
Abstract
Craniosynostosis (CS) refers to the group of craniofacial malformations characterized by the premature closure of one or more cranial sutures. The disorder is clinically and genetically heterogeneous and occurs usually as an isolated trait, but can also be syndromic. In 30–60% of patients, CS is caused by known genetic factors; however, in the rest of the cases, causative molecular lesions remain unknown. In this paper, we report on a sporadic male patient affected by complex CS (metopic and unilateral lambdoid synostosis), muscular hypotonia, psychomotor retardation, and facial dysmorphism. Since a subset of CS results from submicroscopic chromosomal aberrations, we performed array comparative genomic hybridization (array CGH) in order to identify possibly causative copy-number variation. Array CGH followed by breakpoint sequencing revealed a previously unreported de novo 1.26 Mb duplication at chromosome 1q22-q23.1 that encompassed two genes involved in osteoblast differentiation: BGLAP, encoding osteocalcin (OCN), and LMNA, encoding lamin A/C. OCN is a major component of bone extracellular matrix and a marker of osteogenesis, whereas mutations in LMNA cause several genetic disorders called laminopathies, including mandibuloacral dysostosis (MAD) that manifests with low bone mass, severe bone deformities, and delayed closure of the cranial sutures. Since LMNA and BGLAP overexpression promote osteoblast differentiation and calcification, phenotype of our patient may result from misexpression of the genes. Based on our findings, we hypothesize that both LMNA and BGLAP may be implicated in the pathogenesis of CS in humans. However, further studies are needed to establish the exact pathomechanism underlying development of this defect.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Craniosynostosis (CS), the premature fusion of the cranial sutures, occurs in about 1:2000 to 1:2500 live born infants and represents one of the most common congenital craniofacial malformation (Cohen 1979; French et al. 1990; Lajeunie et al. 1995). Premature fusion of one or more sutures leads to the distortions of the skull shape and early closure of the fontanelles, which in approximately 50% of cases results in raised intracranial pressure. Consequently, increased risk for cortex lesion with intellectual disability or visual and hearing impairment is observed (Renier et al. 1982; Morriss-Kay and Wilkie 2005).
CS is a clinically and genetically heterogeneous group of disorders that may either occur in rare syndromic forms or, more frequently, as nonsyndromic isolated trait (Wilkie et al. 2007). The vast majority of CS patients are sporadic; however, 10–15% cases show familial recurrence (Cohen 2002). Syndromic forms comprise over 180 different genetic conditions, in which CS is associated with a broad spectrum of clinical symptoms, including skeletal defects, digital malformations, facial dysmorphism, cardiac and genitourinary defects or other organ abnormalities (Gleeson et al. 2006; OMIM). The most commonly recognized CS syndromes that have well-defined clinical phenotype include Apert (MIM 101200), Crouzon (MIM 123500), Pfeiffer (MIM 101600), Muenke (MIM 602849), Saethre-Chotzen (MIM 101400), Jackson-Weiss (MIM 123150), and Antley-Bixler (MIM 207410) syndromes (Kutkowska-Kazmierczak et al. 2018). The underlying genetic cause of these disorders involves mutations in the FGFR1, FGFR2, FGFR3, and TWIST1 genes (OMIM). Other less frequent disorders result from different mutations in the TCF12, EFNB1, MSX2, ALX3, GLI3, IL11RA, and ERF genes (Jabs et al. 1993; Twigg et al. 2009, 2013; Hurst et al. 2011; Keupp et al. 2013; Sharma et al. 2013; Kutkowska-Kazmierczak et al. 2018). Conversely, little is known about genetic etiology of isolated CS and in the majority of cases the underlying molecular defect remains unidentified. Nonetheless, a couple of studies have demonstrated that complex forms of the disease result from chromosomal microaberrations referred to as copy number variations (CNVs), which may account for up to 10–15% of CS (Stratton et al. 1986; Eshel et al. 2002; Wilkie et al. 2010; Massalska et al. 2014). Lambdoid CS, accounting only for 2–4% of the cases, represents its rarest form with entirely unknown molecular origin. The pathomechanism of CNVs could be explained by either gene dosage effect leading to overexpression or haploinsufficiency of a gene/genes or by cis-regulatory effect that leads to misexpression of a target gene resulting from change of its regulatory landscape (Klopocki et al. 2011).
