
Abstract
It has previously been reported that disulfide and backbone bonds of native intact proteins can be concurrently cleaved using electrospray ionization (ESI) and collision-induced dissociation (CID) tandem mass spectrometry (MS/MS). However, the cleavages of disulfide bonds result in different cysteine modifications in product ions, making it difficult to identify the disulfide-bonded proteins via database search. To solve this identification problem, we have developed a pseudo MS3 approach by combining nozzle-skimmer dissociation (NSD) and CID on a quadrupole time-of-flight (Q-TOF) mass spectrometer using chicken lysozyme as a model. Although many of the product ions were similar to those typically seen in MS/MS spectra of enzymatically derived peptides, additional uncommon product ions were detected including ci-1 ions (the ith residue being aspartic acid, arginine, lysine and dehydroalanine) as well as those from a scrambled sequence. The formation of these uncommon types of product ions, likely caused by the lack of mobile protons, were proposed to involve bond rearrangements via a six-membered ring transition state and/or salt bridge(s). A search of 20 pseudo MS3 spectra against the Gallus gallus (chicken) database using Batch-Tag, a program originally designed for bottom up MS/MS analysis, identified chicken lysozyme as the only hit with the expectation values less than 0.02 for 12 of the spectra. The pseudo MS3 approach may help to identify disulfide-bonded proteins and determine the associated post-translational modifications (PTMs); the confidence in the identification may be improved by incorporating the fragmentation characteristics into currently available search programs.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
Proteins are typically identified and characterized via bottom-up approach, where they are reduced, digested, and analyzed by mass spectrometry (MS) and tandem mass spectrometry (MS/MS) analysis. Recently, the top-down approach [1–4] has started to gain popularity, where intact proteins are analyzed by MS and MS/MS. This top-down approach has the advantages of high throughput, high sequence coverage, and easier identification of protein variants and PTMs [5], however, with limited applications for proteins containing disulfide bonds. Concurrent cleavages of disulfide and protein backbone bonds were thought to be difficult via CID [6, 7]. Other dissociation methods including Electron capture dissociation (ECD) and Electron transfer dissociation (ETD) have been reported to be capable of the concurrent cleavages [7–9]. However, the fragmentation efficiency of ECD is low [10]; and to the best of our knowledge, no application to characterize disulfide-bonded proteins with ETD has been reported, despite its possessing a higher fragmentation efficiency than ECD [11].
Several years ago, a few studies reported on concurrent cleavages of disulfide and backbone bonds via CID in an ion-trap mass spectrometer [12–14]. No follow-up work has been performed, likely due to the small number of backbone cleavages as well as low resolution and low intensities of the product ions under the experimental condition at that time. Recently, using ESI and CID on an LTQ Orbitrap mass spectrometer, we reported that more product ions were yielded at higher resolution and higher intensity for a model protein that contains highly intra-linked disulfide bonds [15]. However, it is still not easy to identify unknown disulfide-bonded proteins with the top-down approach. The difficulty in the identification results from the cleavages of disulfide bonds producing multiple modifications of cysteine residues in product ions. Additionally, these modifications are different from the native oxidized form of cysteine residues, i.e., half-cystine, in precursor ions [15]. Since the same modifications are typically required for database searching with currently available programs, it is impractical to identify proteins from their product ions involving dissociation of disulfide bonds in intact proteins.
The difficulty of identifying disulfide-bonded proteins via database search due to inconsistent modifications could be circumvented via an approach based on MS3, or alternatively pseudo MS3 analysis [16, 17] that combines in-source dissociation such as NSD [18] (also termed as cone fragmentation) and regular CID MS/MS analysis. If the MS3 or pseudo MS3 analysis is performed on the first-generation product ions that do not undergo further dissociation of disulfide bonds, the modifications in the precursor and product ions will be the same. Although the application of MS3 (including pseudo MS3) analysis for identification of proteins has been reported previously [16, 19, 20], to the best of our knowledge, its application to the identification of proteins from product ions involving CID cleavages of disulfide bonds has never been reported.
An additional advantage of the pseudo MS3 approach is that it can identify the proteins of interest with higher confidence compared with typical top-down MS/MS analysis. This is due to the following: (1) the precursor ions, being small fragments of protein ions instead of protein ions themselves, are less affected by unknown modifications [19], which could lead to false identification; (2) monoisotopic peaks of small fragments are much easier to be accurately measured; (3) many more precursors are available (typically only one precursor ion is fragmented in MS/MS top-down analysis). Identification of protein variants and characterization of PTMs may be significantly improved by combining the higher confidence identification via MS3 approach and the overview picture of all fragments from precursor protein ions via top-down MS/MS approach.
To cleave disulfide bonds in proteins, typically the charge state of protein ions is required to be less than the number of Arg residues [15]. As a result, all of the protons are sequestered on the proteins due to the high basicity of arginine side-chain. The well-known mobile proton theory [21], which suggests a proton initially localizes at a basic site of a peptide ion and then mobilizes to a cleavage site (usually amide bond ) inducing a charge-directed dissociation, is thus not applicable. For peptide or protein ions lacking mobile protons, the dissociation mechanism often involves an aspartic acid residue with its side chain carboxyl group attacking its C-terminal amide group [22]. The dissociation of peptides lacking mobile protons can also be induced through proton-driven amide bond-cleavage pathways via salt-bridge [23–26], anhydride [23], or imine/enol intermediates [23], where the former two pathways involve deprotonation of the C-terminal carboxyl group, while the last pathway involves a proton transfer from an enol group (isomerized from an amide group) to a nearby amide nitrogen. All the mechanisms mentioned above are actually charge-directed, including that involving the side chain of aspartic acid where protonation and/or salt bridge play a role [22] even though this type of dissociation is often referred to as “charge-remote.” There are yet other dissociation mechanisms with no charge involved and where the dissociation resulted from bond rearrangements via a six-membered ring transition state. Examples include the formation of dn and cn-1 ions from dissociation of b ions with Asp at the C-terminus [27] and formation of c and z – S ions from dissociation of N–Cα bond on the N-terminal of a dehydroalanine residue (resulted from cleavage of a disulfide bond) [12, 15]. This set of mechanisms has seldom been discussed in the literature.
We report here pseudo MS3 analysis of intact native chicken lysozyme using a Q-TOF mass spectrometer with a maximum resolution around 10,000. The pseudo MS3 analysis was achieved by combining NSD at high cone voltage and CID MS/MS. The fragmentation patterns of the pseudo MS3 spectra for precursor ions lacking mobile protons will be discussed and mechanisms for the formation of uncommon product ions are proposed. We also demonstrate chicken lysozyme can be identified as the only hit by searching the pseudo MS3 spectra against the Gallus gallus (chicken) database using the program Batch-Tag [28] of ProteinProspector [29].
2 Materials and Methods
Chicken lysozyme was purchased from Sigma (St. Louis, MO, USA). HPLC-grade water, acetonitrile and acetic acid were purchased from ThermoFisher Scientific (Waltham, MA, USA). Chicken lysozyme was dissolved in water containing 1%–2% acetic acid at a concentration of 10–20 μM. The sample was analyzed on an ESI-Q-TOF mass spectrometer (Waters Q-TOF II; Milford, MA, USA) via direct infusion at the flow rate of 5–20 μL/min. MS, NSD, CID MS/MS, and pseudo MS3 analyses were performed.
For MS analysis, the typical instrument parameters were set as follows: capillary voltage at 3.2 kV, source temperature at 100 °C, desolvation temperature at 150 °C, cone voltage at 50 V, collision energy at 5 eV (to maximize ion transmission, as recommended by the manufacturer’s manual), and the “high res” and “low res” parameters at 15. The same parameters, except a few changes, were applied to the other analyses. For NSD, the cone voltage was 100–120 V. For CID MS/MS analysis, (M + 9H)9+ ion was isolated and the collision energy was 40 eV. For pseudo MS3 analysis, the cone voltage was 100 or 120 V; 20 peaks of relatively high intensities with a maximum of one disulfide bond cleavage (which was determined from the pattern of an ion cluster [15]) were isolated; and the collision energy was 30–50 eV. The precursor ions for pseudo MS3 analysis were either the same as typical peptide ions (for y precursor ions), or the same as typical peptide ions with C-terminal dehydration (for b and yb internal ions as the precursor ions) or amidation (for c and yc internal ions as the precursor ions). The same scan range m/z 100–2000 was set for all analyses. The acquisition time for each spectrum was typically 4–10 min. Unit isolation resolution of the precursor ions was achieved for CID MS/MS and pseudo MS3 analyses.
3 Data Analysis
3.1 Peak Assignments
The peaks of the CID or NSD tandem mass spectra were assigned with the help of the data analysis program ICR2Ls for main sequence ions as described earlier [15] and MS-Product of ProteinProspector [29] for internal fragments. Unless indicated otherwise, the peaks in pseudo MS3 spectra were assigned from a database search against chicken lysozyme (accession number P00698) using Batch-Tag [29] (with the same parameters as for searches against “Gallus gallus” when the digest was “no enzyme,” see Section Database Search for details), followed by manual validation. For convenience, the nomenclature describing the second-generation product ions in pseudo MS3 spectra discussed in this manuscript follows that of MS/MS product ions from typical peptides. The classification of pseudo MS3 product ions is based only on the cleavage sites regardless of different modifications on the cysteine residue(s) and/or different types of C-termini (resulting from MS/MS product ions, e.g., y, b, or c ions, which become the precursor ions for pseudo MS3 analysis).
