
Abstract
A 69-year-old woman presented with a cortical hand syndrome progressing over several weeks. MRI brain showed characteristic appearances of progressive multifocal leukoencephalopathy (PML), confirmed by detection of the JC virus in CSF, despite the absence of any evidence of immunosuppression. Treatment with mirtazapine, mefloquine and cidofovir did not affect the progression of the disease, which was fatal within 7 months of presentation. This report adds to the small case literature that suggests that PML can occur in immunocompetent people, albeit extremely rarely.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Progressive multifocal leukoencephalopathy (PML) is potentially fatal demyelinating encephalopathy of gradual onset caused by the John-Cunningham virus (JCV). JCV is common in the general population with serum antibody to the virus being detectable in up to 80% of adults. Primary infection is usually asymptomatic and leads to indefinite survival of the virus in the kidney (Sabath and Major 2002). In conditions of immunocompromise, reactivation of JCV may occur leading to PML. Human immunodeficiency virus (HIV) infection is the most typical context in which JCV reactivation, and subsequent development of PML, occurs. However, another more recent but relatively common situation in which JCV reactivation and consequent development of PML can occur, is as a result of treatment of multiple sclerosis (MS) with Natalizumab. Both of these examples are associated with a relatively high occurrence of PML, reaching incidence levels of around 1 in 100 in HIV-infected patients in the years before the development of combined antiretroviral therapy and similarly at present, in patients with MS that are high positive for the JC virus (in peripheral blood) and have been on treatment with Natalizumab for 2 years or more (Pavlovic et al. 2015; Plavina et al. 2014). PML is also rarely associated with a plethora of other conditions, including malignancies; autoimmune diseases, such as Sjogren’s syndrome; granulomatous diseases, such as sarcoidosis; and primary immunodeficiencies such as idiopathic CD4 lymphopenia (Zaheer and Berger 2012). These conditions are associated with PML because of immunosuppressive effects—either through the condition itself or through the treatment of them. However, some cases of PML have been reported in the context of only mild immunosuppression such as in hepatic cirrhosis or liver failure (Gheuens et al. 2010) and very rarely in immunocompetent patients.
Case report
A 69-year-old right-handed retired head teacher presented with fatigue and left hand weakness of insidious onset over 6 weeks. Her past medical history was unremarkable apart from hypertension and mild COPD. She had a 40-pack-year smoking history but stopped smoking 13 years earlier following the diagnosis of COPD. Approximately once a year, she received steroids to treat COPD exacerbations with the last course being 8 months before presentation. She had travelled to Grenada and California with no ill health during these visits.
On examination, she had mild left facial weakness, global weakness of the left hand, increased tone in the left arm and leg, brisk reflexes and a positive Hoffman’s sign on the left, with withdrawal plantar responses. Sensation was normal.
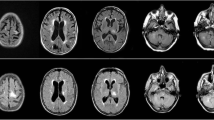
MRI of brain revealed multiple T2-hyperintensities in the right corona radiata with restricted diffusion in a patchy, peripheral pattern on diffusion weighted imaging and matched low signal on the apparent diffusion coefficient sequence. Follow-up imaging 4 weeks later showed progression of these changes without contrast enhancement (Fig. 1). There was no hypointensity on susceptibility weighted imaging. An incidental 7 mm diameter aneurysm of the middle cerebral artery was also found. MRI of cervical spine was unremarkable.

MR imaging of brain demonstrating multifocal areas of high signal on T2 (a, g) and flair (b, h) with restricted patchy and peripheral diffusion on the axial DWI sequence (c, i) matched on the ADC (d, j). T1 images showed low signal (e, k) and post-contrast (f, l) there was no enhancement. The imaging changes progressed over a one-month interval (a–f followed by g–l)
The imaging findings and history of gradually progressive weakness were felt to be strongly suggestive of progressive multifocal leukoencephalopathy (PML), despite the fact that she had no risk factors for HIV and had no history of immunosuppression.
CSF PCR detected JVC and confirmed the diagnosis of PML. The rest of the CSF constituents were normal (protein 0.35 g/L, glucose 3.5 mmol/L, white cell count less than 2/μL). HIV-1 and HIV-2 testing was negative. FBC, ESR (2 mm/h), CRP, renal, liver function, B12 and folate, blood film and LDH were all normal. Hepatitis B and C testing was negative. A CT body did not reveal evidence of an occult malignancy.
Serum electrophoresis showed a polyclonal increase in IgA, a finding which is associated with chronic mucosal inflammation such as in COPD. Immunoglobulin measurements were IgG 12.89 (6.00–16.00 g/l), IgA 6.31 (0.80–4.00 g/l) and IgM 0.73 (0.50–2.00 g/l). IgG subclasses 1–4 were also normal.
She was referred to immunology to assess whether she might have an underlying immunodeficiency. Phenotypic characterisation of peripheral B and T lymphocytes by flow cytometry (of blood) was normal (Table 1). Extensive immunological assessment including human T lymphocyte virus (HTLV), functional assessment of her T cells, T cell spectrotype, T cell receptor excisions circles (TRECS) count (which allows assessment of the thymic output of T cells) and in vitro cytokine production did not reveal any underlying immunodeficiency. There was no family history suggestive of immunodeficiency, although one of her two brothers had died at the age of 8 from disseminated TB.