In this paper, we describe a sporadic male proband affected by complex CS, composed of metopic and lambdoid synostosis, muscular hypotonia, psychomotor retardation, and facial dysmorphism, resulting from a previously unreported de novo 1.26 Mb duplication at chromosome 1q22-q23.1, encompassing two genes involved in osteoblastogenesis: BGLAP encoding osteocalcin (OCN) and LMNA encoding lamin A/C. To our knowledge, this is the first genetic abnormality found in a patient presenting with lambdoid CS and the first report concerning the putative contribution of a CNV affecting BGLAP and LMNA genes to the premature closure of the cranial sutures.
Methods
Array comparative genomic hybridization (array CGH)
Genomic DNA of the index patient and his parents was extracted from peripheral blood leukocytes using standard protocols. Array comparative genomic hybridization (array CGH) was performed with the use of high resolution 1.4 M NimbleGen oligonucleotide array CGH (Roche NimbleGen) according to standard protocols provided by the manufacturers. Analysis was carried out with Deva software (Roche NimbleGen). Analysis settings were as follows: aberration algorithm, ADM-2; threshold, 6.0; window size, 0.2 Mb; filter, five probes, log2ratio = 0.29. The genomic profile was visualized by the SignalMap software (NimbleGen Systems Inc.). The aberrant genomic locus was visualized and analyzed in Cytoscape v3.3.0 (Shannon et al. 2003).
Quantitative real-time PCR (qPCR)
We performed a quantitative real-time PCR (qPCR) in the index patient and his parents in order to confirm array CGH results and to show the inheritance pattern of the identified duplication using ViiA™ 7 Real-Time thermal cycler (Applied Biosystems). The qPCR assay was designed to determine the number of copies within the 1q22-q23.1 locus. Three primer pairs were used to amplify the region of duplication and two primer pairs for the 5′ and 3′ flanking regions. Moreover, we used qPCR assay to narrow down the duplication region prior to breakpoint sequencing. This was carried out with the set of eight primer pairs. All reactions were run in triplicate. The results were normalized to albumin (ALB) and the copy number in each of the analyzed regions was determined by means of comparative DDCt method using healthy control DNA as a calibrator. In order to assure reliability of the assay, we performed sex determination of samples in reference to factor VIII (F8) located on X chromosome. Reaction conditions and primer sequences are available upon request.
Breakpoint sequencing
The exact breakpoints of the rearrangement were determined with the use of polymerase chain reaction (PCR) with primers designed to amplify the DNA fragment spanning the 3′ and 5′ ends of the duplication at chromosome 1q22-q23.1. Sequencing of the PCR product was carried out using dye-terminator chemistry (kit v.3, ABI 3130XL) and run on automated sequencer ABI Prism 3700 DNA Analyzer (Applied Biosystems). Reaction conditions and primer sequences are available upon request.
BGLAP and LMNA genes relative expression
The relative expression level of both BGLAP and LMNA genes in blood was carried out in the proband and five controls by means of comparative DDCt method. Total RNA was extracted from whole blood with the use of PAXgene Blood RNA System (PreAnalytiX), according to standard protocols provided by the manufacturers. One microgram of total RNA was reversely transcribed using random hexamer primer (RevertAid First Strand cDNA Synthesis Kit; ThermoFisher Scientific), according to standard protocols provided by the manufacturers. Quantifications of the target and reference gene in the proband and controls was carried out using ViiA™ 7 Real-Time thermal cycler (Applied Biosystems). For each cDNA sample, all reactions were run in triplicate. Relative expression of BGLAP and LMNA in the proband was normalized to TBP reference transcript and to mean value of five controls. Reaction conditions and primer sequences are available upon request.
Serum levels of bone turnover markers
Biochemical analyses of serum levels of alkaline phosphatase, inorganic phosphate, and total calcium were performed with the use of standard laboratory methods. The serum osteocalcin and intact parathyroid hormone levels were determined using chemiluminescence immunoassay (CLIA) (LIAISON XL; Diasorin and ADIVIA Centaur XP Immunoassay System; Siemens respectively). The results were compared to the laboratory reference ranges.
