9, e1003876 (2013)
C. Esteban-Jurado, M. Vila-Casadesus, P. Garre, J. J. Lozano, A. Pristoupilova, S. Beltran, J. Munoz, T. Ocana, F. Balaguer, M. Lopez-Ceron, M. Cuatrecasas, S. Franch-Exposito, J. M. Pique, A. Castells, A. Carracedo, C. Ruiz-Ponte, A. Abuli, X. Bessa, M. Andreu, L. Bujanda, T. Caldes, S. Castellvi-Bel, Whole-exome sequencing identifies rare pathogenic variants in new predisposition genes for familial colorectal cancer. Genet. Med. 17, 131–142 (2015)
C. G. Smith, M. Naven, R. Harris, J. Colley, H. West, N. Li, Y. Liu, R. Adams, T. S. Maughan, L. Nichols, R. Kaplan, M. J. Wagner, H. L. McLeod, J. P. Cheadle, Exome resequencing identifies potential tumor-suppressor genes that predispose to colorectal cancer. Hum. Mutat. 34, 1026–1034 (2013)
T. Tanskanen, A. E. Gylfe, R. Katainen, M. Taipale, L. Renkonen-Sinisalo, H. Jarvinen, J. P. Mecklin, J. Bohm, O. Kilpivaara, E. Pitkanen, K. Palin, P. Vahteristo, S. Tuupanen, L. A. Aaltonen, Systematic search for rare variants in Finnish early-onset colorectal cancer patients. Cancer Genet. 208, 35–40 (2015)
J. X. Zhang, L. Fu, R. M. de Voer, M. M. Hahn, P. Jin, C. X. Lv, E. T. Verwiel, M. J. Ligtenberg, N. Hoogerbrugge, R. P. Kuiper, J. Q. Sheng, A. Geurts van Kessel, Candidate colorectal cancer predisposing gene variants in Chinese early-onset and familial cases. World J. Gastroenterol. 21, 4136–4149 (2015)
R. M. de Voer, M. M. Hahn, R. D. Weren, A. R. Mensenkamp, C. Gilissen, W. A. van Zelst-Stams, L. Spruijt, C. M. Kets, J. Zhang, H. Venselaar, L. Vreede, N. Schubert, M. Tychon, R. Derks, H. K. Schackert, A. Geurts van Kessel, N. Hoogerbrugge, M. J. Ligtenberg, R. P. Kuiper, Identification of novel candidate genes for early-onset colorectal cancer susceptibility. PLoS Genet. 12, e1005880 (2016)
R. M. de Voer, M. M. Hahn, A. R. Mensenkamp, A. Hoischen, C. Gilissen, A. Henkes, L. Spruijt, W. A. van Zelst-Stams, C. M. Kets, E. T. Verwiel, I. D. Nagtegaal, H. K. Schackert, A. Geurts van Kessel, N. Hoogerbrugge, M. J. Ligtenberg, R. P. Kuiper, Deleterious germline BLM mutations and the risk for early-onset colorectal cancer. Sci. Rep. 5, 14060 (2015)
J. A. Tennessen, A. W. Bigham, T. D. O’Connor, W. Fu, E. E. Kenny, S. Gravel, S. McGee, R. Do, X. Liu, G. Jun, H. M. Kang, D. Jordan, S. M. Leal, S. Gabriel, M. J. Rieder, G. Abecasis, D. Altshuler, D. A. Nickerson, E. Boerwinkle, S. Sunyaev, C. D. Bustamante, M. J. Bamshad, J. M. Akey, G. O. Broad, G. O. Seattle, N. E. S. Project, Evolution and functional impact of rare coding variation from deep sequencing of human exomes. Science 337, 64–69 (2012)
A. Keinan, A. G. Clark, Recent explosive human population growth has resulted in an excess of rare genetic variants. Science 336, 740–743 (2012)
G. T. Marth, F. Yu, A. R. Indap, K. Garimella, S. Gravel, W. F. Leong, C. Tyler-Smith, M. Bainbridge, T. Blackwell, X. Zheng-Bradley, Y. Chen, D. Challis, L. Clarke, E. V. Ball, K. Cibulskis, D. N. Cooper, B. Fulton, C. Hartl, D. Koboldt, D. Muzny, R. Smith, C. Sougnez, C. Stewart, A. Ward, J. Yu, Y. Xue, D. Altshuler, C. D. Bustamante, A. G. Clark, M. Daly, M. DePristo, P. Flicek, S. Gabriel, E. Mardis, A. Palotie, R. Gibbs, P. Genomes, The functional spectrum of low-frequency coding variation. Genome Biol. 12, R84 (2011)
