
Abstract
Lambdoid bacteriophages are excellent models in studies on molecular aspects of virus-host interactions. However, some of them carry genes encoding toxins which are responsible for virulence of pathogenic strains of bacteria. Shiga toxin-converting bacteriophages (Stx phages) encode Shiga toxins that cause virulence of enterohemorrhagic Escherichia coli (EHEC), and their effective production depends on Stx prophage induction. The exo-xis region of the lambdoid phage genome consists of genes which are dispensable for the phage multiplication under laboratory conditions; however, they might modulate the virus development. Nevertheless, their exact effects on the phage and host physiology remained unclear. Here, we present results of complex studies on the role of the exo-xis region of bacteriophage Φ24B, one of Stx2b phages. Transcriptomic analyses, together with proteomic and metabolomic studies, provided the basis for understanding the functions of the exo-xis region. Genes from this region promoted lytic development of the phage over lysogenization. Moreover, expression of the host genes coding for DnaK, DnaJ, GrpE, and GroELS chaperones was impaired in the cells infected with the Δexo-xis phage mutant, relative to the wild-type virus, corroborating the conclusion about lytic development promotion by the exo-xis region. Proteomic and metabolomic analyses indicated also modulation of gad and nrf operons, and levels of amino acids and acylcarnitines, respectively. In conclusion, the exo-xis region controls phage propagation and host metabolism by influencing expression of different phage and bacterial genes, directing the virus to the lytic rather than lysogenic developmental mode.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Bacteriophage λ, a virus infecting Escherichia coli, has been used as a model in molecular biology for over 60 years. Many crucial discoveries were made during studies on this virus (Casjens and Hendrix 2015). Nevertheless, it still plays its role in molecular biology, and perhaps surprisingly, novel exciting discoveries are being made with λ, including discovery of molecular regulatory mechanisms and development of novel methods in genetic engineering and biotechnology (Caldwell and Bell 2019; Zhao et al. 2023).
While most Escherichia coli strains are harmless commensals, the Shiga toxin-producing E. coli (STEC) strains and related enterohemorrhagic E. coli (EHEC) subtypes contribute to food poisoning and kidney disorders in human populations throughout the world (Kaper et al. 2004; Bugarel et al. 2011). In the USA, STEC causes 100,000 illnesses, 3000 hospitalizations, and 90 deaths annually (Al Qabili et al. 2022), with comparable effects in other countries (Auvray et al. 2023; Matussek et al. 2023; Rodwell et al. 2023; Svendsen et al. 2023).
The main virulence factors of STEC are Shiga toxins (Stx), encoded by genes stx1 and stx2, located in genomes of bacteriophages, which occur in bacteria as prophages (Mizutani et al. 1999). Shiga toxin-converting bacteriophages (Stx phages) belong to the lambdoid family of phages (Besser et al. 1999; Schmidt 2001). All lambdoid phages share many similarities in the life cycle and genome organization. These bacteriophages can undergo either a lytic or lysogenic development (Ptashne 2004; Węgrzyn and Węgrzyn 2005; Kędzierska et al. 2007).
When the lysogenic pathway is chosen, phage DNA is incorporated into E. coli genome, forming a prophage that can be maintained in this state for many cell generations. The prophage is replicated together with the bacterial genome and is transmitted to daughter cells at each subsequent cell division. As a result of prophage induction, caused by different factors like low pH, iron ions, antibiotics (Kimmitt et al. 1999, 2000), or hydrogen peroxide (Wagner et al. 2001a; Łoś et al. 2009, 2010), the excision of the phage genome occurs and efficiency of expression of the stx genes increases rapidly. Consequently, at this stage, the viral genomes exist as separate DNA molecules in the bacterial cell and replicate separately from the host nucleoid as extrachromosomal elements (Nejman et al. 2009, 2011).
During the lytic development, there are many round of phage DNA replication, and the genes encoding head, tail, and lysis proteins are expressed. This leads to the assembly of many (100–200, on average, under optimal conditions) new phage particles within the cell. The development ends with disruption of the host cell and liberation of progeny virions.
When E. coli strain infecting human gut contains an Stx prophage, its induction ends with by the release of Stx progeny phages and Shiga toxin molecules, produced at the time of the expression of phage genes. It is crucial to stress that the effective production of Shiga toxins occurs only upon prophage induction and its further lytic development (Wagner et al. 2001b, 2002; Waldor and Friedman 2005). Treatment of infection caused by STEC bacteria is difficult because many therapeutics, including antibiotics, are prophage inducers. Thus, their application increases expression of toxin genes and enhances the severity of the disease (Kimmitt et al. 2000; Gamage et al. 2004; Serna and Boedeker 2008). In the light of this problem, it is obvious that understanding the mechanisms of regulation of development of lambdoid phages is crucial for both basic knowledge and putative further work on prevention and treatment of STEC infections. Although bacteriophage λ, the best investigated member of lambdoid phage family, has served as a model organism in molecular biology for over 60 years (Węgrzyn et al. 2012), the functions of some of its genes are still unknown.
For many years, functions of the region located between exo and xis genes of the phage genome (called the exo-xis region or the bin region) have been almost completely unknown, despite its conservation among lambdoid phages. This region is located in the central part of the phage genome and is transcribed from the early pL promoter which is repressed by the phage-encoded cI protein during lysogeny (Fig. 1). Studies performed by Kourilsky and Knapp (1974) indicated that in the case of bacteriophage λ, transient expression of genes of the pL operon can induce cell-cycle synchrony in a population of host cells. This assumption was confirmed by Sergueev et al. (2002), who showed that induction of the pL operon give rise to two separable effects on the host cell cycle: (i) a temporary block of cell division and, at the same time, (ii) a block of the initiation of DNA replication (hence the term the bin region).

A simplified circular map of bacteriophage Φ24B genome, highlighting phage genes and promoters of the exo-xis region. Promoters are depicted by arrows and thin black lines
Despite the evidence that the exo-xis region modulates the host genome function, the physiological significance of this regulation in the bacteriophage development remains unknown. The prevailing hypothesis since 2002 is that exo-xis region genes regulate the phage decision whether to lysogenize the host cell or to enter the lytic development (Sergueev et al. 2002). It was speculated that transient inhibition of host DNA replication may help phage, which uses the host replication proteins, to maximize its DNA replication during lytic development (Sergueev et al. 2002). However, no experimental data supporting such a hypothesis were presented until 2008, when the first results presenting an evidence for the relation between exo-xis region and phage development were shown (Łoś et al. 2008). These data indicated that the exo-xis region contributes to a decreased transcription from cII-stimulated promoters pI, paQ, and pE. That work displayed also that the exo-xis dependent decrease in the efficiency of λ plating on host strain requires the intact cI gene. However, the absence of cI activity could be compensated by either increased stability of cII or cII-independent transcription of cI, which in turn suggests that transcription in this region, but not necessarily the cI activity, is responsible for this effect (Łoś et al. 2008). Besides, overproduction of cIII significantly enhanced the negative effect of the exo-xis region on λ plating (Łoś et al. 2008).
The ea8.5 gene, located in the exo-xis region, appeared to have the strongest effects on the phage development (Łoś et al. 2008). Other well-known genes involved in the lysis vs. lysogenization decision (cI, cII, and cIII) were shown to participate in the processes leading to phenotypic effects of the exo-xis region. Those results allowed to conclude that the exo-xis region may be involved in the regulation of bacteriophage λ development, most probably at the stage of the lysis vs. lysogenization decision; however, it was not enough knowledge to indicate the mechanism of this phenomenon (Łoś et al. 2008).
In the subsequent work, effects of expression of genes from the exo-xis region on various stages of development of phages λ and Φ24B (an Stx2b phage) were investigated (Bloch et al. 2013). It was demonstrated that the presence of this region on a multicopy plasmid in naive E. coli resulted in impaired lysogenization of host bacteria and more effective induction of prophages, both spontaneous and stimulated by various agents (mitomycin C, H2O2, and UV radiation) (Bloch et al. 2013). Moreover, patterns of expressions of particular genes from the exo-xis region were determined, but unexpectedly, in both phages λ and Ф24B, these patterns were significantly different not only between conditions of the host cell infection by bacteriophages and prophage induction, but also between induction of prophages with various agents (mitomycin C and hydrogen peroxide) (Bloch et al. 2014).