Results
Clinical report
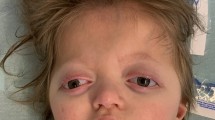
The proband, a 5-month-old male patient of Polish ethnicity, was born by spontaneous delivery after uneventful pregnancy (G1P1) at 38 weeks of gestation to a non-consanguineous and healthy 32-year-old mother and a 33-year-old father. At birth, his weight was 2900 g (3rd–10th percentile), length 57 cm (75th–90th percentile), head circumference 30 cm (below 3rd percentile), and Apgar score was 10. Physical examination after birth showed trigonocephaly and associated facial dysmorphism. Abdominal and transfontanellar ultrasounds performed after birth were unremarkable. Hearing tests and ophthalmologic examinations were carried out during the first week after birth and repeated regularly throughout the first months of life and were normal. The boy was referred to the genetic clinic for diagnosis and first investigated by a clinical geneticist at the age of 5 months. Upon examination, he presented with global developmental delay, severe muscular hypotonia, and craniofacial dysmorphic features comprising prominent metopic ridge, trigonocephaly, high-arched palate, prominent occiput, hypotelorism, shallow orbits, and low-set, posteriorly rotated ears (Fig. 1a, b). His body weight was 5.5 kg (below 3rd percentile). CT scan of the head performed at the age of 7 months showed premature fusion of the metopic suture along with unilateral right-sided lambdoid craniosynostosis (Fig. 1c), as well as thin and hypoplastic corpus callosum, morphological abnormalities of the median brain structures, including abnormal development of the white matter (Fig. 1d), and Chiari malformation (Fig. 1e). 3D modeling of the patient’s skull performed with the use of RadiAnt DICOM Viewer showed posterior plagiocephaly (Fig. 2a) due to prematurely closed right lambdoid suture (Fig. 2c) and prominent metopic ridge due to premature metopic synostosis (Fig. 2a, b), hypotelorism (Fig. 2b), as well as digitate impressions most marked in the bone structures of the right occipito-parietal region (Fig. 2c). Upon neurological assessment at the age of 7 months, the patient presented with psychomotor retardation with global development that amounted to 3 months of age. Conventional chromosomal analysis performed on peripheral blood lymphocytes with a resolution of 550 bands per haploid genome showed normal male karyotype (46,XY).

Facial view of the proband showing craniofacial abnormalities comprising a trigonocephaly with prominent metopic ridge, shallow orbits, proptosis, and hypotelorism and b flat facial profile, prominent occiput, and low-set, posteriorly rotated ears. c CT scan of the head showing premature synostosis of the metopic suture (trigonocephaly) and right lambdoid suture (indicated in red). MS—metopic suture, CS—coronal suture, LS—lambdoid suture. d CT scan of the head (sagittal view) showing thin and hypoplastic corpus callosum (marked with a white arrow) as well as morphological abnormalities of the median brain structures, including abnormal development of the white matter (marked with a yellow arrow). e CT scan of the head (coronal section) showing Chiari malformation; white arrow indicates downward displacement of the right cerebellar tonsil through the foramen magnum

3D modeling of the patient’s skull. a Aerial view presenting asymmetric posterior plagiocephaly and prominent metopic ridge (black arrow) due to premature metopic synostosis. b Frontal view showing prominent metopic ridge (black arrow) and hypotelorism. c Horizontal section presenting prematurely closed right lambdoid suture with digitate impressions (indicated by black arrows) most marked in the right occipito-parietal region
Serum levels of bone turnover markers
Osteocalcin serum level (224 ng/ml) was 3.4-fold higher as compared to the reference range (4.6–65.4 ng/ml). The serum concentrations of alkaline phosphatase (301 U/L), inorganic phosphate (4.6 mg/dl), and total calcium (10.7 mg/dl) were within normal ranges as compared to the reference values (142–335 U/L, 3.3–5.6 mg/dl, 8.8–10.8 mg/dl, respectively), while intact parathyroid hormone was slightly lowered (12.6 pg/ml) in comparison to the reference range (18.5–88.0 pg/ml).
Array comparative genomic hybridization (array CGH)
Array CGH detected a previously unreported duplication at chromosome 1q22-q23.1 (chr1:155927620–157198170; hg19), encompassing 42 protein coding genes, including BGLAP, encoding osteocalcin, and LMNA, encoding lamin A/C (Fig. 3a).