G. M. Cooper, J. Shendure, Needles in stacks of needles: finding disease-causal variants in a wealth of genomic data. Nat. Rev. Genet. 12, 628–640 (2011)
D. G. MacArthur, S. Balasubramanian, A. Frankish, N. Huang, J. Morris, K. Walter, L. Jostins, L. Habegger, J. K. Pickrell, S. B. Montgomery, C. A. Albers, Z. D. Zhang, D. F. Conrad, G. Lunter, H. Zheng, Q. Ayub, M. A. DePristo, E. Banks, M. Hu, R. E. Handsaker, J. A. Rosenfeld, M. Fromer, M. Jin, X. J. Mu, E. Khurana, K. Ye, M. Kay, G. I. Saunders, M. M. Suner, T. Hunt, I. H. Barnes, C. Amid, D. R. Carvalho-Silva, A. H. Bignell, C. Snow, B. Yngvadottir, S. Bumpstead, D. N. Cooper, Y. Xue, I. G. Romero, C. Genomes Project, J. Wang, Y. Li, R. A. Gibbs, S. A. McCarroll, E. T. Dermitzakis, J. K. Pritchard, J. C. Barrett, J. Harrow, M. E. Hurles, M. B. Gerstein, C. Tyler-Smith, A systematic survey of loss-of-function variants in human protein-coding genes. Science 335, 823–828 (2012)
D. F. Gudbjartsson, H. Helgason, S. A. Gudjonsson, F. Zink, A. Oddson, A. Gylfason, S. Besenbacher, G. Magnusson, B. V. Halldorsson, E. Hjartarson, G. T. Sigurdsson, S. N. Stacey, M. L. Frigge, H. Holm, J. Saemundsdottir, H. T. Helgadottir, H. Johannsdottir, G. Sigfusson, G. Thorgeirsson, J. T. Sverrisson, S. Gretarsdottir, G. B. Walters, T. Rafnar, B. Thjodleifsson, E. S. Bjornsson, S. Olafsson, H. Thorarinsdottir, T. Steingrimsdottir, T. S. Gudmundsdottir, A. Theodors, J. G. Jonasson, A. Sigurdsson, G. Bjornsdottir, J. J. Jonsson, O. Thorarensen, P. Ludvigsson, H. Gudbjartsson, G. I. Eyjolfsson, O. Sigurdardottir, I. Olafsson, D. O. Arnar, O. T. Magnusson, A. Kong, G. Masson, U. Thorsteinsdottir, A. Helgason, P. Sulem, K. Stefansson, Large-scale whole-genome sequencing of the Icelandic population. Nat. Genet. 47, 435–444 (2015)
S. Jones, R. H. Hruban, M. Kamiyama, M. Borges, X. Zhang, D. W. Parsons, J. C. Lin, E. Palmisano, K. Brune, E. M. Jaffee, C. A. Iacobuzio-Donahue, A. Maitra, G. Parmigiani, S. E. Kern, V. E. Velculescu, K. W. Kinzler, B. Vogelstein, J. R. Eshleman, M. Goggins, A. P. Klein, Exomic sequencing identifies PALB2 as a pancreatic cancer susceptibility gene. Science 324, 217 (2009)
P. Ostergaard, M. A. Simpson, F. C. Connell, C. G. Steward, G. Brice, W. J. Woollard, D. Dafou, T. Kilo, S. Smithson, P. Lunt, V. A. Murday, S. Hodgson, R. Keenan, D. T. Pilz, I. Martinez-Corral, T. Makinen, P. S. Mortimer, S. Jeffery, R. C. Trembath, S. Mansour, Mutations in GATA2 cause primary lymphedema associated with a predisposition to acute myeloid leukemia (Emberger syndrome). Nat. Genet. 43, 929–931 (2011)
I. Comino-Mendez, F. J. Gracia-Aznarez, F. Schiavi, I. Landa, L. J. Leandro-Garcia, R. Leton, E. Honrado, R. Ramos-Medina, D. Caronia, G. Pita, A. Gomez-Grana, A. A. de Cubas, L. Inglada-Perez, A. Maliszewska, E. Taschin, S. Bobisse, G. Pica, P. Loli, R. Hernandez-Lavado, J. A. Diaz, M. Gomez-Morales, A. Gonzalez-Neira, G. Roncador, C. Rodriguez-Antona, J. Benitez, M. Mannelli, G. Opocher, M. Robledo, A. Cascon, Exome sequencing identifies MAX mutations as a cause of hereditary pheochromocytoma. Nat. Genet. 43, 663–667 (2011)