Intriguingly, in hydrogen peroxide-treated E. coli cells lysogenic for either λ or Φ24B, deletion of the exo-xis region resulted in a significant decrease in the levels of expression of the S.O.S. regulon genes (Licznerska et al. 2016). Moreover, under these conditions, a dramatic decrease in the levels of expression of phage genes crucial for lytic development (particularly xis, exo, N, cro, O, Q, and R) could be observed in Φ24B-, but not in λ-bearing cells (Licznerska et al. 2016). That finding indicated that genes located in the exo-xis region are necessary for efficient expression of both host S.O.S. regulon in lysogenic bacteria and regulatory genes of Shiga toxin-converting bacteriophage Φ24B.
The studies described briefly above indicated intriguing regulatory effects of the exo-xis region. However, despite further charactering principles of structures and basic functions of various products of genes from this region, like Ea8.5 (Kwan et al. 2013), Orf63 (Dydecka et al. 2017), Orf60a and Orf61 (Dydecka et al. 2018), Orf73 (Zdrojewska et al. 2019), and Ea22 (Dydecka et al. 2020; Tong et al. 2020), the role of the exo-xis region in modulating phage development and influencing host physiology remains unclear. Therefore, the aim of this work was to determine effects of the exo-xis region on development of the Stx phage Φ24B and physiology and metabolism of its host. This aim was realized by employing transcriptomic, proteomic, and metabolomic approaches which allow to analyze global changes in gene expression, protein content, and metabolic status in the investigated cells under specific conditions, in this case upon infection with the studied bacteriophages.
Materials and methods
Bacterial strains, plasmids, and bacteriophages
E. coli MG1655 strain, its derivatives, bacteriophages, and plasmids are presented in Table 1.
The deletion mutant of Φ24B phage, lacking the sequence of the region located between exo and xis genes, was constructed as described previously (Licznerska et al. 2016), by using E. coli MG1655 (Φ24B) strain and the Quick and Easy E. coli Gene Deletion Kit (Gene Bridges, Heidelberg, Germany). Plasmid pUC18 (Thermo Fisher Scientific Inc., Waltham, MA, USA) was employed as a control variant in all experiments presented in this work. For construction of the plasmid pUC18_exo-xis_Φ24B, nucleotide sequences of all orfs and genes localized between exo and xis genes from the genome of phage Φ24B were amplified by the PCR method with primers F_Φ24B_exo-xis_HindIII (5′GTC AAG CTT GAA GGC GGT TGT TAG) and R_Φ24B_exo-xis_EcoRI (5′TGA GGA TCC GTA TAT GGG GAG CAA TG). Specific oligonucleotide primers were developed by Primer3web version 4.1.0 and synthesized by GENOMED S.A. (Warsaw, Poland). Phage DNA was isolated by using MasterPure™ Complete DNA and RNA Purification Ki (Biosearch Technologies, Hoddesdon, Great Britain). Following digestion with HindIII and BamHI restriction endonucleases (Thermo Fisher Scientific Inc., Waltham, MA, USA), the Φ24B exo-xis region was ligated with the HindIII-BamHI fragment of plasmid pUC18 bearing an ampicillin-resistant gene and the plac promoter. Ligation was conducted by using the T4 DNA ligase (Thermo Fisher Scientific Inc., Waltham, MA, USA) according the manufacturer’s protocol. The construction of the pUC18_exo-xis_Φ24 was confirmed by DNA sequencing (GENOMED, Warsaw, Poland).
Bacterial growth media and cultures
Bacteria were routinely cultured in the Luria-Bertani (LB) medium (BioShop, Burlington, ON, Canada), supplemented with 10 mM CaCl2 (Chempur, Piekary Śląskie, Poland) and 10 mM MgSO4 (Chempur, Piekary Śląskie, Poland). Where appropriate, the following antibiotics were added: chloramphenicol up to 2.5 μg/ml (BioShop, Burlington, ON, Canada) or ampicillin up to 50 μg/ml (BioShop, Burlington, ON, Canada). The LB broth supplemented with 1.5% agar (LA; BTL, Łódź, Poland) was used as a bottom agar for pouring the Petri dishes. Top agar consisted of LB medium and 0.7% bacteriological agar (BTL, Łódź, Poland). The host bacteria were grown under aeration conditions, achieved by shaking, at 30 °C (most experiments) or 37 °C (preparation of phage lysate). The Petri dishes with bacterial strains were incubated overnight at 37 °C.
Propagation of bacterial viruses
Bacteria lysogenic with tested bacteriophages, Φ24B or Φ24BΔexo-xis, were grown in LB medium to OD600 of 0.1. Prophage induction was provoked by addition of mitomycin C (BioShop, Burlington, ON, Canada) to a final concentration of 1 μg/ml. The mixture was incubated at 37 °C with shaking until lysis occurred. The obtained lysate was treated with 4% chloroform for 15 min (Chempur, Piekary Śląskie, Poland) and cleared of cellular debris by centrifugation (2000 × g, 10 min, 4 °C). In the next step, polyethylene glycol 8000 (PEG8000; BioShop, Burlington, ON, Canada) was added to a final concentration of 10% and stirred overnight at 4 °C. The precipitate was collected by centrifugation (8000 × g, 20 min, 4 °C) and suspended in TM buffer (10 mM Tris-HCl, 10 mM MgSO4; pH 7.2). To recover virions, the suspension was treated with equal volume of chloroform and centrifuged (2000 × g, 10 min, 4 °C). The extraction procedure was repeated until no white interface between the aqueous and organic phases was visible. Finally, the upper phase with phage particles was collected and the titer of the phage lysate was determined by using a plaque assay described below.
Bacteriophage lysate titration
Bacteriophage titration procedure was performed using standard Petri dishes filled with 25 ml of LA medium. Soft agar (0.7%), supplemented with 10 mM CaCl2 and 10 mM MgSO4, was maintained at 60 °C before use. The top layer was prepared by mixing 1 ml of an overnight bacterial host culture with 2 ml of the soft agar. The tube with the mixture was slowly rotated for 10–15 s and then immediately poured onto LA bottom agar supplemented with 2.5 μg/ml of chloramphenicol. To determine the number of phage particles per 1 ml of suspension (plaque forming units per ml, PFU/ml), the obtained phage lysate was serially 10-fold diluted in TM buffer (10 mM Tris-HCl, 10 mM MgSO4; pH 7.2) and then 2.5 μl of each dilution was spotted on the surface of the top agar. The Petri dishes were incubated at 37 °C for 20 h. The next day, single plaques were counted, and the phage titer was calculated.
One-step growth experiment
To assess the kinetics of intracellular phage development, one-step growth experiment was performed according to the previously described procedure (Bloch et al. 2014). Briefly, host bacteria were grown in LB medium at 30 °C to an OD600 of 0.2. Then, 10 ml of bacterial culture was centrifuged (2000 × g, 10 min, 4 °C) and the pellet was suspended in 1 ml of LB medium supplemented with 3 mM sodium azide (Merck, Darmstadt, Germany), 10 mM CaCl2, and 10 mM MgSO4. Then, phage lysate was added to the sample to multiplicity of infection (m.o.i.) of 0.05 and allowed to adsorb on the bacterial cells surface for 10 min at 30 °C. After incubation, unadsorbed phages were removed by 3-times washing in LB medium with 3 mM sodium azide and centrifugation (2000 × g, 10 min, 4 °C). In the next step, 25 μl of the mixture was transferred to the flask filled with 25 ml of fresh, pre-warmed to 30 °C LB medium (time 0). The infected bacterial cultures were aerated in an incubator shaker at 30 °C. The number of infective centers was estimated from nine samples taken in the interval of 0–15 min after the dilution and plating at the permissive conditions. Samples withdrawn at later times were shaken vigorously with equal volume of chloroform and cleared by centrifugation (2000 × g, 5 min, RT). The number of intracellular progeny phages was estimated by plating on appropriate indicator E. coli MG1655 strain. Plates were incubated overnight at 37 °C and then burst size was estimated as a ratio of phage titer at particular time points to the titer of infection centers.
Efficiency of lysogenization
To estimate the efficiency of lysogenization, the procedure described earlier (Dydecka et al. 2017) was used, with some modifications. Briefly, host bacteria were cultivated in LB medium with shaking at 30 °C to OD600 of 0.2. Then, 1 ml of the sample was centrifuged (2000 × g, 10 min, 4 °C) and the bacterial pellet was washed 2 times with TCM buffer (10 mM Tris-HCl, 10 mM MgSO4, 10 mM CaCl2; pH 7.2). Finally, the obtained pellet was suspended in 1 mL of TCM buffer and phage lysate was added to the mixture to an m.o.i. of 5. After incubation of the sample at 30°, the serial 10-fold dilutions in TCM buffer (10 mM Tris-HCl, 10 mM MgSO4, 10 mM CaCl2; pH 7.2) were prepared and 20 μl of each dilution was plated on LA plates. Following overnight incubation at 37 °C, 96 bacterial colonies were cultivated in a multi-well plate containing 200 μl of LB. Each plate was shaken at 37 °C until OD600 reached 0.1. Then, the putative lysogens were treated with ultraviolet light at 50 J/m2 for 20 s and cultivated with shaking for the next 2 h at 37 °C. Afterward, 10 μl of chloroform was added to each well, the plates were centrifugated (2000 × g, 10 min, 4 °C) and 2.5 μL of the supernatant was spotted onto double-layer LB agar plates with 2.5 μg/ml of chloramphenicol. Plates were incubated overnight at 37 °C, and then, the efficiency of prophage formation was calculated as a percentage of lysogens among all tested 96 bacterial colonies.