a Interstitial duplication in the index at 1q22-q23.1 encompassing LMNA and BGLAP genes, identified by means of array CGH (Roche NimbleGen). b Breakpoint sequencing results: a 1.26 Mb duplication (chr1:155931219–157187216; hg19), and an insertion of four nucleotides (TTCT) at one of the breakpoints
Quantitative real-time PCR (qPCR) and breakpoint analysis
qPCR confirmed the 1q22-q23.1 duplication in the proband and excluded its presence in both unaffected parents. Subsequent rounds of qPCR narrowed down the region of duplication and allowed for the design of primers for breakpoint sequencing. Sequencing of the breakpoints revealed an insertion of four nucleotides (TTCT) at one of the breakpoints. The exact size of the duplication was 1,255,998 bp (chr1:155931219–157187216; hg19). Breakpoint sequencing results were shown in Fig. 3b.
BGLAP and LMNA relative expression
The analysis of BGLAP and LMNA relative expression performed in blood samples of the proband and five controls revealed 1.8-fold increase of BGLAP expression and 1.4-fold increase of LMNA expression in the proband in reference to the mean value of controls (Fig. 4).

Relative expression levels of BGLAP and LMNA genes in blood samples of the proband and controls indicating 1.8-fold increase of BGLAP expression and 1.4-fold increase of LMNA expression in the proband compared to the mean value of five healthy controls. Error bars represent standard deviation
Discussion
Genetic changes underlying most of the CS types, especially other than coronal synostosis, remain largely unknown. A small proportion of metopic synostosis is caused by 9p22 deletions and point mutations in FREM1 (Vissers et al. 2011). However, most of the cases remain undiagnosed at a molecular level. Complex CS may rarely result from mutations in a recently discovered gene ERF, which encodes for a transcription factor (Twigg et al. 2013). Furthermore, complex forms of the disease may be also caused by chromosomal rearrangements, including small submicroscopic segmental aberrations referred to as copy number variations (CNVs), which are estimated to account for 10–15% of CS (Stratton et al. 1986; Eshel et al. 2002; Wilkie et al. 2010; Massalska et al. 2014). Several interesting examples of CNVs resulting in craniosynostosis include IHH regulatory mutations (Klopocki et al. 2011), RUNX2 duplications (Mefford et al. 2010), and MSX2 duplications (Kariminejad et al. 2009). The fusion of cranial sutures is orchestrated by an interplay between collagen fibers and mesenchymal cells that differentiate into osteoblasts, osteoclasts and osteocytes upon mechanical stress. The molecular origin of CS has been linked to mutations in genes encoding osteoblastogenic proteins that trigger the premature osteogenesis in cranial sutures via different molecular pathways (Katsianou et al. 2016).
In this paper, we describe a previously unreported de novo 1.26 Mb duplication at chromosome 1q22-q23.1 identified in a sporadic male proband presenting with complex CS (metopic and right-sided lambdoid synostosis), muscular hypotonia, psychomotor retardation, and facial dysmorphism. The duplication encompasses 42 protein coding genes, including BGLAP and LMNA, both implicated in osteoblast differentiation, which are most probably involved in the pathogenesis of the clinical phenotype observed in our patient.