M. J. Smith, J. O’Sullivan, S. S. Bhaskar, K. D. Hadfield, G. Poke, J. Caird, S. Sharif, D. Eccles, D. Fitzpatrick, D. Rawluk, D. du Plessis, W. G. Newman, D. G. Evans, Loss-of-function mutations in SMARCE1 cause an inherited disorder of multiple spinal meningiomas. Nat. Genet. 45, 295–298 (2013)
T. Wiesner, A. C. Obenauf, R. Murali, I. Fried, K. G. Griewank, P. Ulz, C. Windpassinger, W. Wackernagel, S. Loy, I. Wolf, A. Viale, A. E. Lash, M. Pirun, N. D. Socci, A. Rutten, G. Palmedo, D. Abramson, K. Offit, A. Ott, J. C. Becker, L. Cerroni, H. Kutzner, B. C. Bastian, M. R. Speicher, Germline mutations in BAP1 predispose to melanocytic tumors. Nat. Genet. 43, 1018–1021 (2011)
M. Bogliolo, B. Schuster, C. Stoepker, B. Derkunt, Y. Su, A. Raams, J. P. Trujillo, J. Minguillon, M. J. Ramirez, R. Pujol, J. A. Casado, R. Banos, P. Rio, K. Knies, S. Zuniga, J. Benitez, J. A. Bueren, N. G. Jaspers, O. D. Scharer, J. P. de Winter, D. Schindler, J. Surralles, Mutations in ERCC4, encoding the DNA-repair endonuclease XPF, cause Fanconi anemia. Am. J. Hum. Genet. 92, 800–806 (2013)
D. G. MacArthur, T. A. Manolio, D. P. Dimmock, H. L. Rehm, J. Shendure, G. R. Abecasis, D. R. Adams, R. B. Altman, S. E. Antonarakis, E. A. Ashley, J. C. Barrett, L. G. Biesecker, D. F. Conrad, G. M. Cooper, N. J. Cox, M. J. Daly, M. B. Gerstein, D. B. Goldstein, J. N. Hirschhorn, S. M. Leal, L. A. Pennacchio, J. A. Stamatoyannopoulos, S. R. Sunyaev, D. Valle, B. F. Voight, W. Winckler, C. Gunter, Guidelines for investigating causality of sequence variants in human disease. Nature 508, 469–476 (2014)
S. Hanks, E. R. Perdeaux, S. Seal, E. Ruark, S. S. Mahamdallie, A. Murray, E. Ramsay, S. Del Vecchio Duarte, A. Zachariou, B. de Souza, M. Warren-Perry, A. Elliott, A. Davidson, H. Price, C. Stiller, K. Pritchard-Jones, N. Rahman, Germline mutations in the PAF1 complex gene CTR9 predispose to Wilms tumour. Nat. Commun. 5, 4398 (2014)
J. Shi, X. R. Yang, B. Ballew, M. Rotunno, D. Calista, M. C. Fargnoli, P. Ghiorzo, B. Bressac-de Paillerets, E. Nagore, M. F. Avril, N. E. Caporaso, M. L. McMaster, M. Cullen, Z. Wang, X. Zhang, W. Bruno, L. Pastorino, P. Queirolo, J. Banuls-Roca, Z. Garcia-Casado, A. Vaysse, H. Mohamdi, Y. Riazalhosseini, M. Foglio, F. Jouenne, X. Hua, P. L. Hyland, J. Yin, H. Vallabhaneni, W. Chai, P. Minghetti, C. Pellegrini, S. Ravichandran, A. Eggermont, M. Lathrop, K. Peris, G. B. Scarra, G. Landi, S. A. Savage, J. N. Sampson, J. He, M. Yeager, L. R. Goldin, F. Demenais, S. J. Chanock, M. A. Tucker, A. M. Goldstein, Y. Liu, M. T. Landi, Rare missense variants in POT1 predispose to familial cutaneous malignant melanoma. Nat. Genet. 46, 482–486 (2014)
S. Bao, R. Jiang, W. Kwan, B. Wang, X. Ma, Y. Q. Song, Evaluation of next-generation sequencing software in mapping and assembly. J. Hum. Genet. 56, 406–414 (2011)
D. B. Goldstein, A. Allen, J. Keebler, E. H. Margulies, S. Petrou, S. Petrovski, S. Sunyaev, Sequencing studies in human genetics: design and interpretation. Nat. Rev. Genet. 14, 460–470 (2013)
C. Gilissen, J. Y. Hehir-Kwa, D. T. Thung, M. van de Vorst, B. W. van Bon, M. H. Willemsen, M. Kwint, I. M. Janssen, A. Hoischen, A. Schenck, R. Leach, R. Klein, R. Tearle, T. Bo, R. Pfundt, H. G. Yntema, B. B. de Vries, T. Kleefstra, H. G. Brunner, L. E. Vissers, J. A. Veltman, Genome sequencing identifies major causes of severe intellectual disability. Nature 511, 344–347 (2014)
