
Abstract
In our previous study, we introduced a new hybrid approach to effectively approximate the total force on each ion during a trajectory calculation in mass spectrometry device simulations, and the algorithm worked successfully with SIMION. We took one step further and applied the method in massively parallel general-purpose computing with GPU (GPGPU) to test its performance in simulations with thousands to over a million ions. We took extra care to minimize the barrier synchronization and data transfer between the host (CPU) and the device (GPU) memory, and took full advantage of the latency hiding. Parallel codes were written in CUDA C++ and implemented to SIMION via the user-defined Lua program. In this study, we tested the parallel hybrid algorithm with a couple of basic models and analyzed the performance by comparing it to that of the original, fully-explicit method written in serial code. The Coulomb explosion simulation with 128,000 ions was completed in 309 s, over 700 times faster than the 63 h taken by the original explicit method in which we evaluated two-body Coulomb interactions explicitly on one ion with each of all the other ions. The simulation of 1,024,000 ions was completed in 2650 s. In another example, we applied the hybrid method on a simulation of ions in a simple quadrupole ion storage model with 100,000 ions, and it only took less than 10 d. Based on our estimate, the same simulation is expected to take 5–7 y by the explicit method in serial code.
Similar content being viewed by others


1 Introduction
The quality of computational modeling of instrumentation designs depends on how accurately the model reflects reality. In general, models of a complicated process taking place in a mass spectrometer are often oversimplified because of their high computational cost. The major cause is the number of ions involved in the process. When there are n ions present, there will be operations in the scale of n 2/2 depending on the efficiency of the algorithm. To complete trajectory calculations, we need to calculate the total force on each ion by evaluating its interactions with every other ion, and update the profile of all ions at every time step. Obviously, as the technology used in mass spectrometry advances, increased sophistication in multi-physics modeling and efficiency in a computational method becomes necessary to simulate the highly sensitive new instruments.
For simulation containing a large number of ions, a massive weight is applied on the evaluation of the total force acting on each ion at every time step. We identified the problem in the evaluation of two-body interactions, and first developed our serial hybrid algorithm to only run on a CPU system [1]. Even without GPU acceleration, we saw an improvement of an order of magnitude [1]. In the second and intermediate step in our GPGPU project with Compute Unified Device Architecture (CUDA) [2], we developed the parallel code in CUDA C++ to be used with SIMION [3–5]. SIMION is an ion optics simulation program that is a highly versatile, Windows-based computer-simulation program, which has been used to optimize the design of scientific instruments such as mass spectrometers and ion mobility devices. We have added one of the most popular GPU (<$150) in the similar NVIDIA product line to the desktop system, similar to the one we used in the previous study. Our final goal for the project was to develop a code specific to the system of multiple GPUs we are building for designing and optimizing the new instruments, with additional new modeling techniques.
2 Computational Method
All GPGPU simulations were performed on a 64-bit, Windows-based personal computer system with a 3.4 GHz Intel Core i7 quad processor with 4 GB of RAM, and equipped with a NVIDIA GForce GTX460 (Fermi) graphic processor unit with 1 GB of RAM. As mentioned earlier, the parallel codes are written in CUDA C++ and implemented to the SIMION ver. 8.1 through user-defined “Lua” [6] program (the embedded scripting language in SIMION). A single core was assigned to process the code with the highest priority for accurate measurement of CPU time including GPU waiting time.
We evaluated the performance by comparing the CPU time. Two basic designs, (1) a Coulomb explosion model without electrodes, and (2) a model of a quadrupole ion storage device were tested, and their performance was compared to that of other methods.
3 Hybrid Process and Application in Massively Parallel Architecture
The complete discussion on how the hybrid process works can be found in our previous work [1]. For the convenience of readers, we would like to give a brief overview of the key concepts used in the hybrid method. In the hybrid approach, the computational load was reduced by efficiently approximating the total force acting on an ion by not explicitly calculating its interactions with all other ions. These other ions were first divided into groups depending on their distance from the ion under consideration. For those in the short-range, interactions are treated as explicit two-body interactions represented by Coulomb’s law (equation 1)
where the vector F i represents the exact total interactions between ion i and other ions j; q i is the charge of ion i; r is the separation of two charges carried by ions i and j; \( {\widehat{r}_{{ji}}} \) is the unit directional vector from the charge j to i; and ϵ 0 is the vacuum permittivity. Obviously, the acceleration of an ion is largely influenced by nearby ions. When considered individually, the interaction between the current ion and one far enough away is much less important, if not negligible, compared with the interaction with one in the neighborhood. However, a collection of these long-range interactions can significantly contribute to the ion’s trajectory. In the hybrid method, we took a collection of these ions within a certain range and treated it by approximating its influence on the current ion as a Coulomb force between the ion and a conducting spherical surface that we called a “charge diffused cloud.” We defined a charge diffused cloud as a collective charge distribution of ions in a cubic or rectangular space that we called a block. The number of blocks and their sizes are specified at the beginning of the simulation and fixed in space during the simulation. As the charge distribution in a block changes over time as ions flow in and out, the property of the charge diffused cloud has to be updated for all blocks at every time step. Depending on the characteristic of a problem, it may so happen that there are ions that are far away enough that they make a negligible contribution to the total force. In such cases, the distinction can be made by defining an “active region,” the union of the short-range and the long-range, and the outside of the active region. The sizes of short- and long-range interaction regions are defined by the block dimensions and control parameters l and m, respectively. Subscripts may be used when they vary depending on coordinates. Short-range interactions are confined in the region of (2l + 1)3 cubic block edge lengths centered at the block in which the current ion is located. Long-range interactions are confined in the region of (2m + 1)3 cubic block edge lengths outside of short-range interaction region. For example, suppose the set {l i = 1, m i = 7 for i = x, y, z} defines the active region. We omit the subscripts now because they take the same values for x, y, and z. If 1 mm3 cubic blocks are used for the hybrid calculation purpose, the short-range interaction region is the 3 mm3 cube, and the outer boundary of the long-range interaction region, which is also the boundary of the active region, consists of the outer edges of the 15 mm3 cube. In the previous study, we demonstrated the dependency of the properties defining the active region on the simulation result.
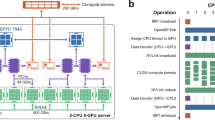
In the SIMION trajectory simulation program, a user may make changes and additions through user-defined Lua program that can call for processes run outside SIMION. In a parallel version of the hybrid algorithm, there are two main processes carried out on GPU: (1) Coulomb interactions (ion–ion and ion–cloud interactions), and (2) the evaluation of the profiles of charge diffused clouds by blocks. For either process, ions are divided into groups and their profiles are called in to the specified GPU kernel. In developing parallel CUDA C++ codes it is of extreme importance to reduce the time used in the barrier synchronization to optimize the code’s overall performance and to minimize data transfer between the host (CPU) memory and device (GPU) memory. In addition, we took full advantage of latency hiding to eliminate the process waiting time. These techniques are critical for efficient parallel executions on massively parallel computing systems. After repeatedly testing the codes, we have reached the significant improvements that we discuss below. Figure 1 shows how it communicates with the SIMION program, and Figure 2 shows the flow of the main processes in the parallel codes.