Survival of host bacteria after phage infection
To determine the survival of host bacteria after phage infection, the procedure described earlier (Dydecka et al. 2017) was used. Briefly, bacteria were grown in LB medium at 30 °C to OD600 of 0.2. Sample of 4 ml was centrifuged (2000 × g, 10 min, 4 °C) and the obtained bacterial pellet was washed (2000 × g, 10 min, 4 °C) twice with cold 0.85% sodium chloride (Chempur, Piekary Śląskie, Poland). Finally, the bacterial pellet was suspended in 1 ml of LB medium supplemented with 10 mM CaCl2 and 10 mM MgSO4 and then phage lysate was added to an m.o.i. of 5. After incubation at 30 °C, serial dilutions in 0.85% sodium chloride were prepared and 40 μl were spread onto Petri dishes filled with the solid LA medium. Plates were incubated overnight at 37 °C. Percentage of survivors was calculated relative to bacterial sample in which TM buffer was added instead of phage virions.
Efficiency of phage adsorption
The rate of adsorption of virions on the surface of host cells was determined according to the procedure described previously (Bloch et al. 2013), with some modifications. Briefly, host bacteria were grown in LB medium at 30 °C until OD600 reached 0.1. Sample of 1 ml was centrifuged (2000 × g, 10 min, 4 °C) and the pellet was washed 2 times with 0.85% sodium chloride (2000 × g, 10 min, 4 °C). Finally, the bacterial pellet was suspended in 150 μl of LB medium supplemented with 10 mM CaCl2 and 10 mM MgSO4. Tested bacteriophages were added to the bacterial samples to an m.o.i. of 0.05, and these mixtures were incubated at 30 °C for 20 min. During incubation, the phage titer was determined at indicated times. Petri dishes were incubated at 37 °C for 18 h. A bacterial sample withdrawn immediately after addition of phage virions to bacterial host suspension (time 0) was considered as 100% of non-adsorbed bacteriophages and other values were calculated relative to them.
Bacteriophage infection procedure
Infection of host bacteria with tested phages was prepared according to the procedure described previously (Bloch et al. 2014). Briefly, E. coli MG1655 was grown in LB medium with aeration at 30 °C to OD600 of 0.3. After centrifugation of 120 ml of bacterial culture (2000 × g, 10 min, 4 °C), the obtained pellet was suspended in 36 ml of LB medium with 10 mM CaCl2 and 10 mM MgSO4. Then, phage lysate was added to the sample to an m.o.i. of 5. Following incubation on ice, infected host bacteria were cultivated with shaking at 30 °C. At indicated times, bacterial cells were harvested and treated with the killing buffer (200 mM NaN3, 50 mM MgCl2, 200 mM Tris-HCl; pH 8.0) to stop their growth. After centrifugation (2000 × g, 10 min, 4 °C), bacterial pellet was frozen in liquid nitrogen and then stored at −80 °C for further analysis.
Isolation of total RNA from bacterial cells for microarray analysis and RT-qPCR
For microarray analysis, total RNA was isolated from 7.5 × 108 bacterial cells harvested at 30 min after phage infection. RNA used for RT-qPCR was the same RNA sample as for the microarray analysis; however, to determine the expression patterns of particular genes during the infection process of host bacteria with tested phages, RNA was also extracted from cells at additional time points: 0, 7.5, 15, 45, and 60 min. Ribonucleic acids were isolated using the RNeasy Mini kit (Qiagen, Hilden, Germany). To remove DNA contaminants, the TURBO DNA-free™ Kit (Thermo Fischer Scientific, Waltham, MA, USA) was employed. The quality of the obtained RNA samples was tested with the Nano Chips RNA kit (Agilent Technologies, Santa Clara, CA, USA) using the Agilent 2100 Bioanalyzer System.
Microarray analysis
Gene expression microarray analyses were carried out commercially by the Agilent Technologies, Inc. (Santa Clara, CA, USA) using Agilent’s E. coli Microarray Kit 8x15K, P/N G4813A (Agilent Microarray Design ID 020097). Two-color (Cy3 and Cy5) RNA spike-ins were added to the RNA samples from the Two-Color RNA Spike-In kit (P/N 5188-5279). The labeling and hybridizations of target RNAs were performed by employing the Two-Color (Cy3 and Cy5) Low Input Quick Amp Gene expression kit and the Gene Expression Hybridization kit according to patented protocol of the Agilent Technologies, Inc. Each microarray was composed of 15,208 probes representing the complete genomes of four E. coli strains: K-12 MG1655, O157:H7 VT2-Sakai, CFT073, and EDL933 (Guernec et al. 2013). The biological replicates of prepared samples were randomly separated onto two microarray slides. The slides were scanned with the Agilent High Resolution C Scanner (G2656CA) according to the two-color microarray assay scanning protocol (Agilent Technologies, Inc.) and raw microarray image files were generated. Agilent Feature Extraction Image Analysis Software (Version 10.7.3) was involved to extract raw microarray data image files. The microarray data were analyzed for gene expression using the Gene Expression workflow in GeneSpring GX (Version 13.0) Software. Default flag settings were used to make the detection calls. Signal intensities for each probe were normalized to the 75th percentile without baseline transformation. Dye swap arrays were identified for the control vs. treated experiments. The software was used to visualize QC metrics, and the signal values were transferred to SpotFire for correlation analysis. Analysis was carried out an entity list consisting of “detected” probes only. The analysis was carried out using this filtered entity list, using a t-test unpaired statistical method with Benjamini Hochberg FDR method. The p-values were computed asymptotically. Statistical significance was assessed at FDR < 0.05 and p < 0.05.
Relative RT-qPCR and data analysis
Preparation of cDNAs from 1.25 μg of RNA samples for RT-qPCR was performed with Transcriptor Reverse Transcriptase (Roche Diagnostics International, Rotkreuz, Switzerland) and random hexamer primers (Roche Diagnostics International, Rotkreuz, Switzerland), according to the manufacturer’s instructions. All obtained cDNAs were 10-fold diluted in DEPC-treated water (Thermo Fischer Scientific, Waltham, MA, USA) and tested in RT-qPCR. Gene expression patterns were determined by RT-qPCR as described previously (Bloch et al. 2014). All experiments were performed by using the LightCycler® 480 Real-Time PCR System (Roche Diagnostics International, Rotkreuz, Switzerland). Each reaction mixture consisted of 2× SYBR Green I Master Mix, 6.25 ng/μl of cDNA, and 200-nM specific oligonucleotide primers. Amplification was performed according to following program: 95 °C for 5 min, 55 cycles of 95 °C for 10 s, 60 °C for 15 s, and 72 °C for 15 s. All primers (Table 2), amplifying 100–150 nt of target genes, were designed by Primer3web version 4.1.0 and synthetized by GENOMED S.A. (Warsaw, Poland). To confirm the specificity of created primers, melting curve for each product was analyzed. The housekeeping gene icdA was employed as a reference for normalization of samples. The samples harvested before addition of phage particles (time point 0) were applied as a calibrator. The relative changes in the level of gene expression were determined with LinRegPCR using the E-Method with efficiency correction (Bloch et al. 2014).