M. K. Gala, Y. Mizukami, L. P. Le, K. Moriichi, T. Austin, M. Yamamoto, G. Y. Lauwers, N. Bardeesy, D. C. Chung, Germline mutations in oncogene-induced senescence pathways are associated with multiple sessile serrated adenomas. Gastroenterology 146, 520–529 (2014)
D. R. Goudie, M. D’Alessandro, B. Merriman, H. Lee, I. Szeverenyi, S. Avery, B. D. O’Connor, S. F. Nelson, S. E. Coats, A. Stewart, L. Christie, G. Pichert, J. Friedel, I. Hayes, N. Burrows, S. Whittaker, A. M. Gerdes, S. Broesby-Olsen, M. A. Ferguson-Smith, C. Verma, D. P. Lunny, B. Reversade, E. B. Lane, Multiple self-healing squamous epithelioma is caused by a disease-specific spectrum of mutations in TGFBR1. Nat. Genet. 43, 365–369 (2011)
E. Ruark, K. Snape, P. Humburg, C. Loveday, I. Bajrami, R. Brough, D. N. Rodrigues, A. Renwick, S. Seal, E. Ramsay, V. Duarte Sdel, M. A. Rivas, M. Warren-Perry, A. Zachariou, A. Campion-Flora, S. Hanks, A. Murray, N. Ansari Pour, J. Douglas, L. Gregory, A. Rimmer, N. M. Walker, T. P. Yang, J. W. Adlard, J. Barwell, J. Berg, A. F. Brady, C. Brewer, G. Brice, C. Chapman, J. Cook, R. Davidson, A. Donaldson, F. Douglas, D. Eccles, D. G. Evans, L. Greenhalgh, A. Henderson, L. Izatt, A. Kumar, F. Lalloo, Z. Miedzybrodzka, P. J. Morrison, J. Paterson, M. Porteous, M. T. Rogers, S. Shanley, L. Walker, M. Gore, R. Houlston, M. A. Brown, M. J. Caufield, P. Deloukas, M. I. McCarthy, J. A. Todd, Breast, C. Ovarian Cancer Susceptibility, C. Wellcome Trust Case Control, C. Turnbull, J. S. Reis-Filho, A. Ashworth, A. C. Antoniou, C. J. Lord, P. Donnelly, N. Rahman, Mosaic PPM1D mutations are associated with predisposition to breast and ovarian cancer. Nature 493, 406–410 (2013)
M. Morak, T. Massdorf, H. Sykora, M. Kerscher, E. Holinski-Feder, First evidence for digenic inheritance in hereditary colorectal cancer by mutations in the base excision repair genes. Eur. J. Cancer 47, 1046–1055 (2011)
H. Okkels, L. Sunde, K. Lindorff-Larsen, O. Thorlacius-Ussing, P. Gandrup, J. Lindebjerg, P. Stubbeteglbjaerg, J. R. Oestergaard, F. C. Nielsen, H. B. Krarup, Polyposis and early cancer in a patient with low penetrant mutations in MSH6 and APC: hereditary colorectal cancer as a polygenic trait. Int. J. Color Dis. 21, 847–850 (2006)
N. Uhrhammer, Y. J. Bignon, Report of a family segregating mutations in both the APC and MSH2 genes: juvenile onset of colorectal cancer in a double heterozygote. Int. J. Color Dis. 23, 1131–1135 (2008)
A. G. Knudson Jr., Mutation and cancer: statistical study of retinoblastoma. Proc. Natl. Acad. Sci. U S A 68, 820–823 (1971)
A. H. Berger, A. G. Knudson, P. P. Pandolfi, A continuum model for tumour suppression. Nature 476, 163–169 (2011)
S. Seshagiri, E. W. Stawiski, S. Durinck, Z. Modrusan, E. E. Storm, C. B. Conboy, S. Chaudhuri, Y. Guan, V. Janakiraman, B. S. Jaiswal, J. Guillory, C. Ha, G. J. Dijkgraaf, J. Stinson, F. Gnad, M. A. Huntley, J. D. Degenhardt, P. M. Haverty, R. Bourgon, W. Wang, H. Koeppen, R. Gentleman, T. K. Starr, Z. Zhang, D. A. Largaespada, T. D. Wu, F. J. de Sauvage, Recurrent R-spondin fusions in colon cancer. Nature 488, 660–664 (2012)
N. Cancer Genome Atlas Research, J. N. Weinstein, E. A. Collisson, G. B. Mills, K. R. Shaw, B. A. Ozenberger, K. Ellrott, I. Shmulevich, C. Sander, J. M. Stuart, The Cancer Genome Atlas Pan-Cancer analysis project. Nat. Genet. 45, 1113–1120 (2013)