Schematic flow diagram describing our implementation of the parallel CUDA C++ hybrid codes to the SIMION trajectory calculation via the user-defined Lua program. SIMION communicates with CUDA kernels on GPU via the segment call, segment.accel_adjust. Massively parallel computations occur in the sections in blue in the box on the right

Schematic process flow diagram of major processes in the parallel hybrid algorithm
4 Results and Discussion
In the following two sections we, would like to discuss details of the simulations and improvements we made with GPU acceleration by using the massively parallel computational method.
4.1 Simulation #1: Coulomb Explosion
To study how simulation time grows with the number of ions, we ran Coulomb explosion simulations of: 1000, 8000, 16,000, 32,000, 64,000, 128,000, 512,000, and 1,024,000 singly charged ions with each ion carrying a mass of 100 u. We varied the size of the cubic simulation domain by 40, 80, 98, 124, 160, 196, 320, and 392 mm3 with respect to these cases. For every case, we used 20 × 20 × 20 blocks spanning the entire simulation domain, regardless of its size. Ions were given zero kinetic energy and initially normally distributed at the coordinate origin with standard deviations of 1.00, 2.00, 2.52, 3.17, 4.00, 5.04, 8.00, and 10.08 mm in any coordinate direction for the cases of 1000, 8000, 16000, 64,000, 128,000, 512,000, and 1,024,000 ions, respectively. Short-range interactions were restricted within the center block in which the current ion resides by setting the parameters to l x = l y = l z = 0 (i.e., no blocks outside the center block are included in the short range). The active range was defined by the boundary set by other control parameters, m x = m y = m z = 9, (i.e., the long range was limited to ±9 blocks in the x-, y-, and z-directions from the center block; hence the entire domain is covered when the ion is in the center block located at the origin. We measured the time it took for each ion to reach the domain boundary, and the simulation was considered “complete” when 90 % of the ions had achieved this. A uniform time step of 50 μs was used.
Figure 3 shows that the arrival time distributions for the simulations by the parallel hybrid algorithm were all similar to those produced by the fully explicit method, regardless of the number of ions. Figure 4 shows the similarlity between the evolution of the radial distributions of 64,000 ions obtained by the explit and hybrid methods in which data was taken every 50 μs. As expected, the radial distributions gradually lose their intensity as the ions spread outward. Radial distributions obtained by both methods show fairly persistent tails extending toward the origin. These slow moving ions are not affected by the outer layer of ions due to the spherical symmetry imposed on the initial distribution. Figure 5 shows that the effect of GPU acceleration is more apparent when a larger number of ions were involved. The case of 128,000 ions only took 309 s to complete, whereas with the explicit method—where all two-body interactions were evaluated (serial codes)—the simulation took 63 h. That is an over 700 times faster performance. In the case of 1,024,000 ions, the simulation took only 2650 s by using the massively parallel hybrid algorithm. Accouting for the fact that the CPU time required for a simulation increases much more rapidly as the number of ions flowing in the simulations increases, a simulation of such magnitude would take more than a month under the same conditions when CPU alone is used.