Proteome analysis using two-dimensional electrophoresis (2D-PAGE)
Samples for proteomic analyses were prepared according to the manual provided by the Kendrick Laboratories (Madison, USA). Following 30-min incubation at 30 °C with tested bacteriophages, 5 × 1010 bacterial cells were harvested by centrifugation (4500 × g, 5 min, 4 °C). Then, bacterial pellet was deeply frozen in liquid nitrogen and washed 2 times (4500 × g, 5 min, 4 °C) with a buffer containing 10 mM Tris-HCl, pH 8.0 (Merck, Darmstadt, Germany), 1 mM KH2PO4 (Avantor Performance Materials Poland S.A., Gliwice, Poland), 68 mM NaCl (Chempur, Piekary Śląskie, Poland), and 9 mM NaH2PO4 (Avantor Performance Materials Poland S.A., Gliwice, Poland). Finally, the obtained bacterial pellet was resuspended in 1 ml of a buffer containing 10 mM Tris-HCl, pH 8.0, 1.5 mM MgCl2, 10 mM KCl (Chempur, Piekary Śląskie, Poland), 0.2% SDS (Merck, Darmstadt, Germany), supplemented with 1× Halt Protease Inhibitor Cocktail, EDTA-Free (Thermo Fisher Scientific Inc., Waltham, MA, USA). Cells were then disrupted by ultrasonication for 10 min in Omni-Ruptor 4000 apparatus (OMNI International, Kennesaw, USA), in an ice bath. In the next step, the mixture was incubated at 4 °C for 15 min with DNase (Merck, Darmstadt, Germany) and RNase (Merck, Darmstadt, Germany), added to the final concentration of 100 μg/ml and 50 μg/ml, respectively. Sample was diluted in the proportion 1:1 in boiling buffer containing 5% SDS (Merck, Darmstadt, Germany), 10% glycerol, and 60 mM Tris-HCl, pH 6.8, and then incubated at 95 °C for 10 min. The soluble protein fraction was separated from cell remnants by centrifugation (20,000 × g, 30 min, 20 °C). Afterward, the concentration of proteins was determined by using the Pierce™ BCA Protein Assay kit (Thermo Fisher Scientific Inc., Waltham, MA, USA) as recommended by the manufacturer. All prepared samples were deeply frozen in liquid nitrogen and analyzed commercially by the Kendrick Laboratories (Madison, USA).
Proteins that were separated by SDS-PAGE/2D-PAGE and stained by Coomassie dye were excised, washed, and the proteins from the gel were treated according to previously published protocols (Shevchenko et al. 1996; Darie et al. 2011; Sokolowska et al. 2012). Briefly, the gel pieces were washed in high-purity, high-performance liquid chromatography HPLC-grade water; dehydrated and cut into small pieces; and destained by incubating in 50 mM ammonium bicarbonate, 50 mM ammonium bicarbonate/50% acetonitrile, and 100% acetonitrile under moderate shaking, followed by drying in a speed-vac concentrator. The gel bands were then rehydrated with 50 mM ammonium bicarbonate. The procedure was repeated twice. The gel bands were then rehydrated in 50 mM ammonium bicarbonate containing 10 mM DTT and incubated at 56 °C for 45 min. The DTT solution was then replaced by 50 mM ammonium bicarbonate containing 100 mM iodoacetamide for 45 min in the dark, with occasional vortexing. The gel pieces were then re-incubated in 50 mM ammonium bicarbonate/50% acetonitrile, and 100% acetonitrile under moderate shaking, followed by drying in speed-vac concentrator. The dry gel pieces were then rehydrated using 50 mM ammonium bicarbonate containing 10 ng/μl trypsin, and incubated overnight at 37 °C with low-intensity shaking. The resulting peptides were extracted twice with 5% formic acid/50 mM ammonium bicarbonate/50% acetonitrile and once with 100% acetonitrile under moderate shaking. Peptide mixture was then dried in a speed-vac, solubilized in 20 μl of 0.1% formic acid/2% acetonitrile.
The peptide mixture was analyzed by reversed phase nanoliquid chromatography (LC) and MS (LC-MS/MS) using a NanoAcuity UPLC (Micromass/Waters, Milford, MA) coupled to a Q-TOF Xevo G2 mass spectrometer (Micromass/Waters, Milford, MA), according to published procedures (Shevchenko et al. 1996; Darie et al. 2011; Sokolowska et al. 2012). Briefly, the peptides were loaded onto a 100 μm × 10 mm NanoAquity BEH130 C18 1.7-μm UPLC column (Waters, Milford, MA) and eluted over a 60-min gradient of 2–80% organic solvent (ACN containing 0.1% FA) at a flow rate of 400 nl/min. The aqueous solvent was 0.1% FA in HPLC water. The column was coupled to a Picotip Emitter Silicatip nanoelectrospray needle (New Objective, Woburn, MA). MS data acquisition involved survey MS scans and automatic data-dependent analysis (DDA) of the top six ions with the highest-intensity ions with the charge of 2+, 3+, or 4+ ions. The MS/MS was triggered when the MS signal intensity exceeded 250 counts/s. In survey MS scans, the three most intense peaks were selected for collision-induced dissociation (CID) and fragmented until the total MS/MS ion counts reached 10,000 or for up to 6 s each. The entire procedure used was previously described (Shevchenko et al. 1996; Darie et al. 2011; Sokolowska et al. 2012). Calibration was performed for both precursor and product ions using 1-pmol GluFib (Glu1-Fibrinopeptide B) standard peptide with the sequence EGVNDNEEGFFSAR and the monoisotopic doubly-charged peak with m/z of 785.84. The raw data were processed using ProteinLynx Global Server (PLGS, version 2.4) software. The following parameters were used: background subtraction of polynomial order 5 adaptive with a threshold of 30%, two smoothings with a window of three channels in Savitzky-Golay mode, and centroid calculation of top 80% of peaks based on a minimum peak width of 4 channels at half height. The resulting pkl files were submitted for database search and protein identification to the in-house Mascot server (http://www.matrixscience.com, Matrix Science, London, UK) for database search using the following parameters: databases from NCBI (bacteria and viruses), parent mass error of 0.5 Da with 1 13C, product ion error of 0.8 Da, and enzyme used: trypsin, three missed cleavages, propionamide as cysteine fixed modification, and methionine oxidized as variable modification. To identify the false negative results, we used additional parameters such as different databases or organisms, a narrower error window for the parent mass error (1.2 and then 0.2 Da) and for the product ion error (0.6 Da), and up to two missed cleavage sites for trypsin. In addition, the pkl files were also searched against in-house PLGS database version 2.4 (http://www.waters.com) using searching parameters similar to the ones used for Mascot search. The Mascot and PLGS database search provided a list of proteins for each gel band. To eliminate false positive results, for the proteins identified by either one peptide or a mascot score lower than 25, we verified the MS/MS spectra that led to identification of a protein. Additionally, image analysis was performed with the use of the DECODON Delta 2D software, version 4.0 (DECODON), which is based on the dual-channel image analysis technique (Bernhardt et al. 1999).
Extraction and quantification of endogenous metabolites
Samples for endogenous metabolite analyses were prepared according to the procedure described for bacteriophage infection. Following 30-min incubation at 30 °C with tested bacteriophage, 1 × 109 bacterial cells were harvested by centrifugation (2000 × g, 10 min, 4 °C). To release metabolites from E. coli cells, the cold methanol extraction method (Maharjan and Ferenci 2003) was used with some modifications. Briefly, the obtained pellet was suspended in 1 ml of 50% cold (−20 °C) methanol (Merck, Darmstadt, Germany). After rapid mixing, the sample was frozen in liquid nitrogen, and then thawed on a dry ice. The freeze-thaw cycle was performed 3 times in order to permeabilize the cells, and then the mixture was centrifuged (16,000 × g, 10 min, 4 °C). The supernatant was transferred to a new tube and stored in dry ice. The pellet was suspended in 0.5 ml of 50% cold (−20 °C) methanol, intensively mixed, frozen in liquid nitrogen, and then thawed on a dry ice. The procedure of freeze-thawing was repeated 3 times. Following the centrifugation (16,000 × g, 10 min, 4 °C), the first and second extracts were combined in a new tube. Finally, the samples were concentrated for 4–6 h in the vacuum centrifugation at 30 °C to approximately 40 μl. Finally, samples from three biological replicates were pooled into two groups.
Absolute IDQ® p180 kit plates (Biocrates Life Science, Innsbruck, Austria) were used for the quantification of amino acids, biogenic amines, acylcarnitines, and hexoses in prepared samples. The analysis has been carried out commercially by Biocrates Life Science (Innsbruck, Austria). The fully automated assay was based on phenylisothiocyanate (PITC) derivatization in the presence of internal standards, followed by flow injection analysis-tandem mass spectrometry (FIA-MS/MS; for the analysis of acylcarnitines and hexoses) and liquid chromatography-tandem mass spectrometry (LC-MS/MS; for the analysis of amino acids and biogenic amines) using 4000 QTRAP® (AB Sciex, Darmstadt, Germany) and a Xevo TQ-S micro (Waters, Vienna, Austria) instrument with an electrospray ionization (ESI) source. The experimental metabolomic measurement technique was described previously (Ramsay et al. 2005a, 2005b). Data were quantified using a mass spectrometry software (Sciex Analyst® and Waters MassLynx™) and imported into Biocrates MetIDQ™ software for further analysis.