D. C. Blaydon, S. L. Etheridge, J. M. Risk, H. C. Hennies, L. J. Gay, R. Carroll, V. Plagnol, F. E. McRonald, H. P. Stevens, N. K. Spurr, D. T. Bishop, A. Ellis, J. Jankowski, J. K. Field, I. M. Leigh, A. P. South, D. P. Kelsell, RHBDF2 mutations are associated with tylosis, a familial esophageal cancer syndrome. Am. J. Hum. Genet. 90, 340–346 (2012)
J. B. Courcet, S. C. Elalaoui, L. Duplomb, M. Tajir, J. B. Riviere, J. Thevenon, N. Gigot, N. Marle, B. Aral, Y. Duffourd, A. Sarasin, V. Naim, E. Courcet-Degrolard, M. H. Aubriot-Lorton, L. Martin, J. E. Abrid, C. Thauvin, A. Sefiani, P. Vabres, L. Faivre, Autosomal-recessive SASH1 variants associated with a new genodermatosis with pigmentation defects, palmoplantar keratoderma and skin carcinoma. Eur. J. Hum. Genet. 23, 957–962 (2015)
Y. Sei, X. Zhao, J. Forbes, S. Szymczak, Q. Li, A. Trivedi, M. Voellinger, G. Joy, J. Feng, M. Whatley, M. S. Jones, U. L. Harper, S. J. Marx, A. M. Venkatesan, S. C. Chandrasekharappa, M. Raffeld, M. M. Quezado, A. Louie, C. C. Chen, R. M. Lim, R. Agarwala, A. A. Schaffer, M. S. Hughes, J. E. Bailey-Wilson, S. A. Wank, A hereditary form of small intestinal carcinoid associated with a germline mutation in inositol polyphosphate Multikinase. Gastroenterology 149, 67–78 (2015)
J. H. Nadeau, Modifier genes in mice and humans. Nat. Rev. Genet. 2, 165–174 (2001)
A. R. Moser, W. F. Dove, K. A. Roth, J. I. Gordon, The Min (multiple intestinal neoplasia) mutation: its effect on gut epithelial cell differentiation and interaction with a modifier system. J. Cell Biol. 116, 1517–1526 (1992)
W. F. Dietrich, E. S. Lander, J. S. Smith, A. R. Moser, K. A. Gould, C. Luongo, N. Borenstein, W. Dove, Genetic identification of mom-1, a major modifier locus affecting Min-induced intestinal neoplasia in the mouse. Cell 75, 631–639 (1993)
A. E. McCart, N. K. Vickaryous, A. Silver, Apc mice: models, modifiers and mutants. Pathol. Res. Pract. 204, 479–490 (2008)
T. K. Starr, R. Allaei, K. A. Silverstein, R. A. Staggs, A. L. Sarver, T. L. Bergemann, M. Gupta, M. G. O’Sullivan, I. Matise, A. J. Dupuy, L. S. Collier, S. Powers, A. L. Oberg, Y. W. Asmann, S. N. Thibodeau, L. Tessarollo, N. G. Copeland, N. A. Jenkins, R. T. Cormier, D. A. Largaespada, A transposon-based genetic screen in mice identifies genes altered in colorectal cancer. Science 323, 1747–1750 (2009)
H. N. March, A. G. Rust, N. A. Wright, J. ten Hoeve, J. de Ridder, M. Eldridge, L. van der Weyden, A. Berns, J. Gadiot, A. Uren, R. Kemp, M. J. Arends, L. F. Wessels, D. J. Winton, D. J. Adams, Insertional mutagenesis identifies multiple networks of cooperating genes driving intestinal tumorigenesis. Nat. Genet. 43, 1202–1209 (2011)
K. H. Goss, M. A. Risinger, J. J. Kordich, M. M. Sanz, J. E. Straughen, L. E. Slovek, A. J. Capobianco, J. German, G. P. Boivin, J. Groden, Enhanced tumor formation in mice heterozygous for Blm mutation. Science 297, 2051–2053 (2002)
N. Chester, F. Kuo, C. Kozak, C. D. O’Hara, P. Leder, Stage-specific apoptosis, developmental delay, and embryonic lethality in mice homozygous for a targeted disruption in the murine Bloom’s syndrome gene. Genes Dev. 12, 3382–3393 (1998)