Normalized arrival time distribution for Coulomb explosion simulations on GPGPU by the parallel hybrid method and the parallel explicit two-body calculation method. The same number of blocks (20 × 20 × 20) was used for every case for the hybrid method, regardless of the size of the domain; and the active region was similarly defined in every case (see text). As the initial condition, normally distributed, singly-charged ions carrying 100 u each were initially released at the center of the domain in the properly-scaled, field-free region. The number in the legend corresponds to the number of ions released in the simulations. The similarity in results was as expected

Radial distributions taken at every 50 μs for Coulomb explosion simulations of 64,000 ions on GPGPU by the parallel hybrid method and the parallel explicit two-body calculation method. Exactly the same parameters were used for the hybrid method (see text). The similarity in results was as expected

Comparison of computational time for Coulomb explosion simulations. This summary shows all the results from our previous and current studies. The explicit two-body method results are shown with filled symbols, and the hybrid algorithm with open symbols. The effects of varying the size of the active region and the number of blocks, and the performance improvement made by using a GPU, can be seen in the figure. All single thread calculation results from our precious study are shown in solid lines. See text for the specifications of the machines on which these calculations were run
4.2 Simulation #2: Quadrupole Ion Storage Device
For the modeling of a basic device design of a rf-only quadrupole based ion storage, we used rectangular blocks in the rectangular domain space as shown in Figure 6. We assigned the inscribed radius (r in), rod radius (r rod), and length of the rod (l rod) to be 4, 3.6, and 80 mm, respectively. Both the entrance and exit electrode plates were separated from the ends of the rod electrodes by 4 mm, and their voltages were set at 100 V. rf Voltage on the quadrupole rod electrodes was set at 138 V0–p at 1.1 MHz to maximize the pseudopotential. Neutral helium (2 mTorr) was used as the collisional damping gas. Each of the 100,000 ions was assigned the mass of 100 u. The initial kinetic energies of all ions were set to zero, and the ions were distributed uniformally in the 60 mm × 2 mm × 2 mm rectangular space located along the longitudinal (x-) axis (i.e., at the center of the quadrupole configuration at the time of release). Ion-neutral collisions were simulated using the Collision Model HS1 [5]. Briefly, the model is based on the kinetic theory of gases, and the individual collision between ion and gas particles are modeled, in which colliding particles are treated as elastic hard-spheres. Detail of the model can be found in the newer version of the SIMION manual [5]. In this simulation, Mathieu parameter q

Simple quadrupole ion storage device model used in this study. Variables used in the discussion in the text are defined as shown in the figure: r rod is the radius of a rod electrode; l rod is its length; r in is the distance from the center to the edge of a rod; and <r(t)>n is the “average radius,” the measure of the spread of n ions at time t, is the average of ion-to-center distances; b x is the number of blocks in the x-direction. In this particular example, the model has 20 blocks stacked in the x-direction, so b x = 20, while no division is made in the y- and z-directions; hence, b y = b z = 1
is set at 0.7. To avoid confusion, it should be emphasized that all trajectory calculations are performed using direct nuemerical simulations. We would like to point out here that ion-neutral collisions can also be improved by converting the code in parallel. However, we do not expect a great amount of overall performance improvement because the ratio of computational load of the collisions to the Coulumb interaction calculations is in the scale of n to n 2/2. Hence we decided to just parallelize the two main processes in the right box in Figure 1.
Returning to Figure 6, the block dimensions were defined by setting the number of blocks to b x = 20 and b y = b z = 1, and we set the active region parameters to l x = l y = l z = 0 and m x = m y = m z = 1. Hence, the domain contains a single layer of blocks stacked in the x-direction, the ions in the short range are in the same block as the current ion, and the ion–cloud interactions are limited to two blocks immediately adjacent to the center block. At every time step, the ions’ average distance to the x-axis was calculated, and the average was recorded as the “average radius” of the distribution at that time, and the simulation was continued until equillibrium was reached. Initially, ions rapidly oscillate as the amplitude diminishes and the value converges. The average radius of oscillation ranges from 0.7 to 1.5 mm in the first few μs. The progress of the average radius and its convergence to equilibrium for the simulation of 100,000 ions is shown in Figure 7. We note here that the number of ions used in this study is far less than the theoretial threshold, where repulsive interactions overcome the pseudopotential produced in this paricular storage device. By using the formula