3.2 Database Search
The precursor ions subjected to pseudo MS3 analysis were either singly or doubly charged. Converting all peaks of precursor and product ions to single charges was found to significantly increase the confidence in identifying proteins via the database search by decreasing random matches. About 100 most abundant peaks were picked for each spectrum and all peaks of double charges were converted to single charges. For the spectra of singly charged precursor ions, the peak lists were directly generated from smoothed and centered spectra. For doubly charged precursor ions, the spectra were processed with background subtracting, followed by MaxEnt 3 function of MassLynx (Waters, Milford, MA, USA) to convert the product ions to single charges; the m/z values corresponding to singly charged precursor ions were manually calculated. A total of 20 pseudo MS3 spectra were included in an MGF file for the database search. The MGF file was searched against SwissProt2010.03.30 database using Batch-Tag [29].
To test if chicken lysozyme could be identified when it was assumed to be an unknown protein, three types of searches were performed. These types include searching with “no enzyme” against database of “Gallus gallus” and “all species,” and searching with “Full Protein” and “Nonspecific” at “N-term” against database of “Gallus gallus.” The common parameters for all database searches were set as follows: “1+” for precursor charge; “monoisotopic” masses and 100 ppm mass tolerance for both parent and fragment ions. ESI-Q-TOF instrument specific default ions for the search were applied, which included a, b, y, internal, immonium, b + H2O, a loss, b loss, c loss, and internal loss ions, where “loss” means a single loss of NH3 or H2O. The variable modifications were set depending on the type of digest, i.e., “no enzyme” or “Full protein.”
When the digest was “no enzyme”, the variable modifications included “Dehydrated (C-term),” “Amidated (C-term),” “Cys → Dha (C),” “Didehydro (C),” and “Sulfide (C).” The modifications “Dehydrated (C-term)” and “Amidated (C-term)” correspond to precursors being b (including internal yb) type and c (including internal yc) type ions, respectively; while the absence of any one of the two modifications corresponds to precursor being y type ions. The modifications “Cys → Dha (C),” “Didehydro (C),” and “Sulfide (C)” correspond to the changes (− SH, – H, and + SH, respectively) of cysteine residues from the oxidized form upon disulfide bond dissociation; the absence of any one of the three modifications corresponds to the reduced form of cysteine residues (i.e., oxidized form + H) [15]. A maximum of “2” of the five variable modifications were applied.
When the digest was based on “Full Protein”, a maximum of 1 of the three variable modifications for cysteine residue were applied; neither “Amidated (C-term)” nor “Dehydrated (C-term)” was applied as a variable modification. This type of digest is similar to “no enzyme,” but only considers the fragments containing the C-terminus of the protein, which means it is only applicable when precursor ions (the first-generation product ions) are y ions. Since the protein sequences in the database often contain the signal peptides at the N-termini [30], a similar approach is generally not applicable to b precursor ions.
To report only the matches of high confidence, the search results were filtered with appropriate settings for several parameters. These parameters were set as follows: (1) “Min Best Discriminant Score” was set to 0; (2) both “Min Protein Score” and “Min Peptide Score” were set to 8; (3) both expectation values, i.e., “Max E Value Protein” and “Max E Value Peptide,” were set to 10, higher than 0.05—the commonly used one for various search engines [31]. The setting of higher expectation values is because Batch-Tag is not specifically designed for MS3 analysis and the set of many modifications may also break down the independent trials for the statistical analysis.
4 Results and Discussion
4.1 NSD and CID Analysis of Chicken Lysozyme Ions
At the cone voltage of 50 V, when no organic solvent was added to the solution, the chicken lysozyme protein peaks in the MS spectrum were predominantly of the charge states from 8+ to 11+ (Figure S-1a). Since there are altogether 11 Arg residues in chicken lysozyme, all the charges were essentially sequestered by these Arg residues, meeting a condition for the charge-remote cleavage of disulfide bond [15]. When cone voltage was increased to 100 V, fragment ions including those resulted from disulfide bond cleavages were observed (Figure S-1b). An increase of cone voltage to 120 V dissociated precursor ions further (Figure S-1c). The higher cone voltage at 120 V did not increase the signal-to-noise ratios for most of the fragment peaks; however, it did generate some fragments with higher signal-to-noise ratio including those of m/z 605.39 and 828.48. These two fragments were identified as c5 and y10c126 ions, and both involve the cleavage of N–Cα bond, suggesting the dissociation of this type of bond might require a higher energy. Interestingly, c ions have also been reported in in-source decay of peptides and proteins with matrix-assisted laser desorption (MALDI) [32–34]. However, in contrast to what was observed in this current work, the generation of series of c-ions in MALDI often stopped before reaching a disulfide bond [32–34], suggesting different dissociation mechanisms might have been involved during the two ionization processes.
For the dissociation of chicken lysozyme on the Q-TOF mass spectrometer, comparing to CID MS/MS of the protein ions in only one charge state—9+ (Figure 1a, Table S-1), the NSD of the protein ions in different charge states—essentially 8+ to 11+ (Figure 1b) displayed a similar fragmentation pattern. In both cases, many product ions are similar to those acquired on an LTQ Orbitrap mass spectrometer via CID [15]. The peaks corresponding to internal fragments including (y87b48)+, (y88b48)+, and (y89b48)+ (only seen in Figure 1b) are likely originated from the protein ions with 11+ charges, as reported previously [15]. Compared with the LTQ Orbitrap mass spectrometer, the dissociation efficiency (based on the relative intensity of the precursor ions to that of the product ions) in the Q-TOF mass spectrometer appeared lower.

Comparison of (a) collision induced dissociation of (M + 9H)9+ chicken lysozyme precursor ion at the collision energy of 40 eV and (b) nozzle-skimmer dissociation of all charge states (dominantly 8+ to 11+) of chicken lysozyme precursor ions at the cone voltage of 100 V
There are three possibilities for the lower dissociation efficiency of chicken lysozyme in the Q-TOF mass spectrometer compared with the LTQ Orbitrap mass spectrometer [15]. First, with the Q-TOF mass spectrometer, higher collision energy could cause further fragmentation due to continuous collisions and thus decreased signal-to-noise ratio; in contrast, for the LTQ Orbitrap mass spectrometer, since the excitation frequencies for product ions are different from those for precursor ions, the applied higher collision energy theoretically would yield high intensity product ions without causing further dissociation. Second, the rate for the concurrent cleavages of disulfide and backbone bonds could be at the edge of the range sampled by the Q-TOF mass spectrometer (a beam-type mass spectrometer), while within the range sampled by an ion-trap instrument. The dissociation rates sampled by beam-type and ion-trapping type mass spectrometers have been estimated to be greater than 104 and 1–100 s–1, respectively [35]. Third, the nascent protein ions have a high tendency to refold due to much stronger electrostatic intramolecular interactions resulted from solvent removal [6], which could be more serious in the case of this study where there was less Coulombic repulsion with fewer charges carried by the protein ions; the refolding could be prevented or minimized by high temperature of the ion transfer capillary that situates before the skimmer in the LTQ Orbitrap mass spectrometer, which is absent in the Q-TOF instrument setup.
Despite the lower dissociation efficiency in the Q-TOF mass spectrometer, due to the lack of low mass cutoff restriction, more ions including those of low m/z values were detected. This makes it easier to study the fragmentation patterns and obtain higher confidence database search results.
4.2 Uncommon Product Ions in Pseudo MS3 Spectra of Chicken Lysozyme Ions
Twenty of the first-generation product ions from NSD were selected as the precursor ions for CID MS/MS to acquire pseudo MS3 spectra. The spectra were searched against chicken lysozyme using Batch-Tag to determine the precursor ions and assign the pseudo MS3 product ions. Of the 20 pseudo MS3 spectra, 18 matched those of chicken lysozyme with expectation values <0.0032 (data not shown). These two unmatched spectra were manually analyzed and the corresponding precursor ions were determined to be b +5 and sodiated c +5 . The spectrum of b +5 does not match that of chicken lysozyme because the program does not take into consideration precursor ions with short sequence (<m/z 600), while the spectrum of c +5 does not match because the chosen default parameters are only for proton adducts.
The pseudo MS3 spectra of the 20 precursor ions were manually analyzed, with the focus on the unassigned peaks. Many product ions, including a series of b, y, and internal fragment ions, were generated (see following discussions for details). When a pseudo MS3 product ion contained a cysteine residue, the modification of the cysteine residue (− SH, – H, + H, and + SH) was consistent with that in the precursor ion (see Figures S-2 and 3 for examples).