G. Luo, I. M. Santoro, L. D. McDaniel, I. Nishijima, M. Mills, H. Youssoufian, H. Vogel, R. A. Schultz, A. Bradley, Cancer predisposition caused by elevated mitotic recombination in bloom mice. Nat. Genet. 26, 424–429 (2000)
L. D. McDaniel, N. Chester, M. Watson, A. D. Borowsky, P. Leder, R. A. Schultz, Chromosome instability and tumor predisposition inversely correlate with BLM protein levels. DNA repair 2, 1387–1404 (2003)
I. Ezkurdia, D. Juan, J. M. Rodriguez, A. Frankish, M. Diekhans, J. Harrow, J. Vazquez, A. Valencia, M. L. Tress, Multiple evidence strands suggest that there may be as few as 19,000 human protein-coding genes. Hum. Mol. Genet. 23, 5866–5878 (2014)
C. T. Kwok, I. P. Vogelaar, W. A. van Zelst-Stams, A. R. Mensenkamp, M. J. Ligtenberg, R. W. Rapkins, R. L. Ward, N. Chun, J. M. Ford, U. Ladabaum, W. C. McKinnon, M. S. Greenblatt, M. P. Hitchins, The MLH1 c.-27C > A and c.85G > T variants are linked to dominantly inherited MLH1 epimutation and are borne on a European ancestral haplotype. Eur. J. Hum. Genet. 22, 617–624 (2014)
L. B. Hesson, D. Packham, C. T. Kwok, A. C. Nunez, B. Ng, C. Schmidt, M. Fields, J. W. Wong, M. A. Sloane, R. L. Ward, Lynch syndrome associated with two MLH1 promoter variants and allelic imbalance of MLH1 expression. Hum. Mutat. 36, 622–630 (2015)
M. J. Kempers, R. P. Kuiper, C. W. Ockeloen, P. O. Chappuis, P. Hutter, N. Rahner, H. K. Schackert, V. Steinke, E. Holinski-Feder, M. Morak, M. Kloor, R. Buttner, E. T. Verwiel, J. H. van Krieken, I. D. Nagtegaal, M. Goossens, R. S. van der Post, R. C. Niessen, R. H. Sijmons, I. Kluijt, F. B. Hogervorst, E. M. Leter, J. J. Gille, C. M. Aalfs, E. J. Redeker, F. J. Hes, C. M. Tops, B. P. van Nesselrooij, M. E. van Gijn, E. B. Gomez Garcia, D. M. Eccles, D. J. Bunyan, S. Syngal, E. M. Stoffel, J. O. Culver, M. R. Palomares, T. Graham, L. Velsher, J. Papp, E. Olah, T. L. Chan, S. Y. Leung, A. Geurts van Kessel, L. A. Kiemeney, N. Hoogerbrugge, M. J. Ligtenberg, Risk of colorectal and endometrial cancers in EPCAM deletion-positive Lynch syndrome: a cohort study. Lancet Oncol. 12, 49–55 (2011)
M. P. Hitchins, Constitutional epimutation as a mechanism for cancer causality and heritability? Nat. Rev. Cancer 15, 625–634 (2015)
W. N. Cooper, A. Luharia, G. A. Evans, H. Raza, A. C. Haire, R. Grundy, S. C. Bowdin, A. Riccio, G. Sebastio, J. Bliek, P. N. Schofield, W. Reik, F. Macdonald, E. R. Maher, Molecular subtypes and phenotypic expression of Beckwith-Wiedemann syndrome. Eur. J. Hum. Genet. 13, 1025–1032 (2005)
A. Goel, T. P. Nguyen, H. C. Leung, T. Nagasaka, J. Rhees, E. Hotchkiss, M. Arnold, P. Banerji, M. Koi, C. T. Kwok, D. Packham, L. Lipton, C. R. Boland, R. L. Ward, M. P. Hitchins, De novo constitutional MLH1 epimutations confer early-onset colorectal cancer in two new sporadic lynch syndrome cases, with derivation of the epimutation on the paternal allele in one. Int. J. Cancer 128, 869–878 (2011)
M. P. Hitchins, S. Owens, C. T. Kwok, G. Godsmark, U. F. Algar, R. S. Ramesar, Identification of new cases of early-onset colorectal cancer with an MLH1 epimutation in an ethnically diverse south African cohort. Clin. Genet. 80, 428–434 (2011)
M. P. Hitchins, The role of epigenetics in lynch syndrome. Familial Cancer 12, 189–205 (2013)