Statistical analyses
Each experiment was repeated 3 or 4 times. Variation among biological replicates was presented as error bars indicating the standard deviation (SD). Comparison of two average values was performed by using Student’s t-test and the significance differences were marked by asterisks as follows: p < 0.05 (*), p < 0.01 (**), or p < 0.001 (***).
Data availability
Row data of transcriptomic (microarray), proteomic, and metabolomic analyses are presented in the supplementary materials, as Table S1, Table S2, and Table S3, respectively.
Results
Effects of the deletion of the exo-xis region on bacteriophage development of the Φ24B phage
Since most of previously published studies on the role of the exo-xis region of the Stx phage Φ24B were conducted in the experimental system with overexpression of the genes from this region (Bloch et al. 2013, 2014), our first attempt was to determine effects of the deletion of this region on the phage development. When comparing the lytic propagation of Φ24B and Φ24BΔexo-xis (Fig. 1) phages, no significant differences could be observed (Fig. 2a). However, lysogenization efficiency was significantly more effective in the absence of the exo-xis region in the phage genome (Fig. 2b). This was corroborated by the higher fraction of bacteria surviving bacteriophage infection when the Δexo-xis mutant of Φ24B was used (Fig. 2c). The effects of the absence of the exo-xis region on the lysogenization or host survival efficiencies could be significantly reduced or completely reversed, respectively, by the presence of this phage genome region in a plasmid in the host cell (Fig. 2b, c). This finding ensured that the observed changes were specific to the exo-xis region rather than arising from any polar or side effects. Thus, we confirmed that the exo-xis region plays a role in directing the phage Φ24B development into the lytic, rather than lysogenic pathway.

Effects of the exo-xis region on bacteriophage Φ24B development. a Intracellular development of Φ24B (open squares) and Φ24BΔexo-xis (closed squares) bacteriophages after infection of E. coli MG1655 [pUC18] at an m.o.i. of 0.05. The presented results are mean values ± SD from three independent experiments. Results are shown as PFU (plaque forming units) per cell. b Efficiency of lyzogenization after infection of E. coli MG1655 strain bearing pUC18 or pUC18_exo-xis_Φ24B plasmid with Φ24B or Φ24BΔexo-xis bacteriophage. The experiment was conducted in the TCM buffer at 30 °C and at m.o.i. of 5. c Survival of host bacteria (%) after infection of E. coli MG1655 strain bearing pUC18 or pUC18_exo-xis_Φ24B plasmid with Φ24B or Φ24BΔexo-xis bacteriophage. The experiment was conducted in the LB medium at 30 °C and an m.o.i. of 5. d Rate of adsorption of Φ24B (open squares) and Φ24BΔexo-xis (closed squares) bacteriophages on E. coli MG1655 bacteria bearing pUC18 plasmid. The experiment was conducted in the LB medium at 30 °C and an m.o.i. of 0.05. In panels b, c, and d, the results are presented as mean values ± SD from three independent biological experiments. Statistical analysis was performed by using Student’s t-test. The significance of differences between compared experimental variants was marked by asterisks: p < 0.01 (**) or p < 0.001 (***)
Intriguingly, the kinetics of adsorption of the Φ24BΔexo-xis phages on the host cells was significantly slower than that of the wild-type Φ24B phage, though finally, similar efficiency of adsorption was reached by both bacteriophages after 20 min (Fig. 2d). This suggests that expression of the genes located between exo and xis might influence the structure of the phage virion, especially structures involved in the interactions with the host cell receptor.
Transcriptomic analyses of the exo-xis effects on expression of phage and host genes
To learn more about the global effects of the exo-xis region on expression of phage and host genes, we have employed transcriptomics. Host cells were infected with either Φ24B or Φ24BΔexo-xis and transcriptomes were compared at different times after infection. Both phage and bacterial genes were analyzed.
Generally, differences between E. coli cells infected with the wild-type phage and that devoid of the exo-xis region involved several hundred genes which were differentially expressed (Fig. 3 and Table S1). Abundance of selected transcripts, revealing significant differences between E. coli cells infected with Φ24B or Φ24BΔexo-xis, was confirmed by using an independent method, RT-qPCR. When analyzing bacteriophage genes, it was evident that those supporting lysogenization, especially cI and cII, were up-regulated in Φ24BΔexo-xis-infected E. coli cells relative to the wild-type phage-infected bacteria (Fig. 4). This was true throughout the infection period. Contrary to these genes, expression of the N gene, coding for one of major regulators promoting lytic development, was down-regulated in the case of the Φ24BΔexo-xis phage (Fig. 4). These results suggest the mechanism of the exo-xis region-mediated promotion of the lytic development, namely, negative control of the lysogenization-specific genes and positive control of the expression of those involved in the lytic development. Summary of the transcriptomic analyses of the phage genes is shown in Table 3.

Bacterial and phage genes which expression (measured as transcript levels) was significantly changed (FDR < 0.05 and p < 0.05) in E. coli MG1655 infected with phage Φ24BΔexo-xis relative to host bacteria infected with wild-type phage Φ24B, as revealed by transcriptomic (microarray) analysis at time 30 min after infection

Expression patterns of selected genes of phage Φ24B (open columns) and Φ24B∆exo-xis (closed columns) infecting E. coli MG1655 host at 30 °C, assessed by RT-qPCR analysis. Levels of transcripts corresponding to particular genes were determined at following times after infection: 7.5, 15, 30, 45, or 60 min. The presented results are mean values from three independent experiments with error bars indicating SD
When analyzing transcripts of bacterial genes after infection with Φ24B or Φ24BΔexo-xis, it was interesting that those coding for chaperone proteins (DnaK, DnaJ, GrpE, GroELS) were down-regulated in the absence of the exo-xis region (Fig. 5). Since chaperone proteins are required for replication of lambdoid phage DNA and assembly of the progeny virions (Węgrzyn et al. 2012), these results suggest another mechanism by which the exo-xis region may stimulate lytic development, namely, supporting expression of the molecular chaperone genes. Interestingly, other host genes modulated by the exo-xis region were those from the gad and nrf operons. They are involved in the survival of cells under low pH and anaerobic conditions, respectively. These functions are especially important for bacteriophages which lysogenize bacterial cells, as being prophages they are totally dependent on the host survival under various conditions. A summary of the transcriptomic analyses of host genes after infection with Φ24B or Φ24BΔexo-xis is shown in Table 4.

Expression patterns of selected bacterial genes after infection of the E. coli MG1655 host with Φ24B (open columns) and Φ24B∆exo-xis (closed columns) bacteriophages 30 °C, assessed by RT-qPCR analysis. Levels of transcripts corresponding to particular genes were determined at following times after infection: 7.5, 15, 30, 45, or 60 min. The presented results are mean values from three independent experiments with error bars indicating SD
As mentioned above, among several hundred genes whose expressions were changed as revealed by the microarray analysis (Fig. 3), some were selected for more detailed analysis (Figs. 4 and 5) on the basis of the specific fold-change values and potential importance for bacteriophage development and host survival. To estimate general reliability of the transcriptomic data and validity of the choice of the genes for more detailed studies, we have confirmed that results of the gene expression microarray analyses were compatible with those obtained in reverse transcriptase quantitative PCR (RT-qPCR) experiments (Fig. 6). This indicated reliability of the analyzed transcriptomic results.

Comparison of changes in levels of selected bacterial (a) and phage (b) transcripts in E. coli MG1655 infected with phage Φ24BΔexo-xis, assessed by the microarray technique (open columns) and the RT-qPCR (closed columns) method, relative to host bacteria infected with wild-type phage Φ24B at time 30 min after infection
Proteomic analyses of the exo-xis effects on levels of proteins in host cells
Levels of proteins in E. coli cells infected with either Φ24B or Φ24BΔexo-xis were examined in classical proteomic experiments. Significant changes in intensities of specific spots after two-dimensional gel electrophoresis were evident as summarized in Fig. 7 and Table S2, and exemplified in Fig. 8. Identification of proteins included in specific spots visualized in Fig. 8 and their quantitative analyses are shown in Table 5.