In the course of the pseudo MS3 analysis, despite many product ions being similar to those observed in MS/MS spectra of peptide ions, various types of uncommon product ions were also detected (Table 1). These characteristic pseudo MS3 product ions are most likely related to the lack of mobile protons in the precursor ions as well as the high energy applied during the analysis. Some of the uncommon product ions, e.g., c +7 , b +9 , and (P – HN=C=NH)+ detected in pseudo MS3 spectrum of (y10 – SH)+, were also observed in the CID-MS/MS spectrum of (M + 9H)9+ (corresponding to (y10c126)+, (y10b128 – SH)+, and (y10 – HN=C=NH – SH)+ ions, respectively) on an ion-trap Orbitrap mass spectrometer [15] within 5 ppm of theoretical m/z values. It is most likely that similar spectra would be obtained in an ion-trap mass spectrometer with true MS3 analysis. If isolation efficiency is satisfactory, true MS3 analysis should provide “cleaner” spectra for database searching, especially for complex samples. The higher dissociation efficiency in LTQ-Orbitrap instrument could also significantly decrease the acquisition time needed. Further studies are to be conducted to compare the performance on different types of mass spectrometers. The results and discussion of these uncommon ions are as follows.
-
1)
ci-1, di, and bi – CO2 product ions from precursor ions with Asp as the ith residue
Product ci-1, di and bi – CO2 ions were detected from the dissociation of almost all b and yb type precursor ions with Asp as the ith residue in this study (Table 1, Figures 2, 3, and S-4). These types of product ions have previously been reported by Wysocki and coworkers to be generated from MS/MS analysis of peptide ions as well as MS3 analysis of b ions with Asp at the C-terminus [27]. The authors termed these ions as cn-1, dn ,and bn – CO2, where “n” represents the total number of residues in the precursor ions. For the generation of these uncommon product ions, the Asp residue does not have to be at the C-terminus of the precursor ions (see Figure 3b for an example). Therefore, we use the subscript “i” instead of “n” to indicate it is applicable to general cases. However, the initial formation of b ions with Asp as the C-terminus residue does appear to be the first step in the case that Asp is not the C-terminal residue.
Figure 2 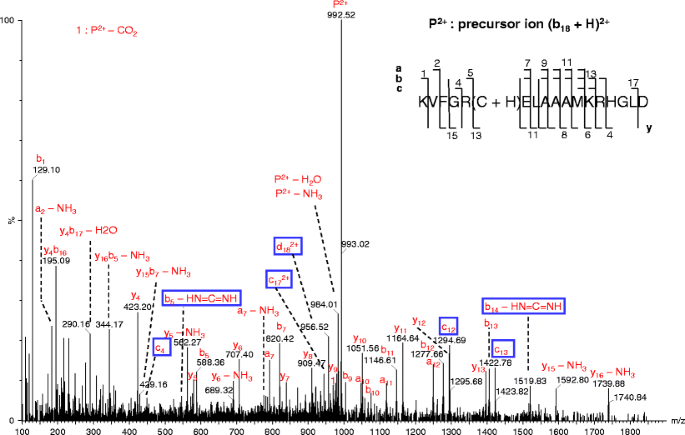
Pseudo MS3 spectra of (b18 + H)2+ ion at the cone voltage of 100 V and collision energy of 50 eV. The uncommon product ions in this and following figures are highlighted by enclosing in open boxes. In this and following figures, the precursor ions are labeled as “P” with the corresponding charges; for clarity, the charge states of product ions are not labeled when they are 1+
Figure 3 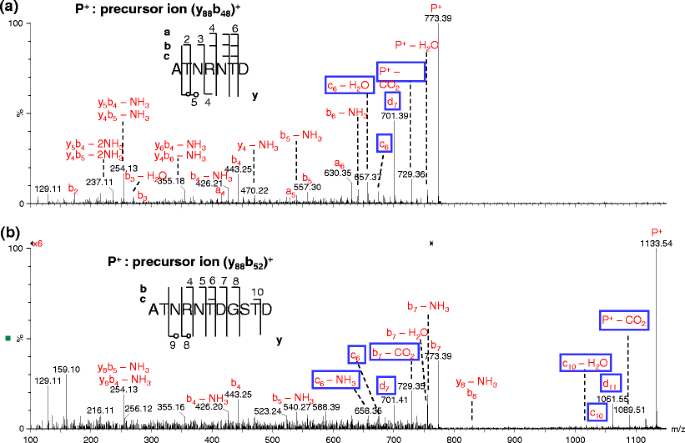
Comparison of pseudo MS3 spectra of (a) (y88b48)+ and (b) (y88b52)+ ions at the cone voltage of 100 V and collision energy of 40 (a) and 50 eV (b), respectively. The labels with empty circles in the sequence map in this and following figures indicate the cleavages only seen in internal fragments
The formation of these ci-1 ions was proposed to involve bond rearrangement [27], while the formation of di ions was proposed to involve concerted dissociation of several bonds [27]. No mechanism was suggested for the formation of bi – CO2 ion. In this work, we propose a different mechanism for the formation of the di ions as well as a novel mechanism for the formation of ions with neutral loss of CO2 (Scheme 1). Both proposed mechanisms involve bond rearrangement of the precursor b ion in a configuration as shown in Scheme 1 [36], which is different from the configuration containing a cyclic anhydride structure [37] that was proposed to result in the formation of di ions [27].
Scheme 1 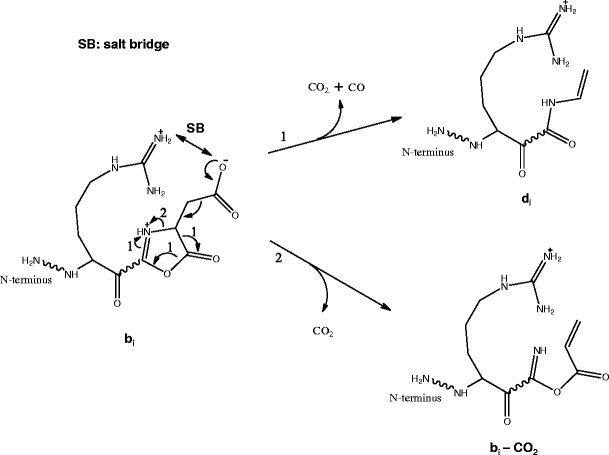
Formation of di and bi – CO2 ions from further dissociation (pseudo MS3) of a bi ion with Asp at the C-terminus
-
2)
ci-1 product ions from precursor ions with Arg or lysine (Lys) as the ith residue
Some ci-1 ions were observed from CID of first-generation product ions when Arg or Lys is the ith residue (Table 1, Figures 2, 4, S-2, S-3, S-5, and S-6). The same type of c ions has previously been reported by Fu et al. [38] (these ions were termed by the authors as cx ions with Arg or Lys as the (x+1)th residue). Fu et al. [38] proposed a mechanism for the formation of this type of c ions directly from the precursor ion of a particular sequence AGHKLL, where nucleophilic attack of Cα of Lys by its side chain amine group was involved.
Figure 4 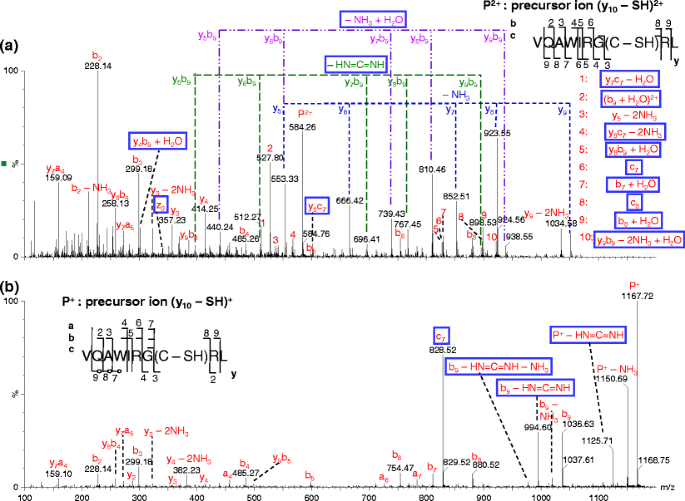
Comparison of pseudo MS3 spectra of (a) (y10 – SH)2+ and (b) (y10 – SH)+. The analysis was performed at the cone voltage of 100 V and collision energy of 30 eV (a) or 50 eV (b)
Alternative mechanisms are proposed here for the formation of such ci-1 ions with the formation of bi ions as the first step (Scheme 2). These mechanisms are based on the lack of mobile protons, the types of corresponding precursors ions and the structures of bi ions with Lys or Arg at the C-terminus [22]. The ci-1 ions are proposed to be formed from bond rearrangement via a six-membered ring transition state resulting from the ring structures of these bi ions.