C. Tomasetti, B. Vogelstein, Cancer etiology. Variation in cancer risk among tissues can be explained by the number of stem cell divisions. Science 347, 78–81 (2015)
C. Tomasetti, L. Marchionni, M. A. Nowak, G. Parmigiani, B. Vogelstein, Only three driver gene mutations are required for the development of lung and colorectal cancers. Proc. Natl. Acad. Sci. U S A 112, 118–123 (2015)
O. Zuk, E. Hechter, S. R. Sunyaev, E. S. Lander, The mystery of missing heritability: genetic interactions create phantom heritability. Proc. Natl. Acad. Sci. U S A 109, 1193–1198 (2012)
J. S. Witte, P. M. Visscher, N. R. Wray, The contribution of genetic variants to disease depends on the ruler. Nat. Rev. Genet. 15, 765–776 (2014)
O. Zuk, S. F. Schaffner, K. Samocha, R. Do, E. Hechter, S. Kathiresan, M. J. Daly, B. M. Neale, S. R. Sunyaev, E. S. Lander, Searching for missing heritability: designing rare variant association studies. Proc. Natl. Acad. Sci. U S A 111, E455–E464 (2014)
L. Moutsianas, V. Agarwala, C. Fuchsberger, J. Flannick, M. A. Rivas, K. J. Gaulton, P. K. Albers, G. McVean, M. Boehnke, D. Altshuler, M. I. McCarthy, The power of gene-based rare variant methods to detect disease-associated variation and test hypotheses about complex disease. PLoS Genet. 11, e1005165 (2015)
A. M. Meynert, M. Ansari, D. R. FitzPatrick, M. S. Taylor, Variant detection sensitivity and biases in whole genome and exome sequencing. BMC Bioinformatics 15, 247 (2014)
A. Belkadi, A. Bolze, Y. Itan, A. Cobat, Q. B. Vincent, A. Antipenko, L. Shang, B. Boisson, J. L. Casanova, L. Abel, Whole-genome sequencing is more powerful than whole-exome sequencing for detecting exome variants. Proc. Natl. Acad. Sci. U S A 112, 5473–5478 (2015)
E. E. Schadt, S. Turner, A. Kasarskis, A window into third-generation sequencing. Hum. Mol. Genet. 19, R227–R240 (2010)
G. F. Schneider, C. Dekker, DNA sequencing with nanopores. Nat. Biotechnol. 30, 326–328 (2012)
M. J. Chaisson, J. Huddleston, M. Y. Dennis, P. H. Sudmant, M. Malig, F. Hormozdiari, F. Antonacci, U. Surti, R. Sandstrom, M. Boitano, J. M. Landolin, J. A. Stamatoyannopoulos, M. W. Hunkapiller, J. Korlach, E. E. Eichler, Resolving the complexity of the human genome using single-molecule sequencing. Nature 517, 608–611 (2015)
E. Khurana, Y. Fu, V. Colonna, X. J. Mu, H. M. Kang, T. Lappalainen, A. Sboner, L. Lochovsky, J. Chen, A. Harmanci, J. Das, A. Abyzov, S. Balasubramanian, K. Beal, D. Chakravarty, D. Challis, Y. Chen, D. Clarke, L. Clarke, F. Cunningham, U. S. Evani, P. Flicek, R. Fragoza, E. Garrison, R. Gibbs, Z. H. Gumus, J. Herrero, N. Kitabayashi, Y. Kong, K. Lage, V. Liluashvili, S. M. Lipkin, D. G. MacArthur, G. Marth, D. Muzny, T. H. Pers, G. R. Ritchie, J. A. Rosenfeld, C. Sisu, X. Wei, M. Wilson, Y. Xue, F. Yu, E. T. Dermitzakis, H. Yu, M. A. Rubin, C. Tyler-Smith, M. Gerstein, Integrative annotation of variants from 1092 humans: application to cancer genomics. Science 342, 1235587 (2013)
M. Kircher, D. M. Witten, P. Jain, B. J. O’Roak, G. M. Cooper, J. Shendure, A general framework for estimating the relative pathogenicity of human genetic variants. Nat. Genet. 46, 310–315 (2014)