The progress of the average radius (see text) of the ion distribution for 0 < time < 600 μs in a simulation with 100,000 ions in a basic rf-only quadrupole ion storage device model. The result is very similar to the simulation of 5000 ions performed with the explicit calculation method in our previous study; and we did not see a significant deviation from the limit observed in the simulation of 5000 ions
it is estimated as 80 million ions for this setup [7–10]. As expected, we observed very small deviation in the limits, the average radius at 600 μs in the simulation with 100,000 ions and that of 5000 ions obtained by the explicit method from our previous study. From this observation, we are expecting this ion storage design to be able to focus on many more ions before we start seeing repulsive interactions effectively defocusing ions. We did not run a larger number of ions in the simulations at this time because it requires substantial computing time on the system based on multiple GPUs.
Parallelization had a much greater effect on the ion storage device than the Coulomb explosion simulation. At 600 μs, the ion storage simulation of 100,000 ions was completed in only about 10 d on our hardware with added GPU power. The total number of time steps (ranging from 0.02 to 0.025 μs) required for the entire process were approximately 25,000 steps, compared with the ~25 steps required for the Coulomb explosion simulation. When the explicit method is used, two-body Coulomb interaction calculations must be repeated approximately 1000 times more for the ion storage simulation than the Coulomb explosion simulation containing same numbers of ions; hence approximately 1000 times more computation time is expected. Based on this basic comparison, we can expect approximately 5 to 7 y to complete the simulation if each and every two-body interaction were to be explicitly calculated on the system without GPU acceleration.
5 Conclusion
The original hybrid algorithm used in our previous study was reconstructured for GPGPU, and our massively parallel hybrid codes were demonstrated to work much more efficiently and as accurately as the fully explicit method on the CPU-only system. The Coulomb explosion simulation of 128,000 ions was over 700 times faster; and we were able to successfully simulate over a million ions within a couple of thousands of s. We also tested the simulation of a quadrupole ion storage device with 100,000 ions; the 600 μs simulation with ~25,000 time steps was completed in only about 10 days, which may take 5 ~ 7 years if done with the explicit method. In this work, we have achieved the goal we set for the project and will continue to the next level by developing parallel codes for the system with multiple GPUs, and for the simulation of much more complicated device designs. Our algorithm can be effective in speeding up SIMION trajectory calculations for any generic device designs that involve large scale calculations of more than one million ions.
References
Saito, K., Reilly, P.T.A., Koizumi, E., Koizumi, H.: A hybrid approach to calculating Coulombic interactions: An effective and efficient method for optimization of simulations of many ions in quadrupole ion storage device with SIMION. Int. J. Mass Spectrom. 315(1), 74–80 (2012)
NVIDIA Corporation: NVIDIA CUDA Programming Guide (2006–2011).
Dahl, D.A., Delmore, J.E., Appelhans, A.D.: SIMION PC/Ps2 electrostatic lens design program. Rev. Sci. Instrum. 61, 607 (1990)
Dahl, D.A.: SIMION for the personal computer in reflection. Int. J. Mass Spectrom. 200, 3–25 (2000)
Manura, D., Dahl, D.: SIMION (R) 8.0 User Manual; Scientific Instrument Services Inc., (2008).
Ierusalimschy, R., de Figueiredo, L.H., Celes, W.: Lua 5.1 Reference Manual, ISBN 85-903798-3-3 (2006).
Douglas, D.J., Frank, A.J., Mao, D.: Linear ion trap in mass spectrometry. Mass Spectrom. Rev. 24, 1–29 (2005)
Parks, J.H., Szoke, A.: Simulation of collisional relaxation of trapped ion clouds in the presence of space charge fields. J. Chem. Phys. 103(4), 1422–1439 (1995)
Grinfeld, D.E., Kopaev, I.A., Makarov, A.A., Monastyrskiy, M.A.: Equilibrium ion distribution modeling in rf ion traps and guides with regard to Coulomb effects. Nucl. Instr. Methods Phys. Res. A 645, 141–145 (2011)
Hager, J.W.A.: New linear ion trap mass spectrometer. Rapid Commun. Mass Spectrom. 16, 512–526 (2002)
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Saito, K., Koizumi, E. & Koizumi, H. Application of Parallel Hybrid Algorithm in Massively Parallel GPGPU—The Improved Effective and Efficient Method for Calculating Coulombic Interactions in Simulations of Many Ions with SIMION. J. Am. Soc. Mass Spectrom. 23, 1609–1615 (2012). https://doi.org/10.1007/s13361-012-0435-6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13361-012-0435-6