Scheme 2 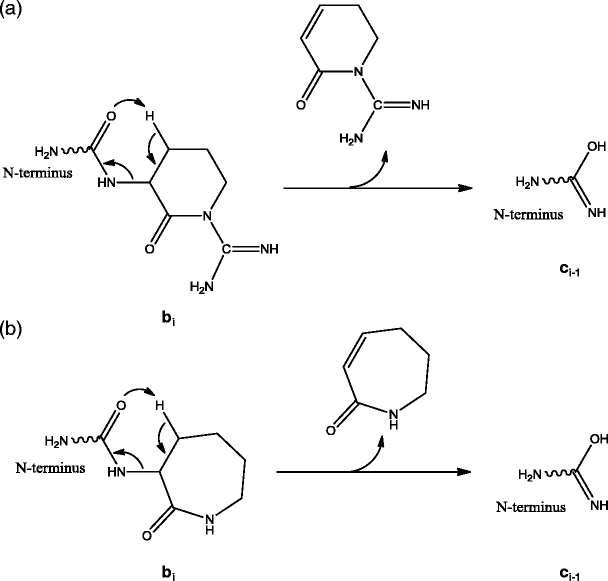
Formation of ci-1 or bi – HN=C=NH ion from bi ions with (a) arginine or (b) lysine as the ith amino acid residue
The ci-1 ion can be detected when there is a charge retained on the N-terminal fragment of the precursor ion. These mechanisms can directly explain the formation of ci-1 ion from b precursor ions with Arg or Lys residue at the C-terminus, e.g., c +4 (Figure S-6a) and c +13 (Figure S-5) product ions from dissociation of b +5 and (b14 + H)2+ ions, respectively. On the other hand, the formation of ci-1 ion from precursor ions of b 2+18 cluster (Figures 2 and S-2), c +5 (Figure S-6b), and y 2+10 cluster (Figures 4a and S-3) likely involves one additional prior step to form the corresponding b ions with Arg or Lys residue at the C-terminus [22] assuming the structure as shown in Scheme 2.
-
3)
bi – HN=C=NH product ions from precursor ions with Arg as the ith residue
Neutral losses corresponding to HN=C=NH (42.02 Da) were observed for product ions in pseudo MS3 spectra of different types of precursor ions (Table 1, Figures 1, 2, 4, 5, S-2, S-3, S-6, and S-7). These product ions with neutral loss of HN=C=NH are b, yb, or equivalent product ions (e.g., the pseudo MS3 product y1-3 ions dissociated from b5 ion have the same structure as b ions) with Arg as the C-terminal residue (Scheme 2a). The bi – HN=C=NH ions probably result from bond rearrangement of the bi ions via another set of six-membered ring transition state that contains a part of the guanidine group and its neighboring carbonyl group, where the C-terminal structure of the bi ions is the same as shown in Scheme 2a. A similar mechanism has been proposed by O’Hair group for loss of HN=C=NH from a b2 ion structure with C-terminal Arg residue (after loss of CH3OH from the C-terminus of N-acyl arginine methyl ester) [39].
Figure 5 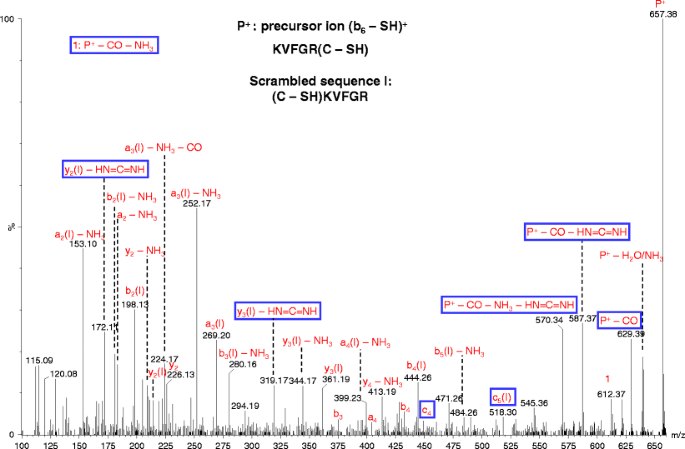
Pseudo MS3 spectrum of (b6 – SH)+. The cone voltage was 100 V and the collision energy was 35 eV. The non-direct sequence ions due to sequence scrambling are indicated by “(I)” following the label for the sequence ions along with the neutral losses (if present)
-
4)
P – HN=C=NH product ions from Arg-containing precursor ions
A peak corresponding to neutral losses of HN=C=NH (42.02 Da) from precursor ion (y10 – SH)+ was also observed (Figure 4b). This precursor ion contains two Arg residues, one protonated and the other non-protonated. Since there is no involvement of the formation of b ion that contains Arg residue at its C-terminus with the structure as shown in Scheme 2a, a different mechanism is proposed for the neutral loss of HN=C=NH (Scheme 3). The mechanism involves transfer of a proton from the protonated Arg residue to a non-protonated amine group of the side chain of another Arg residue. The neutral loss of HN=C=NH from precursor ions have also been reported in MS/MS analysis of singly charged bradykinin and its derivative [40], and MS/MS analysis of peptides Arg-Gly and Gly-Arg [25]. In the case of pseudo MS3 spectrum of c5 ion, the neutral loss seems to be more likely via the mechanism shown in Scheme 2a with ammonia loss and the formation of bi structure as the prior step.
Scheme 3 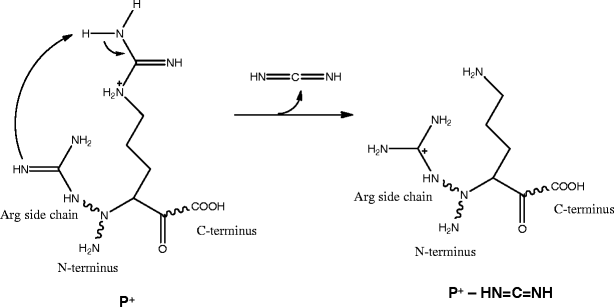
Neutral loss of HN=C=NH from precursor ion (y10 – SH)+
-
5)
ci-1 product ions from precursor ions with dehydroalanine (i.e., Cys – SH ) as the ith residue
Many c ions were previously observed from MS/MS of disulfide-bonded polypeptides by McLuckey and coworkers [12, 14] as well as by our group [15]. Peaks corresponding to the same type of c ions were also observed in pseudo MS3 spectra of several precursor ions in this work (Table 1, Figure 4 and Figure S-2d). Similar to that reported by McLuckey group, the loss of SH from cysteine residue is also found to be required for the formation of these c ion in this study, e.g., c5 ion is only seen in pseudo MS3 spectrum of (b18 – SH)2+ ion but not in the other spectra of the related b18 ions (Figure S-2). Since the corresponding z ions have the same m/z values as y – NH3 (note that the m/z values for these even electron z ions are different from those free radical z ions observed in ECD and ETD analysis [41]), it is often difficult to unambiguously distinguish the two types of ions based on m/z values of peaks (e.g., z +3 ion in Figure 4a); however, the existence of z +13 ion in Figure S-2d is obvious when comparing with Figures S-2a-c. Based on the fact that loss of SH from cysteine residue is the first step for the formation of c ions, the McLuckey group proposed a mechanism that involves bond rearrangement via a six-membered ring for the generation of such c ions [12]. The C=S double bond formation upon disulfide dissociation appears to bring the hydrogen to the appropriate position to form a relatively stable six-membered ring transition state.
-
6)
bn-1 + H2O product ions from precursor y ions containing Arg residues
A peak (b9 + H2O)2+ (a bn-1 + H2O ion) and a series of yb type internal fragment peaks, e.g., (y8b9 – NH3 + H2O)+ (a ymbn-1 – NH3 + H2O ion) from dissociation of (y10 – SH)2+, were observed in pseudo MS3 spectrum of each of the y 2+10 cluster ions (Table 1, Figure 4a and Figure S-3). In addition, a (b8 + H2O)+ peak (a bn-2 + H2O ion) and a (b7 + H2O)+ (a bn-3 + H2O ion) were also observed (Figures 4a and S-3). Interestingly, no such product ions were observed in the pseudo MS3 spectrum of the ion of the same peptide but with a different charge state—(y10 – SH)+ (Figure 4b).
Different mechanisms [42–46] have been proposed for the formation of bn-1 + H2O ions from dissociation of peptide ions; however, none of these mechanisms can explain why the bn-1 + H2O (i.e., (b9 + H2O)2+ ion) and related ions were present in the pseudo MS3 spectrum of (y10 – SH)2+ ion but absent in that of (y10 – SH)+ (Figure 4). A mechanism instead is proposed here for the formation of these ions (Scheme 4), where a mobile proton present in (y10 – SH)2+ along with salt bridge formation is involved. The different fragmentation characteristics between pseudo MS3 spectra of (y10 – SH)2+ and (y10 – SH)+, i.e., many more product ions generated and much lower collision energy required (30 eV versus 50 eV) for the former than for the latter, suggest the possibility of a mobile proton present in the (y10 – SH)2+ ion but absent in the (y10 – SH)+ ion. The presence of a mobile proton could result from the formation of a salt-bridge between the guanidine group at the ninth residue (Arg) and the C-terminal carboxyl group via proton transfer, similar to the case for peptides with C-terminus Arg residue interacting with cysteic acids [47]. Thus for (y10 – SH)2+, only one proton is sequestered by Arg at the sixth residue, while the other proton is mobile. The mobile proton is proposed to initially localize at the N-terminus amine group; the protonated N-terminus amine group then forms a hydrogen bond with the (n – 1)th amide oxygen and induces the nucleophilic attack of the amide carbon center by the negatively charged oxygen of the C-terminal carboxyl group; bond rearrangement follows and leads to the formation of the (b9 + H2O)2+ product ion, which is stabilized by a salt-bridge. Similar reactions account for the formation of the (b8 + H2O)+ and (b7 + H2O)+ product ions. When the formation of (b9 + H2O)2+ product ion is followed (or preceded) by backbone bond cleavages and ammonia loss, (ymb9 + H2O – NH3)+ ions—a series of internal fragments with water adduct and ammonia loss, are formed.