On the basis of the limited current literature, she was started on treatment with mefloquine 250 mg daily for 3 days initially, followed by 250 mg once a week plus mirtazapine 30 mg once a day. Her symptoms continued to progress such that she lost the use of her left arm after a further month, and the progression of weakness in her left leg led to the use of a wheelchair around 3 months later. After review by the immunologists, she was also commenced on cidofovir 5 mg/kg once weekly for 2 weeks, then continued at fortnightly intervals, with no apparent response after 6 weeks of treatment at which point it was discontinued. She developed a dysarthria 7 months after initial presentation and died shortly after that.
Discussion
The clinical features of PML result from lytic infection of oligodendrocytes by JCV, typically as a result of failure of the T cell-mediated immune response, whose role it is to prevent JCV reactivation and subsequent viraemia (Gheuens et al. 2010). This, therefore, is the basis of the thinking that progression to PML requires immunosuppression.
Although PML is relatively commonly seen in immunosuppressed patients, it has, very infrequently, been reported in both mildly immunocompromised (Zaheer and Berger 2012) and apparently immunocompetent (Chang et al. 2007) patients. In the 1980s, it was thought that up to 5% of patients with PML had no discernible immunosuppression (Brooks and Walker 1984). However, understanding of the mechanisms of immune dysfunction has since improved greatly, indicating that many of these patients may have been suffering from immunodeficiencies that were, at that stage, undetectable.
There are two different types of JCV: the archetypal form which is transmitted between people in childhood and resides in the kidney and the neurotropic type which evolves from the archetype and causes PML. It has been proposed that the mutations that transform the archetype virus into the neurotropic form occur in the white blood cells of the bone marrow. These cells are then the source of haematogenous spread of JCV to the brain (Wollebo et al. 2015). It has been postulated that a period of dysfunction in the immune surveillance of the virus’ life cycle could be sufficient to allow the necessary viraemia to occur in otherwise immunocompetent patients (Grewal et al. 2016). It is possible that a transient period of immune suppression may result in this dysfunction in immune surveillance (Chang et al. 2007). Considering this, the history of occasional use of oral steroids to treat exacerbations of COPD may be relevant to this case. However, the ubiquitous nature of both oral steroid use and past infection with JCV renders the exposure to oral steroids an unlikely explanation of her development of PML.
The diagnosis of PML requires a combination of clinical suspicion, imaging, CSF PCR for JCV and occasionally brain biopsy. MRI of brain typically shows asymmetric demyelination of white matter represented by lesions that are hypointense on T1-weighted images and hyperintense on T2-weighted imaging and fluid attenuated inversion recovery images. A rim of high signal on diffusion weighted imaging is characteristic, as in this case. The lesions rarely show marked contrast enhancement (Shah et al. 2010); however, faint contrast enhancement may be seen peripherally in approximately 10% of CT and 15% of MRI of brain scans (Berger et al. 1998). Furthermore, contrast enhancement is a common finding in PML-IRIS (immune reconstitution inflammatory syndrome) in association with natalizumab treatment. CSF PCR for JCV is useful in confirming the diagnosis as it has a sensitivity of 67–78% and a specificity of 93–100% (Brew et al. 2010). It is important to note that the sensitivity of CSF PCR is not sufficient to exclude PML, so if clinical suspicion is high, further investigation with a brain biopsy may be indicated.
In vitro evidence suggesting that 5-HT2a receptors play a role in JCV entry into glial cells has led to the investigation of serotonin receptor antagonists such as mirtazapine, as treatment for PML (Elphick et al. 2004). Mefloquine has been suggested to act synergistically with mirtazapine, whereby viral infection of glial cells is prevented by mirtazapine and viral replication is inhibited by mefloquine (Christakis et al. 2013). Despite the proposed mechanism of interaction of mirtazapine and mefloquine in treating PML, there is limited evidence for their use. Indeed, mefloquine was found to have no treatment effect on PML in one randomised study (Clifford et al. 2013).
While the role of cidofovir in treating PML is yet unproven there have been reports both that its use may improve survival (Viallard et al. 2007) and that these effects may be increased in immunocompetent patients (Naess et al. 2010). However, the latter point of view is seemingly at odds with the case described above where, unfortunately, cidofovir had no effect on progression, despite the fact that the patient had no demonstrable immunodeficiency. The outcome of this case seem to be congruent with both in vitro data suggesting that cidofovir does not prevent viral replication in neuroglial cells (Hou and Major 1998), and in vivo data showing that cidofovir has no impact on mortality related to the development of PML (De Luca et al. 2008).
This case demonstrates the possibility that PML may very rarely occur in apparently immunocompetent individuals. Furthermore, the disappointing lack of efficacy of the currently available treatments for this condition is highlighted in this case. This is further exemplified by the high 3-month mortality rate of between 20 and 50% (Brew et al. 2010), which is most noted in those without any reversible immunodeficiency.