Number of proteins which levels were significantly increased (red columns) or decreased (blue columns) in E. coli MG1655 infected with phage Φ24BΔexo-xis relative to host bacteria infected with wild-type phage Φ24B at time 30 min after infection

Proteins occurring at different levels in E. coli MG1655 infected phage Φ24BΔexo-xis and the wild-type phage Φ24B. Enlarged images of 2D-PAGE fragments show a significant increase (red) or decreases (blue) in protein levels between the cells infected with investigated bacteriophages at time 30 min after infection. Protein spots of note are indicated with green line circles
These analyses confirmed that the exo-xis region has significant effects on expression of genes in the phage-infected host cells at the level of gene products. The proteins which levels were affected by this region included, but were not limited to, those involved in the development of lambdoid bacteriophages, like molecular chaperones, type I restriction-modification system methyltransferase, RecA, and peptidase E. Modulation of levels of the RecA protein by the exo-xis region might be of particular interest, as this protein is crucial for induction of lambdoid prophages (for a review, see Węgrzyn et al. 2012). Upon binding to single-stranded DNA regions (often appearing as a result of DNA damage), RecA is transformed to its active form, called RecA*, which causes the autocleavage of the phage-encoded cI repressor. This results in de-repression of phage promoters, prophage induction, and subsequent lytic development (Węgrzyn et al. 2012). Therefore, these results corroborate the proposal that the exo-xis region has a role in the regulation of phage development in response to various environmental conditions. Similarly, changes in levels of molecular chaperones in response to the presence/absence of the exo-xis region further support the above hypothesis on the biological role of this phage genome region.
Metabolomic analyses of the exo-xis effects on levels of metabolites in host cells
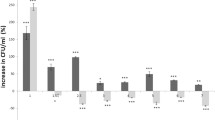
To test effects of the exo-xis region on the cellular metabolism of host cells infected with either Φ24B or Φ24BΔexo-xis, metabolomic analyses were performed (Table S3). The most striking difference between Φ24B- and Φ24BΔexo-xis-infected E. coli was significantly lowered levels of many amin acids and several biogenic amines in the latter host (Fig. 9a). This strongly suggest that the exo-xis region is required to maintain production of amino acids in infected cells which should facilitate effective synthesis of phage-encoded proteins during the lytic development. Interestingly, levels of acylcarnitines and (Fig. 9b) and hexoses (Fig. 9c) were also decreased in Φ24BΔexo-xis-infected cells relative to those infected with the wild-type phage.

Changes in the levels of bacterial metabolites in E. coli MG1655 infected with phage Φ24BΔexo-xis relative to host bacteria infected with wild-type phage Φ24B at time 30 min after infection. Analyses of amino acids and biogenic amines (a), acylcarnitines (b), and hexoses (c) are demonstrated
The results of metabolomic analyses suggest that the exo-xis region of Φ24B supports the bacteriophage lytic development also by stimulating expression of a battery of genes involved in production of compounds which should support effective production of progeny virions, like amino acids and hexoses. This might be another mechanism by which genes included in the investigated region of the Φ24B phage ensure efficient lytic development of this virus.
Discussion
Previous studies suggested that genes from the exo-xis region of lambdoid bacteriophage genome may influence lysogenization and prophage induction processes (Bloch et al. 2013, 2014). Further studies led to the hypothesis that proteins encoded in the exo-xis region play roles in transcription, possibly through regulating RNA polymerase (Kwan et al. 2013; Dydecka et al. 2017, 2018, 2020; Zdrojewska et al. 2019; Tong et al. 2020; Donaldson 2021). In this light, we investigated global effects of the exo-xis region on gene expression and metabolism in E. coli cells infected with the Stx phage Φ24B.
Experiments with the Φ24B bacteriophage devoid of the exo-xis region indicated that the mutant virus preferred lysogenization rather than the lytic development (Fig. 2). These results corroborate the proposal that genes included in this region are important for replication of bacteriophages and production of phage progeny. Interestingly, mutant phages were less effective in adsorption to the host cells, suggesting that the exo-xis region ensures proper assembly of the progeny virions. Although the mechanism of this phenomenon remains to be elucidated, transcriptomic and proteomic studies, performed in this work, provide an interesting possibility. Namely, expression of genes coding for molecular chaperones (DnaK, DnaJ, GrpE, GroELS) was down-regulated in the Φ24BΔexo-xis-infected cells relative to those infected with the wild-type virus. Since chaperones (especially GroELS) are required to assembly phage virions properly (Węgrzyn et al. 2012), their deficit might negatively influence this process, resulting in production of partially defective viral particles. Intriguingly, it was demonstrated that Stx phages are significantly more sensitive to UV irradiation than bacteriophage λ which arose from formation of less stable virions (Bloch et al. 2015). Whether this phenomenon is influenced by the exo-xis functions remains to be tested.
The general conclusion from the transcriptomic, proteomic, and metabolomic studies performed in this work is that the exo-xis region facilitates lytic development of the Φ24B bacteriophage by regulating expression of a battery of phage and host genes involved in the lysis vs. lysogenization decision (especially phage genes cI, cII, and N), coding for molecular chaperones, and required for metabolism of amino acids, hexoses, and some other compounds, like biogenic amines and acyl carnitines. This corroborates recent predictions about the regulation of gene expression by products of genes included in the exo-xis region, based on use of the AlphaFold and RoseTTAFold machine learning algorithms (Donaldson 2021).
The effects of bacteriophage genes or their products on expression of host genes have been reported previously, also for Stx bacteriophages, though such works were not numerous. Microarray analyses of non-lysogenic E. coli and those lysogenic by the Stx phage ΦMin27 indicated that prophage-located genes caused changes in expression of various host genes, leading to enhanced host resistance to low-pH conditions and to more effective motility (Su et al. 2010). Similar results were also obtained in studies with the Φ24B bacteriophage, where transcriptomic analyses identified enhanced expression of the gad operon, involved in the bacteria acid resistance (Veses-Garcia et al. 2015). More recent transcriptomic studies with hosts bearing different Stx prophages (either ϕO104 or ϕPA8) demonstrated significant changes in expression of E. coli genes encoding enzymes involved in source utilization (Berger et al. 2019). Specifically, lysogenic cells revealed enhanced expression of genes which products are involved in mixed acid fermentation, whereas expression of those encoding enzymes of the Krebs cycle, NADH dehydrogenase I, and polypeptides required for the transport and assimilation of carbon sources was impaired, relative to non-lysogenic E. coli (Berger et al. 2019). Hence, it appears that the presence of the Stx prophage causes inhibition of aerobic metabolism while activation of mixed acid fermentation.
Interestingly, the gad operon expression was demonstrated in this work to be modulated by the exo-xis region of this phage (Fig. 5). Moreover, another operon which expression was found to be influenced by exo-xis is nrf (Fig. 5). Expression of this operon enhances bacterial growth under anaerobic conditions, and a promoter of this operon has been demonstrated previously to be more active when E. coli is lysogenic with an Stx phage (Godfrey et al. 2017). Although, to our knowledge, effects of Stx phages on host metabolome were not demonstrated previously, it is worth noting that phage-specific metabolic alterations were reported for six different bacteriophages infecting Pseudomonas aeruginosa (De Smet et al. 2016).
In conclusion, the exo-xis region of the Φ24B bacteriophage genome supports lytic development of the bacteriophage over lysogenization through significantly modulating expression of a battery of phage and host genes. The phage-mediated changes include changes in levels of molecular chaperones, proteins involved in acid tolerance by the host cells, and proteins facilitating E. coli growth under anaerobic conditions. Moreover, the exo-xis region is required for maintaining relatively high levels of amino acids, biogenic amines, acylcarnitines, and hexoses. Therefore, the exo-xis region, playing an important role in the lysis vs. lysogenization decision and the phage lytic development, might be considered as a potential target for anti-STEC/EHEC drugs, as impairment of the propagation of Stx phages should reduce virulence of the E. coli strains producing Shiga toxins due to inhibition of expression of genes coding for these toxins and located in genomes of these phages.