Scheme 4 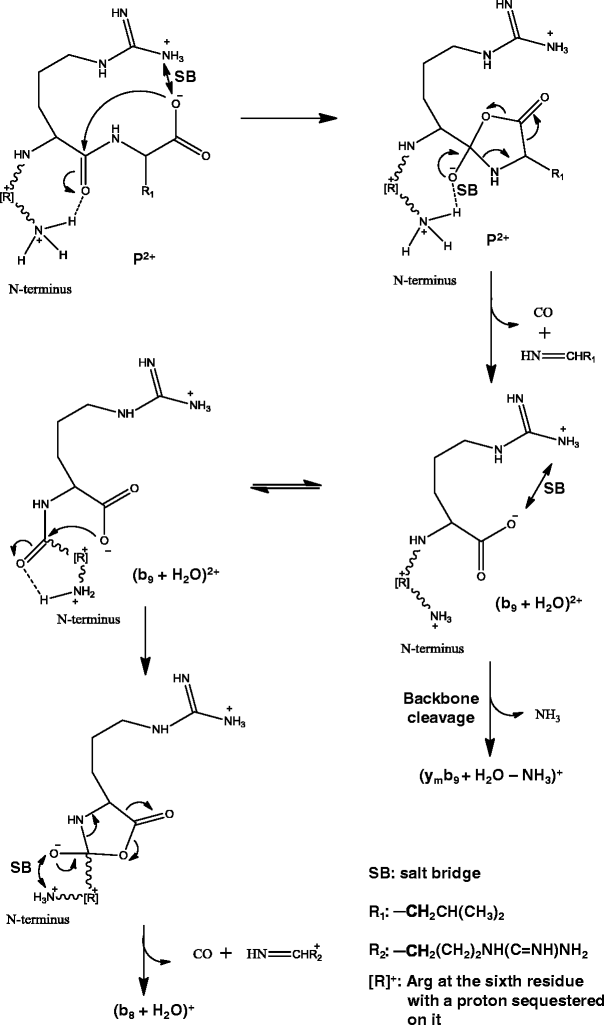
Formation of bn-1 + H2O, bn-2 + H2O and related ions from dissociation of y 2+10 precursor cluster ions with arginine as the 9th amino acid residue
The mechanism proposed here combines features of those previously proposed mechanisms. Such features include the stabilization of deprotonated C-terminal carboxyl group by a salt-bridge [42], the weakening of the (n – 1)th amide bond by a hydrogen bond between protonated Arg and the amide oxygen [43, 45], the nucleophilic attacking of the amide carbon center by the negatively charged oxygen of the C-terminal carboxyl group [25, 42, 46] (which is supported by the disappearance of the bn-1 + H2O ion from dissociation of precursor ion of peptide RYGGFL upon C-terminal methylation [26]), and the stabilization of the transition ion and product ion by a salt-bridge [42]. Our proposed mechanism is very similar to the one proposed by Gonzalez et al. [42], however, with one important difference, i.e., the weakening of the amide bond. Gonzalez et al. [42] proposed “the cationized guanidine group involved in a salt bridge could interact more efficiently with its own carbonyl group via the compact ring formation” for the enhancement of the effect by Arg at n – 1 position; Vachet et al. [45] and She et al. [43] also proposed that the (n – 1)th amide bond was weakened by a hydrogen bond between protonated Arg and the amide oxygen. In contrast, based on the absence of bn-1 + H2O ion in the pseudo MS3 spectrum of (y10 – SH)+, we propose it is the hydrogen bond formed between the protonated N-terminus amine group and the (n – 1)th amide oxygen that weakens the amide bond; the reason is likely that the protonated N-terminus amine group is more flexible and probably undergoes less energy strain and steric hindrance for the hydrogen bond formation compared with the protonated Arg residue.
Our proposed mechanism can also explain the previous different and sometimes even contradictory reports on positions of Arg residue that enhance the formation of b + H2O ions [40, 42–45]. These positions include the (n – 1) position [42], at or near C- [44, 45], or N-terminus [44], or alternatively, a combination of one at the C-terminus and the other somewhere in the peptide backbone [40]. When one Arg residue was at the C-terminus, two of the reports on bn-1 + H2O ions are contradictory: absent in one work [42], while significantly present in another [44], with the former report partially supported by a systematic study on the formation of bn-1 + H2O ions, where a non-C-terminal basic residue was a proposed requirement [43]. These different observations can be rationalized based on Scheme 4. An Arg residue at or close to the C-terminus [42, 44, 45] can more easily abstract a proton from the C-terminal carboxyl group to result in nucleophilic negatively-charged-oxygen, while an Arg residue on N-terminus[44] or non-C-terminus[43] or somewhere in the middle of the sequence [40], which will be on the N-terminal fragment of the precursor ion, can retain a charge and thus makes the bn-1 + H2O ion observable. An Arg residue on N-terminus [44] may also be more flexible than that in the middle of the sequence and thus more readily form a salt-bridge with the C-terminal carboxyl group. When an Arg residue is at the C-terminus [42–44], the loss of an Arg residue will carry away a charge due to its high basicity (Scheme 4), thus the bn-1 + H2O ion will not be observed if the precursor ion contains only one charge [42] unless there is another Arg residue [43, 44] or other basic residues [43] in the sequence to compete for a proton.
-
7)
Non-direct sequence product ions from precursor b ion with protonated oxazolone structure
In the pseudo MS3 spectrum of (b6 – SH)+, other than several peaks identified as the sequence ions (including sequence ions with neutral losses), most of peaks correspond to non-direct sequence ions of a scrambled sequence or the precursor ion with neutral losses (Figure 5). Sequence scrambling has previously been reported for MS3 analysis of b ions and the corresponding mechanism has been proposed with the involvement of multiple sequences generated from ring opening of macrocyclic b isomers [48–50]. In contrast, only one scrambled sequence appears to be dominant in this study. It has also been reported by Stipdonk and coworkers that an Arg residue apparently inhibits the sequence scrambling [51]; however, in this study, the Arg residue does not appear to have such an effect
A mechanism is proposed for the formation of this scrambled sequence (Scheme 5). The precursor ion (b6 – SH)+ is proposed to contain an oxazolone structure formed through a pathway involving the protonation of the N-terminal amide nitrogen of the glutamic acid residue by its side chain carboxyl group (Scheme 5). This step is similar to the dissociation mechanism proposed by Paizs and coworkers with the mobilization of a proton from the C-terminal carboxyl group [23], The next step is the formation of a head-to-tail macrocyclic b isomer. The dominant scrambled sequence (C – SH)KVFGR is then formed due to the preferential nucleophilic attack by the guanidine group of the Arg residue to its C-terminal amide carbon atom. The likelihood of this scrambled sequence was supported by the peaks corresponding to (y2 – HN=C=NH)+ and (y3 – HN=C=NH)+ for the scrambled sequence, which could have been generated from a precursor b ion with Arg as the C-terminus residue (see above discussion).
Scheme 5 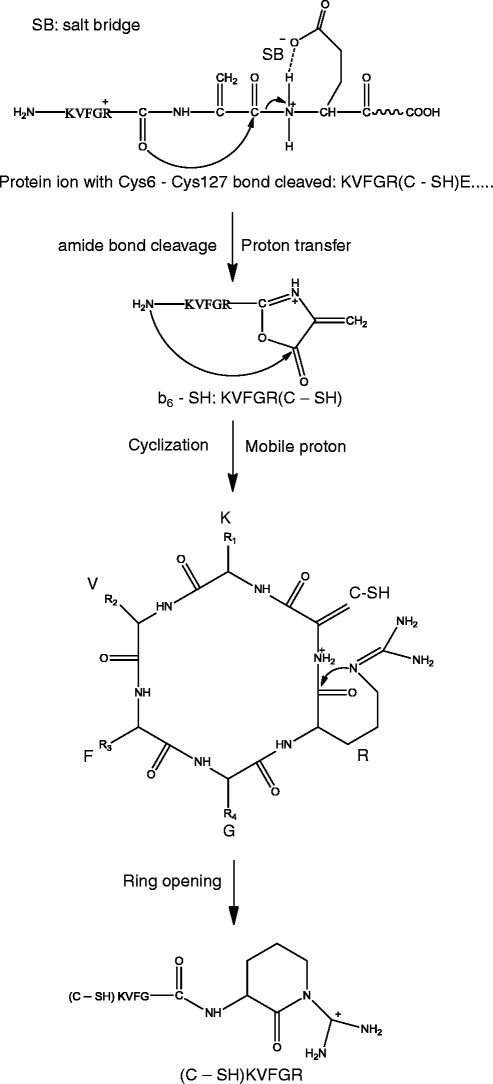
Formation of a scrambled sequence of b6 – SH ion
The proposed mechanism may also explain the reason for the difference between our observation and that by Stipdonk and coworkers [51]. In Scheme 5, the oxazolone structure retains a proton that can induce the macrocyclic ring structure formation; while in the scheme proposed by Stipdonk et al. [51], with the dissociation via the imine-enol pathway [23], no proton is retained on the oxazolone structure. Sequence scrambling was not pronounced for pseudo MS3 analysis of all other b and yb precursor ions discussed in this manuscript, probably because no significant amount of protonated oxazolone structure is formed for these other ions.