References
Berger JR, Pall L, Lanska D, Whiteman M (1998) Progressive multifocal leukoencephalopathy in patients with HIV infection. J Neuro-Oncol 4:59–68
Brew BJ, Davies NW, Cinque P, Clifford DB, Nath A (2010) Progressive multifocal leukoencephalopathy and other forms of JC virus disease. Nat Rev Neurol 6:667–679
Brooks BR, Walker DL (1984) Progressive multifocal leukoencephalopathy. Neurol Clin 2(2):299–313
Chang YY, Lan MY, Peng CH, Wu HS, Chang D, Liu JS (2007) Progressive multifocal leukoencephalopathy in an immunocompetent Taiwanese patient. J Formos Med Assoc 106(2 Suppl):S60–S64
Christakis PG, Okin D, Huttner AJ, Baehring JM (2013) Progressive multifocal leukoencephalopathy in an immunocompetent patient. J Neurol Sci 326(1–2):107–110
Clifford D, Nath A, Cinque P, Brew B, Zivadinov R, Gorelik L (2013) A study of mefloquine treatment for progressive multifocal leukoencephalopathy: results and exploration of predictors of PML outcomes. J Neuro-Oncol 19:351–358
De Luca A, Ammassari A, Pezzotti P et al (2008) Cidofovir in addition to antiretroviral treatment is not effective for AIDS-associated progressive multifocal leukoencephalopathy: a multicohort analysis. AIDS 22:1759–1767
Elphick GF, Querbes W, Jordan JA et al (2004) The human polyomavirus, JCV, uses serotonin receptors to infect cells. Science 306:1380–1383
Gheuens S, Pierone G, Peeters P, Koralnik IJ (2010) Progressive multifocal leukoencephalopathy in individuals with minimal or occult immunosuppression. J Neurol Neurosurg Psychiatry 81(3):247–254
Grewal J, Dalal P, Bowman M, Kaya B, Otero JJ, Imitola J (2016) Progressive multifocal leukoencephalopathy in a patient without apparent immunosuppression. J Neuro-Oncol 22(5):683–687
Hou J, Major EO (1998) The efficacy of nucleoside analogs against JC virus multiplication in a persistently infected human fetal brain cell line. J Neuro-Oncol 4(4):451–456
Naess H, Glad S, Storstein A, Rinaldo CH, Mørk SJ, Myhr KM, Hirsch H (2010) Progressive multifocal leucoencephalopathy in an immunocompetent patient with favourable outcome. BMC Neurol 10:32
Pavlovic D, Patera AC, Nyberg F, Gerber M, Liu M (2015) Progressive multifocal leukoencephalopathy: current treatment options and future perspectives. Ther Adv Neurol Disord 8(6):255–273
Plavina T, Subramanyam M, Bloomgren G, Richman S, Pace A, Lee S, Schlain B, Campagnolo D, Belachew S, Ticho B (2014) Anti–JC virus antibody levels in serum or plasma further define risk of natalizumab-associated progressive multifocal leukoencephalopathy. Ann Neurol 76(6):802–812
Sabath BF, Major EO (2002) Traffic of JC virus from sites of initial infection to the brain: the path to progressive multifocal leukoencephalopathy. J Infect Dis 186:S180–S186
Shah R, Bag AK, Chapman PR, Cure JK (2010) Imaging manifestations of progressive multifocal leukoencephalopathy. Clin Radiol 65:431–439
Viallard JF, Lazaro E, Ellie E, Eimer S, Camou F, Caubet O, Lafon ME, Fleury H, Pellegrin JL (2007) Improvement of progressive multifocal leukoencephalopathy after cidofovir therapy in a patient with a destructive polyarthritis. Infection 35(1):33–36. https://doi.org/10.1007/s15010-006-5103-y
Wollebo HS et al (2015) Persistence and pathogenesis of the neurotropic polyomavirus JC. Ann Neurol 77(4):560–570
Zaheer F, Berger JR (2012) Treatment-related progressive multifocal leukoencephalopathy: current understanding and future steps. Ther Adv Drug Saf 3(5):227–239
Acknowledgements
We are grateful to Dr. Daniel Scoffings, consultant neuroradiologist, for his interpretation of the MR imaging. We also thank Dr. Gkrania-Klotsas and Dr. Kumararatne, consultant immunologists, for their thorough assessment of the patient and helpful advice on management.
Author information
Authors and Affiliations
Contributions
BZ prepared the case report. SRLS is the consultant neurologist responsible for the care of the patient and contributed to the writing of the report.
Corresponding author
Ethics declarations
Competing interests
The authors declare that they have no competing interests.
Statement of informed consent
Consent for publication was provided by the patient’s husband.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Zucker, B.E., Stacpoole, S.R.L. Progressive multifocal leukoencephalopathy in the absence of immunosuppression. J. Neurovirol. 24, 119–122 (2018). https://doi.org/10.1007/s13365-017-0592-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13365-017-0592-2