References
Al Qabili DMA, Aboueisha AM, Ibrahim GA, Youssef AI, El-Mahallawy HS (2022) Virulence and antimicrobial-resistance of shiga toxin-producing E. coli (STEC) isolated from edible shellfish and its public health significance. Arch Microbiol 204:510. https://doi.org/10.1007/s00203-022-03114-2
Allison HE, Sergeant MJ, James CE, Saunders JR, Smith DL, Sharp RJ, Marks TS, McCarthy AJ (2003) Immunity profiles of wild-type and recombinant Shiga-like toxin-encoding bacteriophages and characterization of novel double lysogens. Infect Immun 71:3409–3418. https://doi.org/10.1128/iai.71.6.3409-3418.2003
Auvray F, Bièche-Terrier C, Um MM, Dupouy V, Nzuzi N, David L, Allais L, Drouet M, Oswald E, Bibbal D, Brugère H (2023) Prevalence and characterization of the seven major serotypes of Shiga toxin-producing Escherichia coli (STEC) in veal calves slaughtered in France. Vet Microbiol 282:109754. https://doi.org/10.1016/j.vetmic.2023.109754
Berger P, Kouzel IU, Berger M, Haarmann N, Dobrindt U, Koudelka GB, Mellmann A (2019) Carriage of Shiga toxin phage profoundly affects Escherichia coli gene expression and carbon source utilization. BMC Genomics 20:504. https://doi.org/10.1186/s12864-019-5892-x
Bernhardt J, Buttner K, Scharf C, Hecker M (1999) Dual channel imaging of two-dimensional electropherograms in Bacillus subtilis. Electrophoresis 20:2225–2240. https://doi.org/10.1002/(SICI)1522-2683(19990801)20:11%3C2225::AID-ELPS2225%3E3.0.CO;2-8
Besser RE, Griffin PM, Slutsker L (1999) Escherichia coli O157:H7 gastroenteritis and the hemolytic uremic syndrome: an emerging infectious disease. Annu Rev Med 50:355–367. https://doi.org/10.1146/annurev.med.50.1.355
Bloch S, Nejman-Faleńczyk B, Łoś JM, Barańska S, Łepek K, Felczykowska A, Łoś M, Węgrzyn G, Węgrzyn A (2013) Genes from the exo-xis region of λ and Shiga toxin-converting bacteriophages influence lysogenization and prophage induction. Arch Microbiol 195:693–703. https://doi.org/10.1007/s00203-013-0920-8
Bloch S, Nejman-Faleńczyk B, Dydecka A, Łoś JM, Felczykowska A, Węgrzyn A, Węgrzyn G (2014) Different expression patterns of genes from the exo-xis region of bacteriophage λ and Shiga toxin-converting bacteriophage Ф24B following infection or prophage induction in Escherichia coli. PLoS One 9:e108233. https://doi.org/10.1371/journal.pone.0108233
Bloch S, Nejman-Faleńczyk B, Topka G, Dydecka A, Licznerska K, Narajczyk M, Necel A, Węgrzyn A, Węgrzyn G (2015) UV-sensitivity of Shiga toxin-converting bacteriophage virions Φ24B, 933W, P22, P27 and P32. Toxins 7:3727–3739. https://doi.org/10.3390/toxins7093727
Bugarel M, Martin A, Fach P, Beutin L (2011) Virulence gene profiling of enterohemorrhagic (EHEC) and enteropathogenic (EPEC) Escherichia coli strains: a basis for molecular risk assessment of typical and atypical EPEC strains. BMC Microbiol 11:142. https://doi.org/10.1186/1471-2180-11-142
Caldwell BJ, Bell CE (2019) Structure and mechanism of the Red recombination system of bacteriophage λ. Prog Biophys Mol Biol 147:33–46. https://doi.org/10.1016/j.pbiomolbio.2019.03.005
Casjens SR, Hendrix RW (2015) Bacteriophage lambda: early pioneer and still relevant. Virology 479–480:310–330. https://doi.org/10.1016/j.virol.2015.02.010
Darie CC, Deinhardt K, Zhang G, Cardasis HS, Chao MV, Neubert TA (2011) Identifying transient protein-protein interactions in EphB2 signaling by blue native PAGE and mass spectrometry. Proteomics 11:4514–4528. https://doi.org/10.1002/pmic.201000819
De Smet J, Zimmermann M, Kogadeeva M, Ceyssens PJ, Vermaelen W, Blasdel B, Bin Jang H, Sauer U, Lavigne R (2016) High coverage metabolomics analysis reveals phage-specific alterations to Pseudomonas aeruginosa physiology during infection. ISME J 10:1823–1835. https://doi.org/10.1038/ismej.2016.3
Donaldson LW (2021) Molecular modeling the proteins from the exo-xis region of lambda and shigatoxigenic bacteriophages. Antibiotics 10:1282. https://doi.org/10.3390/antibiotics10111282
Dydecka A, Bloch S, Rizvi A, Perez S, Nejman-Faleńczyk B, Topka G, Gąsior T, Necel A, Węgrzyn G, Donaldson LW, Węgrzyn A (2017) Bad phages in good bacteria: role of the mysterious orf63 of λ and Shiga toxin-converting Φ24B bacteriophages. Front Microbiol 8:1618. https://doi.org/10.3389/fmicb.2017.01618
Dydecka A, Nejman-Faleńczyk B, Bloch S, Topka G, Necel A, Donaldson LW, Węgrzyn G, Węgrzyn A (2018) Roles of orf60a and orf61 in development of bacteriophages λ and Φ24B. Viruses 10:553. https://doi.org/10.3390/v10100553
Dydecka A, Bloch S, Necel A, Topka G, Węgrzyn A, Tong J, Donaldson LW, Węgrzyn G, Nejman-Faleńczyk B (2020) The ea22 gene of lambdoid phages: preserved prolysogenic function despite of high sequence diversity. Virus Genes 56:266–277. https://doi.org/10.1007/s11262-020-01734-8
Gamage SD, Patton AK, Hanson JF, Weiss AA (2004) Diversity and host range of Shiga toxin-encoding phage. Infect Immun 72:7131–7139. https://doi.org/10.1128/iai.72.12.7131-7139.2004
Godfrey RE, Lee DJ, Busby SJW, Browning DF (2017) Regulation of nrf operon expression in pathogenic enteric bacteria: sequence divergence reveals new regulatory complexity. Mol Microbiol 104:580–594. https://doi.org/10.1111/mmi.13647
Guernec A, Robichaud-Rincon P, Saucier L (2013) Whole-genome transcriptional analysis of Escherichia coli during heat inactivation processes related to industrial cooking. Appl Environ Microbiol 79:4940–4950. https://doi.org/10.1128/AEM.00958-13
Jensen KF (1993) The Escherichia coli K-12 “wild types” W3110 and MG1655 have an rph frameshift mutation that leads to pyrimidine starvation due to low pyrE expression levels. J Bacteriol 175:3401–3407. https://doi.org/10.1128/jb.175.11.3401-3407.1993
Kaper JB, Nataro JP, Mobley HL (2004) Pathogenic Escherichia coli. Nat Rev Microbiol 2:123–140. https://doi.org/10.1038/nrmicro818
Kędzierska B, Szambowska A, Herman-Antosiewicz A, Lee DJ, Busby SJW, Węgrzyn G, Thomas MS (2007) The C-terminal domain of the Escherichia coli RNA polymerase subunit plays a role in the CI-dependent activation of the bacteriophage pM promoter. Nucleic Acids Res 35:2311–2320. https://doi.org/10.1093/nar/gkm123
Kimmitt PT, Harwood CR, Barer MR (1999) Induction of type 2 Shiga toxin synthesis in Escherichia coli O157 by 4-quinolones. Lancet 353:1588–1589. https://doi.org/10.1016/s0140-6736(99)00621-2
Kimmitt PT, Harwood CR, Barer MR (2000) Toxin gene expression by Shiga toxin-producing Escherichia coli: the role of antibiotics and the bacterial SOS response. Emerg Infect Dis 6:458–465. https://doi.org/10.3201/eid0605.000503
Kourilsky P, Knapp A (1974) Lysogenization by bacteriophage λ.: III. Multiplicity dependent phenomena occurring upon infection by λ. Biochimie 56:1517–1523. https://doi.org/10.1016/S0300-9084(75)80275-6
Kwan JJ, Smirnova E, Khazai S, Evanics F, Maxwell KL, Donaldson LW (2013) The solution structures of two prophage homologues of the bacteriophage λ Ea8.5 protein reveal a newly discovered hybrid homeodomain/zinc-finger fold. Biochemistry 52:3612–3614. https://doi.org/10.1021/bi400543w
Licznerska K, Dydecka A, Bloch S, Topka G, Nejman-Faleńczyk B, Węgrzyn A, Węgrzyn G (2016) The role of the exo-xis region in oxidative stress-mediated induction of Shiga toxin-converting prophages. Oxid Med Cell Longev 2016:8453135. https://doi.org/10.1155/2016/8453135
Łoś JM, Łoś M, Wegrzyn A, Wegrzyn G (2008) Role of the bacteriophage λ exo-xis region in the virus development. Folia Microbiol 53:443–450. https://doi.org/10.1007/s12223-008-0068-0