For the peaks corresponding to neutral losses from the precursor ion, including (P – CO)+, (P – CO – HN=C=NH)+ and (P – CO – NH3 – HN=C=NH)+, their formation could involve another pathway of macrocyclization, where the head-to-tail macrocyclic b isomer was formed by nucleophilic attack of the N-terminal amine to the Cα of the C – SH residue (Scheme S-1) instead of the carbonyl C of the oxazolone ring (Scheme 5). The loss of HN=C=NH is likely caused by a proton transfer from Arg residue to the side chain amine group of Lys residue, similar as illustrated in Scheme 3.
-
8)
cn-1* (sodiated cn-1) and neutral loss of HN=C(NH2)2 product ions from cn* (sodiated cn) ion
All the precursor ions for the pseudo MS3 analysis in this study were protonated except for one case—sodiated c +5 (c5*+, where “*” indicates the ion is sodiated; for convenience, the same label will apply to other sodiated ions). The sodium ions could originate either from solvents or from glass [52]. Pseudo MS3 analysis of c *+5 ion generated fragments corresponding to the precursor ions with neutral loss of NH3, NH=C=NH or HN=C(NH2)2 ions as well as a series of y*+, a*+, b*+, and c*+ ions (Figure 6). Highly abundant y* and a* ions derived from sodiated peptides have also been reported previously [53, 54]. The frequently reported [b + OH + Na]+ ions [53, 55–57] from dissociation of sodiated peptide ions were not observed in this study, probably because the C-terminus of c *+5 is an amide group instead of a carboxyl group. The mechanism for the neutral loss of HN=C=NH is likely similar to that proposed in Scheme 3. The mechanisms for the formation of the other major ions are discussed as follows.
Figure 6 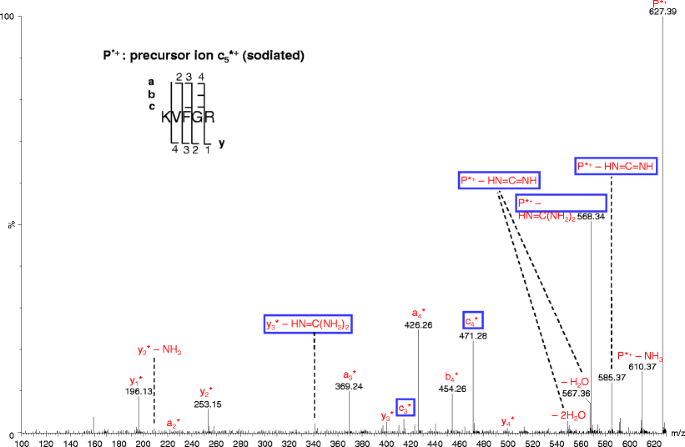
Pseudo MS3 spectrum of sodiated c +5 ions. The cone voltage was 100 V and the collision energy was 35 eV. All the fragments are sodiated ions and indicated with the asterisks (*)
The mechanisms for generation of cn-1*+ and (P* – HN=C(NH2)2)+ ions from dissociation of c5*+ are proposed in Scheme 6, where a salt bridge between protonated Arg and deprotonated imidic acid is involved. Deprotonation of amides has been reported previously [58]; the imidic acid structure of the corresponding c ion [12] could have made deprotonation even easier. The neutral loss of HN=C(NH2)2 is proposed to involve the nucleophilic attack of Cδ atom by the negatively charged oxygen of the C-terminal carboxyl group. The neutral loss of HN=C(NH2)2 has also been reported in MS/MS analysis of Arg derivatives [59], and MS/MS analysis of peptides Arg–Gly and Gly–Arg [25]. The HN=C(NH2)2 loss did not show up for y10 cluster ions, which could be due to higher energy required for the formation of a nine-membered ring transition state compared with a six-membered ring. On the other hand, in the pseudo MS3 spectrum of c +5 ion (Figure S-6b), there is a peak corresponding to neutral loss of HN=C(NH2)2 from the precursor ion; however, this peak seems to be more likely caused by a loss of NH3 followed by a loss of HN=C=NH via the mechanism discussed above for bi – HN=C=NH product ions.
Scheme 6 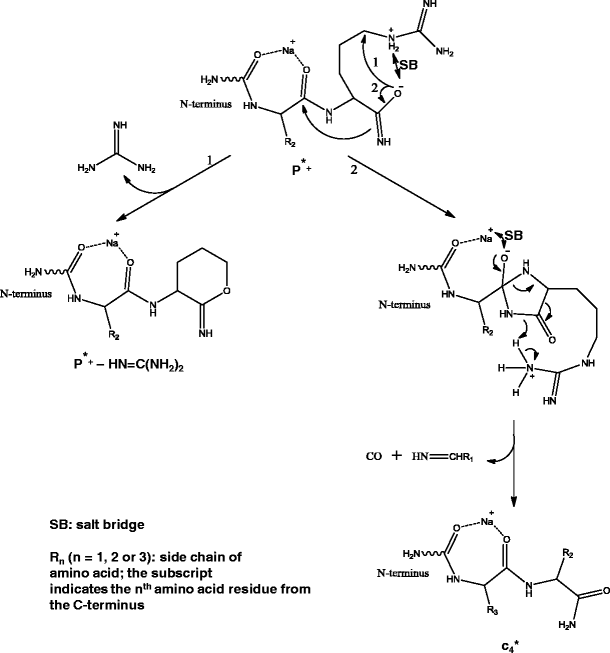
Formation of major product ions from dissociation of c *5 (sodiated c5) ion
The mechanism for the formation of cn-1* ion from c5*+ (Scheme 6) is proposed to be analogous to that for the formation of bn-1 + H2O ion from y 2+10 cluster ions (Scheme 4), where the salt bridge in c5*+ was formed via a proton transferred to the Arg side chain from the imidic acid group [12] instead of from the C-terminal carboxyl group, while the amide bond was weakened by coordination between the sodium ion and the amide oxygen instead of by a hydrogen bond between the protonated N-terminal amine and the amide oxygen. The mechanism for the formation of often reported [b + OH + Na]+ ions from peptide ions is likely similar to what is proposed here for the formation of cn-1* ions, except that the nucleophilic attack was by carboxyl oxygen instead of imidic nitrogen.
-
9)
Summary of fragmentation characteristics
Although the 20 precursor ions for the pseudo MS3 analysis discussed in this study appear to lack mobile protons, many high-intensity product ions were observed. This could be due to the release of a proton from the formation of a salt bridge between a protonated arginine residue and a deprotonated carboxyl group (side chains of glutamic acid and aspartic acid residues, or C-terminal carboxyl group) or imidic acid (C-terminus of ci-1 ions, where ith residue being dehydroalanine, Arg or Lys residues, as discussed above) residue. For instance, for b18 precursor cluster ions, a salt bridge could form between the protonated Arg at the fifth residue and the deprotonated glutamine (Glu) at the seventh residue; while for (y10c126)+ ion, a salt bridge could form between the protonated Arg and the deprotonated imidic acid at the C-terminus. The released proton result from the salt bridge formation then induces dissociation via the mobile proton model. Multiple ion structures, including regular structures (with no salt-bridge formation) and those containing a salt-bridge (and thus mobile protons), could co-exist for precursor ions of the same sequence and charge state. The intensities of product ions could be affected by the relative abundances of the salt-bridged structures. The salt-bridge formation could also lead to some uncommon product ions as discussed above.
Depending on the ion structure formed, dissociation may involve a six-membered ring transition state followed by bond rearrangements under the influence of lone pair electrons on the hetero-atoms. Due to the large size of sulfur atoms compared with carbon atoms, bond rearrangement can also occur via a four-membered ring transition state when sulfur atom is involved, e.g., disulfide bond cleavages from MS/MS analysis of chicken lysozyme protein ions [15].











4.3 Identification of Chicken Lysozyme via Database Search
Using Batch-Tag, the 20 pseudo MS3 spectra (Chicken lysozyme was assumed to be an unknown protein) were searched against database of the Gallus gallus or all species, with the digest “no enzyme” (for both databases) or “full protein” (for the Gallus gallus database). The results are presented in Table 2. All searches identified lysozyme of chicken (or along with lysozyme of three other relevant species when searched against all species) as the only match.
The search against “Gallus gallus” with “no enzyme” identified chicken lysozyme as the only hit with 14 unique spectra matching to those of chicken lysozyme, seven of which with the expectation value <0.05. The matches include the spectra of eight b and yb precursor ions, two c and yc precursor ions, and five y precursor ions (Table 2). The searches for the specific types of precursor ions were also tried by adjusting the parameters; however, no significant improvements were achieved. In contrast, the search against “Gallus gallus” with “Full Protein” and “Nonspecific” at “N-term” identified chicken lysozyme as the only hit with all five spectra of y precursor ions matching to those of chicken lysozyme; the corresponding expectation values are more than three orders lower compared with those from the search with “no enzyme” (Table 2). Combining the two types of search results against the Gallus gallus database, i.e., the search with “Full Protein” and “Nonspecific” at “N-term” (applicable to spectra of y precursor ions only) and the search for all types of precursor ions with “no enzyme,” 12 pseudo MS3 spectra match those of chicken lysozyme with the expectation values <0.02.