D. Lee, D. U. Gorkin, M. Baker, B. J. Strober, A. L. Asoni, A. S. McCallion, M. A. Beer, A method to predict the impact of regulatory variants from DNA sequence. Nat. Genet. 47, 955–961 (2015)
D. Carroll, Genome engineering with targetable nucleases. Annu. Rev. Biochem. 83, 409–439 (2014)
P. D. Hsu, E. S. Lander, F. Zhang, Development and applications of CRISPR-Cas9 for genome engineering. Cell 157, 1262–1278 (2014)
G. M. Findlay, E. A. Boyle, R. J. Hause, J. C. Klein, J. Shendure, Saturation editing of genomic regions by multiplex homology-directed repair. Nature 513, 120–123 (2014)
T. Sato, R. G. Vries, H. J. Snippert, M. van de Wetering, N. Barker, D. E. Stange, J. H. van Es, A. Abo, P. Kujala, P. J. Peters, H. Clevers, Single Lgr5 stem cells build crypt-villus structures in vitro without a mesenchymal niche. Nature 459, 262–265 (2009)
J. Drost, R. H. van Jaarsveld, B. Ponsioen, C. Zimberlin, R. van Boxtel, A. Buijs, N. Sachs, R. M. Overmeer, G. J. Offerhaus, H. Begthel, J. Korving, M. van de Wetering, G. Schwank, M. Logtenberg, E. Cuppen, H. J. Snippert, J. P. Medema, G. J. Kops, H. Clevers, Sequential cancer mutations in cultured human intestinal stem cells. Nature 521, 43–47 (2015)
M. Matano, S. Date, M. Shimokawa, A. Takano, M. Fujii, Y. Ohta, T. Watanabe, T. Kanai, T. Sato, Modeling colorectal cancer using CRISPR-Cas9-mediated engineering of human intestinal organoids. Nat. Med. 21, 256–262 (2015)
M. van de Wetering, H. E. Francies, J. M. Francis, G. Bounova, F. Iorio, A. Pronk, W. van Houdt, J. van Gorp, A. Taylor-Weiner, L. Kester, A. McLaren-Douglas, J. Blokker, S. Jaksani, S. Bartfeld, R. Volckman, P. van Sluis, V. S. Li, S. Seepo, C. Sekhar Pedamallu, K. Cibulskis, S. L. Carter, A. McKenna, M. S. Lawrence, L. Lichtenstein, C. Stewart, J. Koster, R. Versteeg, A. van Oudenaarden, J. Saez-Rodriguez, R. G. Vries, G. Getz, L. Wessels, M. R. Stratton, U. McDermott, M. Meyerson, M. J. Garnett, H. Clevers, Prospective derivation of a living organoid biobank of colorectal cancer patients. Cell 161, 933–945 (2015)
N. Rahman, Realizing the promise of cancer predisposition genes. Nature 505, 302–308 (2014)
H. S. Feigelson, K. A. Goddard, C. Hollombe, S. R. Tingle, E. M. Gillanders, L. E. Mechanic, S. A. Nelson, Approaches to integrating germline and tumor genomic data in cancer research. Carcinogenesis 35, 2157–2163 (2014)
J. R. Pon, M. A. Marra, Driver and passenger mutations in cancer. Annu. Rev. Pathol. 10, 25–50 (2015)
H. Peng, J. Huang, Y. Hu, Y. Wei, H. Liu, M. Huang, L. Wang, J. Wang, Associations between polymorphisms in the SYK promoter and susceptibility to sporadic colorectal cancer in a southern Han Chinese population - a short report. Cell. Oncol. 38, 165–172 (2015)
Acknowledgments
This work was supported by research grants from the Dutch Cancer Society (KWF, grants 2009-4335 and 2014-6666), and the Netherlands Organization for Scientific Research (NWO, grant 917-10-358).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflict of interest.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Hahn, M.M., de Voer, R.M., Hoogerbrugge, N. et al. The genetic heterogeneity of colorectal cancer predisposition - guidelines for gene discovery. Cell Oncol. 39, 491–510 (2016). https://doi.org/10.1007/s13402-016-0284-6
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13402-016-0284-6