Łoś JM, Łoś M, Węgrzyn G, Węgrzyn A (2009) Differential efficiency of induction of various lambdoid prophages responsible for production of Shiga toxins in response to different induction agents. Microb Pathog 47:289–298. https://doi.org/10.1016/j.micpath.2009.09.006
Łoś JM, Łoś M, Węgrzyn A, Węgrzyn G (2010) Hydrogen peroxide-mediated induction of the Shiga toxin-converting lambdoid prophage ST2-8624 in E. coli O157:H7. FEMS Immunol Med Microbiol 58:322–329. https://doi.org/10.1111/j.1574-695x.2009.00644.x
Maharjan RP, Ferenci T (2003) Global metabolite analysis: the influence of extraction methodology on metabolome profiles of Escherichia coli. Anal Biochem 313:145–154. https://doi.org/10.1016/s0003-2697(02)00536-5
Matussek A, Mernelius S, Chromek M, Zhang J, Frykman A, Hansson S, Georgieva V, Xiong Y, Bai X (2023) Genome-wide association study of hemolytic uremic syndrome causing Shiga toxin-producing Escherichia coli from Sweden, 1994-2018. Eur J Clin Microbiol Infect Dis 42:771779. https://doi.org/10.1007/s10096-023-04600-1
Mizutani S, Nakazono N, Sugino Y (1999) The so-called chromosomal verotoxin genes are actually carried by defective prophages. DNA Res 6:141–143. https://doi.org/10.1093/dnares/6.2.141
Nejman B, Łoś JM, Łoś M, Węgrzyn G, Węgrzyn A (2009) Plasmids derived from lambdoid bacteriophages as models for studying replication of mobile genetic elements responsible for the production of Shiga toxins by pathogenic Escherichia coli strains. J Mol Microbiol Biotechnol 17:211–220. https://doi.org/10.1159/000242447
Nejman B, Nadratowska-Wesołowska B, Szalewska-Pałasz A, Węgrzyn A, Węgrzyn G (2011) Replication of plasmids derived from Shiga toxin-converting bacteriophages in starved E. coli. Microbiology 157:220–233. https://doi.org/10.1099/mic.0.042820-0
Ptashne M (2004) Genetic switch: phage lambda revisited, 2nd edn. Cold Spring Harbor Laboratory Press, New York, USA
Ramsay SL, Stoeggl WM, Weinberger KM, Graber A, Guggenbichler W (2005a) Apparatus for analyzing a metabolite profile. EP 1:897014B1
Ramsay SL, Stoeggl WM, Weinberger KM, Graber A, Guggenbichler W (2005b) Device for quantitative analysis of metabolite profile. EP 1:875 401 B1
Rodwell EV, Simpson A, Chan YW, Godbole G, McCarthy ND, Jenkins C (2023) The epidemiology of Shiga toxin-producing Escherichia coli O26:H11 (clonal complex 29) in England, 2014-2021. J Infect 86:552–562. https://doi.org/10.1016/j.jinf.2023.04.006
Schmidt H (2001) Shiga-toxin-converting bacteriophages. Res Microbiol 152:687–695. https://doi.org/10.1016/S0923-2508(01)01249-9
Sergueev K, Court D, Reaves L, Austin S (2002) E. coli cell-cycle regulation by bacteriophage λ. J Mol Biol 324:297–307. https://doi.org/10.1016/s0022-2836(02)01037-9
Serna A, Boedeker EC (2008) Pathogenesis and treatment of Shiga toxin-producing Escherichia coli infections. Curr Opin Gastroenterol 24:38–47. https://doi.org/10.1097/mog.0b013e3282f2dfb8
Shevchenko A, Wilm M, Vorm O, Mann M (1996) Mass spectrometric sequencing of proteins silver-stained polyacrylamide gels. Anal Chem 68:850–358. https://doi.org/10.1021/ac950914h
Sokolowska I, Dorobantu C, Woods AG, Macovei A, Branza-Nichita N, Darie CC (2012) Proteomic analysis of plasma membranes isolated from undifferentiated and differentiated HepaRG cells. Proteome Sci 10:47. https://doi.org/10.1186/1477-5956-10-47
Su LK, Lu CP, Wang Y, Cao DM, Sun JH, Yan YX (2010) Lysogenic infection of a Shiga toxin 2-converting bacteriophage changes host gene expression, enhances host acid resistance and motility. Mol Biol 44:54–66
Svendsen AT, Nielsen HL, Bytzer P, Coia JE, Engberg J, Holt HM, Lemming L, Lomborg S, Marmolin ES, Olesen BS, Andersen LP, Ethelberg S, Engsbro AL (2023) The incidence of laboratory-confirmed cases of enteric pathogens in Denmark 2018: a national observational study. Infect Dis (Lond) 55:340–350. https://doi.org/10.1080/23744235.2023.2183253
Tong J, Nejman-Faleńczyk B, Bloch S, Węgrzyn A, Węgrzyn G, Donaldson LW (2020) Ea22 proteins from lambda and Shiga toxin-producing bacteriophages balance structural diversity with functional similarity. ACS Omega 5:12236–12244. https://doi.org/10.1021/acsomega.0c00894
Veses-Garcia M, Liu X, Rigden DJ, Kenny JG, McCarthy AJ, Allison HE (2015) Transcriptomic analysis of Shiga-toxigenic bacteriophage carriage reveals a profound regulatory effect on acid resistance in Escherichia coli. Appl Environ Microbiol 81:8118–8125. https://doi.org/10.1128/aem.02034-15
Waldor MK, Friedman DI (2005) Phage regulatory circuits and virulence gene expression. Curr Opin Microbiol 8:459–465. https://doi.org/10.1016/j.mib.2005.06.001
Wagner MK, Neely MN, Zhang X, Acheson DWK, Waldor MK, Friedman DI (2001a) Role for a phage promoter in Shiga toxin 2 expression from pathogenic Escherichia coli strain. J Bacteriol 183:2081–2085. https://doi.org/10.1128/jb.183.6.2081-2085.2001
Wagner PL, Acheson DW, Waldor MK (2001b) Human neutrophils and their products induce Shiga toxin production by enterohemorrhagic Escherichia coli. Infect Immun 69:1934–1937. https://doi.org/10.1128/iai.69.3.1934-1937.2001
Wagner PL, Livny J, Neely MN, David WK, Acheson DWK, Friedman DI, Waldor MK (2002) Bacteriophage control of Shiga toxin 1 production and release by E. coli. Mol Microbiol 44:957–970. https://doi.org/10.1046/j.1365-2958.2002.02950.x
Węgrzyn G, Węgrzyn A (2005) Genetic switches during bacteriophage λ development. Prog Nucleic Acid Res Mol Biol 79:1–48. https://doi.org/10.1016/s0079-6603(04)79001-7
Węgrzyn G, Licznerska K, Węgrzyn A (2012) Phage λ — new insights into regulatory circuits. Adv Virus Res 82:155–178. https://doi.org/10.1016/b978-0-12-394621-8.00016-9
Zdrojewska K, Dydecka A, Nejman-Faleńczyk B, Topka G, Necel A, Węgrzyn A, Węgrzyn G, Bloch S (2019) Role of orf73 in the development of lambdoid bacteriophages during infection of the Escherichia coli host. Acta Biochim Pol 66:589–596. https://doi.org/10.18388/abp.2019_2886
Zhao M, Gao B, Wen A, Feng Y, Lu YQ (2023) Structural basis of λCII-dependent transcription activation. Structure S0969–2126(23):00164–00168. https://doi.org/10.1016/j.str.2023.05.008
Acknowledgements
The authors thank Tomasz Gąsior for his assistance at the early stage of this project.
Funding
This work was supported by National Science Center (Poland) within project grants no. 2013/09/B/NZ2/02366 to A. W. and 2018/02/X/NZ1/02680 to B. N.-F.
Author information
Authors and Affiliations
Contributions
Conceptualization: Sylwia Bloch, Bożena Nejman-Faleńczyk, Alicja Węgrzyn, Grzegorz Węgrzyn; investigation: Sylwia Bloch, Bożena Nejman-Faleńczyk, Katarzyna Licznerska, Aleksandra Dydecka, Gracja Topka-Bielecka, Agnieszka Necel; methodology: Sylwia Bloch, Bożena Nejman-Faleńczyk, Alicja Węgrzyn, Grzegorz Węgrzyn; visualization: Sylwia Bloch, Bożena Nejmna-Faleńczyk, Agnieszka Necel; funding acquisition: Bożena Nejmna-Faleńczyk, Alicja Węgrzyn; project administration: Bożena Nejmna-Faleńczyk, Alicja Węgrzyn; supervision: Alicja Węgrzyn, Grzegorz Węgrzyn; writing — original draft preparation: Grzegorz Węgrzyn; writing — review and editing: Sylwia Bloch, Bożena Nejman-Faleńczyk, Alicja Węgrzyn, Grzegorz Węgrzyn.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Communicated by: Agnieszka Szalewska-Palasz
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Bloch, S., Nejman-Faleńczyk, B., Licznerska, K. et al. Complex effects of the exo-xis region of the Shiga toxin-converting bacteriophage Φ24B genome on the phage development and the Escherichia coli host physiology. J Appl Genetics 65, 191–211 (2024). https://doi.org/10.1007/s13353-023-00799-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13353-023-00799-z