The search of the 20 spectra against all species identified lysozyme of four different but relevant species, i.e., CHICK, COLVI, LOPCA, and NUMME, as the only hits. The lysozyme of the former and latter two species were matched by 6 and 7 spectra, respectively. The expectation values for three of these spectra are less than 0.02. These three spectra correspond to ions of the b 2+18 cluster, which is not surprising since the chance of random matches is low for fragments with long sequences. In contrast, the spectra of precursor ion m/z 605.41 matched amidated KVFGR (corresponding to the c +5 ion) of 51 different proteins, indicating more random matches for short sequence ions. The confidence in identification of proteins may be improved by separating the maximum numbers of the variable modifications for C-terminus and for cysteine residues—in this work they were combined, which might have decreased specificity.
This database search method is not limited to disulfide-bonded proteins. For proteins without the need of disulfide bond cleavages, it is not necessary to perform MS3 or pseudo MS3 analysis of the precursor ions in the absence of mobile protons. Therefore, the corresponding MS3 or pseudo MS3 spectra could share more similarity with MS/MS spectra in a typical bottom up approach and, thus, be more suitable for the search with Batch-Tag. Internal disulfide bonds in the pseudo MS3 precursor ions were not considered in this study. To take internal disulfide bonds into consideration, a variable modification of “Cys → Dehydro” may need to be included into the search parameters along with an increase of the maximum number of modifications accordingly.
Examination of the database search results for these spectra revealed some relatively high intensity unassigned fragment peaks. Some of these missed assignments are due to uncommon ions (including the ones discussed above) not considered by Batch-Tag for database searching, as well as imperfection in spectra processing by the MassLynx program, including incomplete conversion of doubly charged precursor ions to single charges and incorrect de-isotoping for peaks with 1 Da apart. The confidence in protein identification could be increased by the incorporation of the uncommon types of ions in search programs and improvement of programs for spectra processing.
5 Conclusions
The pseudo MS3 approach described in this report can be applied for the identification of disulfide-bonded proteins or proteins in general. Unlike conventional bottom-up methods, many labor-intensive procedures (e.g., proteolytic digestion, reduction, and alkylation) are not required with this approach. Similar results are expected for true MS3 analysis of intact protein (not necessarily disulfide-bonded) ions. This approach may benefit PTM characterization of proteins, especially when combined with top-down MS/MS analysis. On the other hand, this pseudo MS3 (and true MS3) approach could also be applied for identification of components of disulfide-bonded peptides (generated from proteolytic digestion without reduction) to assist in the determination of disulfide bond linkages in proteins [60].
Various types of uncommon product ions were observed in the pseudo MS3 spectra in this study. These uncommon product ions may also be present in MS/MS analysis of proteolytic peptides, especially in the case when the precursor ions lack mobile protons. Consideration of these types of uncommon product ions may help interpret spectra, confirm identification, and determine false identification.
References
Kelleher, N.L.: Top-down proteomics. Anal. Chem. 76, 197A–203A (2004)
McLafferty, F.W., Breuker, K., Jin, M., Han, X., Infusini, G., Jiang, H., Kong, X., Begley, T.P.: Top-down MS, a powerful complement to the high capabilities of proteolysis proteomics. FEBS J. 274, 6256–6268 (2007)
Reid, G.E., McLuckey, S.A.: 'Top down' protein characterization via tandem mass spectrometry. J. Mass Spectrom. 37, 663–675 (2002)
Whitelegge, J., Halgand, F., Souda, P., Zabrouskov, V.: Top-down mass spectrometry of integral membrane proteins. Expert Rev. Proteomics 3, 585–596 (2006)
Chait, B.T.: Chemistry. Mass spectrometry: Bottom-up or top-down? Science 314, 65–66 (2006)
Han, X.M., Jin, M., Breuker, K., McLafferty, F.W.: Extending top-down mass spectrometry to proteins with masses greater than 200 kilodaltons. Science 314, 109–112 (2006)
Zubarev, R.A., Kruger, N.A., Fridriksson, E.K., Lewis, M.A., Horn, D.M., Carpenter, B.K., McLafferty, F.W.: Electron capture dissociation of gaseous multiply-charged proteins is favored at disulfide bonds and other sites of high hydrogen atom affinity. J. Am. Chem. Soc. 121, 2857–2862 (1999)
Chrisman, P.A., Pitteri, S.J., Hogan, J.M., McLuckey, S.A.: SO2 -electron transfer ion/ion reactions with disulfide linked polypeptide ions. J. Am. Soc. Mass Spectrom. 16, 1020–1030 (2005)
Gunawardena, H.P., Gorenstein, L., Erickson, D.E., Xia, Y., McLuckey, S.A.: Electron transfer dissociation of multiply protonated and fixed charge disulfide linked polypeptides. Int. J. Mass Spectrom. 265, 130–138 (2007)
Cooper, H.J., Hakansson, K., Marshall, A.G.: The role of electron capture dissociation in biomolecular analysis. Mass Spectrom. Rev. 24, 201–222 (2005)
Wiesner, J., Premsler, T., Sickmann, A.: Application of electron transfer dissociation (ETD) for the analysis of posttranslational modifications. Proteomics 8, 4466–4483 (2008)
Chrisman, P.A., McLuckey, S.A.: Dissociations of disulfide-linked gaseous polypeptide/protein anions: Ion chemistry with implications for protein identification and characterization. J. Proteome Res. 1, 549–557 (2002)
Stephenson Jr., J.L., Cargile, B.J., McLuckey, S.A.: Ion trap collisional activation of disulfide linkage intact and reduced multiply protonated polypeptides. Rapid Commun. Mass Spectrom. 13, 2040–2048 (1999)
Wells, J.M., Stephenson, J.L., McLuckey, S.A.: Charge dependence of protonated insulin decompositions. Int. J. Mass Spectrom. 203, A1–A9 (2000)
Chen, J., Shiyanov, P., Zhang, L., Schlager, J.J., Green-Church, K.B.: Top-down characterization of a native highly intralinked protein: Concurrent cleavages of disulfide and protein backbone bonds. Anal. Chem. 82, 6079–6089 (2010)
Ginter, J.M., Zhou, F., Johnston, M.V.: Generating protein sequence tags by combining cone and conventional collision induced dissociation in a quadrupole time-of-flight mass spectrometer. J. Am. Soc. Mass Spectrom. 15, 1478–1486 (2004)
Raska, C.S., Parker, C.E., Huang, C., Han, J., Glish, G.L., Pope, M., Borchers, C.H.: Pseudo-MS3 in a MALDI orthogonal quadrupole-time of flight mass spectrometer. J. Am. Soc. Mass Spectrom. 13, 1034–1041 (2002)
Loo, J.A., Edmonds, C.G., Smith, R.D.: Tandem mass-spectrometry of very large molecules—serum-albumin sequence information from multiply charged ions formed by electrospray ionization. Anal. Chem. 63, 2488–2499 (1991)
Macek, B., Waanders, L.F., Olsen, J.V., Mann, M.: Top-down protein sequencing and MS3 on a hybrid linear quadrupole ion trap-Orbitrap mass spectrometer. Mol. Cell. Proteomics 5, 949–958 (2006)
Zabrouskov, V., Senko, M.W., Du, Y., Leduc, R.D., Kelleher, N.L.: New and automated MSn approaches for top-down identification of modified proteins. J Am. Soc. Mass Spectrom. 16, 2027–2038 (2005)
Boyd, R., Somogyi, A.: The mobile proton hypothesis in fragmentation of protonated peptides: A perspective. J. Am. Soc. Mass Spectrom. 21, 1275–1278 (2010)
Paizs, B., Suhai, S.: Fragmentation pathways of protonated peptides. Mass Spectrom. Rev. 24, 508–548 (2005)
Bythell, B.J., Suhai, S., Somogyi, A., Paizs, B.: Proton-driven amide bond-cleavage pathways of gas-phase peptide ions lacking mobile protons. J. Am. Chem. Soc. 131, 14057–14065 (2009)
Feng, W.Y., Gronert, C., Fletcher, K.A., Warres, A., Lebrilla, C.B.: The mechanism of C-terminal fragmentations in alkali metal ion complexes of peptides. Int. J. Mass Spectrom. 222, 117–134 (2003)
Farrugia, J.M., O'Hair, R.A.J.: Involvement of salt bridges in a novel gas phase rearrangement of protonated arginine-containing dipeptides which precedes fragmentation. Int. J. Mass Spectrom. 222, 229–242 (2003)
Laskin, J., Yang, Z., Song, T., Lam, C., Chu, I.K.: Effect of the basic residue on the energetics, dynamics, and mechanisms of gas-phase fragmentation of protonated peptides. J. Am. Chem. Soc. 132, 16006–16016 (2010)
Gu, C., Tsaprailis, G., Breci, L., Wysocki, V.H.: Selective gas-phase cleavage at the peptide bond C-terminal to aspartic acid in fixed-charge derivatives of Asp-containing peptides. Anal. Chem. 72, 5804–5813 (2000)
Chalkley, R.J., Baker, P.R., Medzihradszky, K.F., Lynn, A.J., Burlingame, A.L.: In-depth analysis of tandem mass spectrometry data from disparate instrument types. Mol. Cell. Proteomics 7, 2386–2398 (2008)
Baker, P. R., Clauser, K. R.: http://prospector.ucsf.edu/prospector/mshome.htm, accessed 14 Nov 2011
Nesvizhskii, A.I., Aebersold, R.: Interpretation of shotgun proteomic data: The protein inference problem. Mol. Cell. Proteomics 4, 1419–1440 (2005)
Lynn, A. J., Chalkley, R. J., Baker, P. R., Segal, M. R., Burlingame, A. L.:Protein prospector and ways of calculating expectation values. In: Proceedings of the 54th ASMS Conference on Mass Spectrometry and Allied Topics. Seattle, WA, Bioinformatics I–351 (2006)
Demeure, K., Gabelica, V., De Pauw, E.A.: New advances in the understanding of the in-source decay fragmentation of peptides in MALDI-TOF-MS. J. Am. Soc. Mass Spectrom. 21, 1906–1917 (2010)
Suckau, D., Resemann, A.: T3-sequencing: Targeted characterization of the N- and C-termini of undigested proteins by mass spectrometry. Anal. Chem. 75, 5817–5824 (2003)
Brown, R.S., Lennon, J.J.: Sequence-specific fragmentation of matrix-assisted laser-desorbed protein peptide ions. Anal. Chem. 67, 3990–3999 (1995)
Xia, Y., Liang, X., McLuckey, S.A.: Ion trap versus low-energy beam-type collision-induced dissociation of protonated ubiquitin ions. Anal. Chem. 78, 1218–1227 (2006)
Paizs, B., Suhai, S., Hargittai, B., Hruby, V., Somogyi, A.: Ab initio and MS/MS studies on protonated peptides containing basic and acidic amino acid residues I. Solvated proton versus salt-bridged structures and the cleavage of the terminal amide bond of protonated RD-NH2. Int. J. Mass Spectrom. 219, 203–232 (2002)
Yu, W., Vath, J.E., Huberty, M.C., Martin, S.A.: Identification of the facile gas-phase cleavage of the Asp-Pro and Asp-Xxx peptide bonds in matrix-assisted laser desorption time-of-flight mass spectrometry. Anal. Chem. 65, 3015–3023 (1993)
Fu, Q., Li, L.: Neutral loss of water from the b ions with histidine at the C-terminus and formation of the c ions involving lysine side chains. J. Mass Spectrom. 41, 1600–1607 (2006)
Farrugia, J.M., O'Hair, R.A.J., Reid, G.E.: Do all b2 ions have oxazolone structures? Multistage mass spectrometry and ab initio studies on protonated N-acyl amino acid methyl ester model systems. Int. J. Mass Spectrom. 210, 71–87 (2001)
Deery, M.J., Summerfield, S.G., Buzy, A., Jennings, K.R.: A mechanism for the loss of 60 u from peptides containing an arginine residue at the C-terminus. J. Am. Soc. Mass Spectrom. 8, 253–261 (1997)
http://prospector.ucsf.edu/prospector/html/misc/faq.htm#z_ions, accessed 14 Nov 2011
Gonzalez, J., Besada, V., Garay, H., Reyes, O., Padron, G., Tambara, Y., Takao, T., Shimonishi, Y.: Effect of the position of a basic amino acid on C-terminal rearrangement of protonated peptides upon collision-induced dissociation. J. Mass Spectrom. 31, 150–158 (1996)
She, Y.M., Krokhin, O., Spicer, V., Loboda, A., Garland, G., Ens, W., Standing, K.G., Westmore, J.B.: Formation of (bn-1 + H2O) ions by collisional activation of MALDI-formed peptide [M + H]+ ions in a QqTOF mass spectrometer. J. Am. Soc. Mass Spectrom. 18, 1024–1037 (2007)
Thorne, G.C., Ballard, K.D., Gaskell, S.J.: Metastable decomposition of peptide [M + H] + ions via rearrangement involving loss of the C-terminal amino-acid residue. J. Am. Soc. Mass Spectrom. 1, 249–257 (1990)
Vachet, R.W., Asam, M.R., Glish, G.L.: Secondary interactions affecting the dissociation patterns of arginine-containing peptide ions. J. Am. Chem. Soc. 118, 6252–6256 (1996)
Bythell, B.J., Csonka, I.P., Suhai, S., Barofsky, D.F., Paizs, B.: Gas-phase structure and fragmentation pathways of singly protonated peptides with N-terminal arginine. J. Phys. Chem. B 114, 15092–15105 (2010)
Burlet, O., Yang, C.Y., Gaskell, S.J.: Influence of cysteine to cysteic acid oxidation on the collision-activated decomposition of protonated peptides—evidence for intra-ionic interactions. J. Am. Soc. Mass Spectrom. 3, 337–344 (1992)
Erlekam, U., Bythell, B.J., Scuderi, D., Van Stipdonk, M., Paizs, B., Maitre, P.: Infrared spectroscopy of fragments of protonated peptides: Direct evidence for macrocyclic structures of b5 ions. J. Am. Chem. Soc. 131, 11503–11508 (2009)
Bleiholder, C., Osburn, S., Williams, T.D., Suhai, S., Van Stipdonk, M., Harrison, A.G., Paizs, B.: Sequence-scrambling fragmentation pathways of protonated peptides. J. Am. Chem. Soc. 130, 17774–17789 (2008)
Harrison, A.G., Young, A.B., Bleiholder, C., Suhai, S., Paizs, B.: Scrambling of sequence information in collision-induced dissociation of peptides. J. Am. Chem. Soc. 128, 10364–10365 (2006)
Molesworth, S.P., Van Stipdonk, M.J.: Apparent inhibition by arginine of macrocyclic b ion formation from singly charged protonated peptides. J. Am. Soc. Mass Spectrom. 21, 1322–1328 (2010)
Leitner, A., Emmert, J., Boerner, K., Lindner, W.: Influence of solvent additive composition on chromatographic separation and sodium adduct formation of peptides in HPLC-ESI MS. Chromatographia 65, 649–653 (2007)
Kish, M.M., Wesdemiotis, C.: Selective cleavage at internal lysine residues in protonated versus metalated peptides. Int. J. Mass Spectrom. 227, 191–203 (2003)
Sabareesh, V., Balaram, P.: Tandem electrospray mass spectrometric studies of proton and sodium ion adducts of neutral peptides with modified N- and C-termini: Synthetic model peptides and microheterogeneous peptaibol antibiotics. Rapid Commun. Mass Spectrom. 20, 618–628 (2006)
Barr, J.M., Van Stipdonk, M.J.: Multi-stage tandem mass spectrometry of metal cationized leucine enkephalin and leucine enkephalin amide. Rapid Commun. Mass Spectrom. 16, 566–578 (2002)
Lin, T., Payne, A.H., Glish, G.L.: Dissociation pathways of alkali-cationized peptides: Opportunities for C-terminal peptide sequencing. J. Am. Soc. Mass Spectrom. 12, 497–504 (2001)
Newton, K.A., McLuckey, S.A.: Generation and manipulation of sodium cationized peptides in the gas phase. J. Am. Soc. Mass Spectrom. 15, 607–615 (2004)
Chiu, F.C., Lo, C.M.: Observation of amide anions in solution by electrospray ionization mass spectrometry. J. Am. Soc. Mass Spectrom. 11, 1061–1064 (2000)
Shek, P.Y., Zhao, J., Ke, Y., Siu, K.W., Hopkinson, A.C.: Fragmentations of protonated arginine, lysine. and their methylated derivatives: Concomitant losses of carbon monoxide or carbon dioxide and an amine. J. Phys. Chem. A 110, 8282–8296 (2006)
Gorman, J.J., Wallis, T.P., Pitt, J.J.: Protein disulfide bond determination by mass spectrometry. Mass Spectrom. Rev. 21, 183–216 (2002)
Acknowledgments
The authors thank Dr. Peter Baker and Dr. Robert Chalkley for their support on the program Batch-Tag; Dr. Gary Glish and the reviewers for helpful comments; and John Shapiro for proofreading the manuscript.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
(PDF 848 kb)
Rights and permissions
About this article
Cite this article
Chen, J., Shiyanov, P., Schlager, J.J. et al. A Pseudo MS3 Approach for Identification of Disulfide-Bonded Proteins: Uncommon Product Ions and Database Search. J. Am. Soc. Mass Spectrom. 23, 225–243 (2012). https://doi.org/10.1007/s13361-011-0294-6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13361-011-0294-6
Key words
- MS/MS
- MS3
- Intact protein
- Top-down
- Disulfide bond cleavage
- Lysozyme
- Collision-induced dissociation
- Nozzle-skimmer dissociation
- Database search
- ProteinProspector
- Mobile proton
- Uncommon product ions
- Quadrupole time-of-flight
- Electrospray ionization
- c ion
- d ion
- b+H2O ion
- Neutral loss
- Oxazolone structure
- Sequence scrambling
- Sodiated ion
- HN=C=NH
- Salt bridge
- six-membered ring
- Bond rearrangement
- Arginine residue
- Lysine residue
- Aspartic acid residue
- Cysteine residue