BGLAP encodes osteocalcin (OCN), the most abundant non-collagen component of bone extracellular matrix. The protein promotes matrix mineralization by high binding properties for calcium and hydroxyapatite, thus having a pivotal role in osteogenesis (Kruse and Kracht 1986; Sandberg et al. 1993; McGuigan et al. 2010). There are several signal transduction cascades involved in osteogenesis during cranial suture development, including the MAPK/ERK pathway (activated by FGFRs), SMAD signaling network, and Wnt/β-catenin pathway. Hyperactivation of these pathways results in premature closure of sutures leading to craniofacial abnormalities (Katsianou et al. 2016). Two major downstream target proteins for these signaling cascades, Runx2 and Msx2, are required as molecular switches for Bglap expression. The gene’s activity during osteogenesis reflects the stages of osteoblast differentiation. Bglap is transcriptionally repressed in proliferating cells by Msx2, whereas in mature osteoblasts Dlx3, Dlx5, and Runx2 are recruited to initiate the gene transcription. Thus, OCN is considered to be a major marker of mature osteoblasts (Hassan et al. 2004). Of note, studies on rat models demonstrated that Bglap expression level (as well as other bone-associated extracellular matrix molecules) is up-regulated in calvarial dura matter, directly underlying fusion of sutures (Greenwald et al. 2000). To date, genetic abnormalities directly affecting the BGLAP have not been reported to cause premature closure of sutures. However, several studies have determined how mutations in key elements of the aforementioned signal transduction pathways alter the BGLAP expression pattern in osteoblast derived from fused sutures. According to these reports, the gene expression was elevated in patients with Apert syndrome, as well as in mice harboring Fgfr1 Pro250Arg Pfeiffer syndrome mutation, thus confirming the direct correlation between the increased level of osteogenic proteins and the pathological ossification of sutures in both human and mice (Lemonnier et al. 2000, 2001; Zhou et al. 2000). These findings are consistent with the results of our study in which we observed an elevated blood expression of BGLAP and increased serum osteocalcin level in the proband, probably due to the identified duplication. Of note, the serum alkaline phosphatase (ALP) level was within a normal range. Based on our observations, we hypothesize that the CNV results in the general overexpression of BGLAP that may have occurred also locally during osteogenesis in fusing cranial sutures, leading to the craniosynostosis observed in our patient.
Another interesting gene included in the duplication found in our proband was LMNA. LMNA encodes lamin A/C, a nuclear intermediate filament protein that plays a major role in the structural organization of the nucleus and regulation of gene expression via interaction with signaling molecules and transcription factors (Aebi et al. 1986). The role of lamin A/C in osteoblastogenesis is associated with its potential to control the fate of mesenchymal stem cells (MSCs), which may differentiate into either osteoblast or adipocytes. Different mutations in LMNA disrupt the integrity of the nuclear envelope, mostly in the cells subjected to mechanical stress, leading to a group of disorders known as laminopathies (Manilal et al. 1999; Maraldi et al. 2006). Based on clinical features, laminopathies could be organized into four groups: muscle disorders, lipodystrophies, neuropathies, and accelerated aging disorders. The explanation of pleiotropic effect of various mutations in a single gene is attributed to the differences in the molecular mechanisms via which the variants affect the protein function. These include abnormal protein structure, alteration of its charge, or failure of the post-transcriptional processing of LMNA. Unprocessed protein is accumulated in the cells and incorporated into the nuclei where it interferes with the nuclear integrity, resulting in destabilization of the nucleus structure (Worman and Bonne 2007).
The pathomechanism underlying a variety of phenotypes found in human laminopathies has been investigated in both in vitro and in vivo studies. The features associated with LMNA loss-of function mutations arise from primarily affected MSCs and include reduction in muscle mass, defects of bone formation, and craniofacial symptoms, such as delayed closure of sutures as seen in mandibuloacral dysplasia (MAD) and Hutchinson-Gilford progeria (Novelli et al. 2002; De Sandre-Giovannoli et al. 2003; Shen et al. 2003; Yang et al. 2006; Kim et al. 2011). Studies using the spontaneous Lmna mutation (Dhe) mouse, which serves as a naturally occurring animal model for laminopathies, show that the cranial suture tissue that fails to fuse exhibits abnormal nuclear morphology, hypomineralization, and low level of collagen I and III expression (Odgren et al. 2010). Furthermore, the important role of LMNA in the formation of mineralized matrix has been demonstrated in vitro using human osteoblasts and MSCs. Lamin A/C knock-down in these cell lines resulted in impaired osteoblastogenesis, enhanced osteoclast formation, and increased adipogenesis, which was associated with reduced RUNX2, BGLAP, and ALP expression levels, as well as the impaired RUNX2 nuclear binding activity (Akter et al. 2009; Rauner et al. 2009). Consistent with these data, studies using Lmna−/− mice showed decreased bone volume and muscular atrophy that occurred concomitantly with increased bone marrow and inter-myofiber fat infiltration, in the 4-week-old mutants as compared to controls (Tong et al. 2011). Since progressive muscle weakness is a principal sign in a number of laminopathies, including Emery-Dreifuss muscular dystrophy, limb-girdle muscular dystrophy, and Charcot-Marie-Tooth type 2B1 and has been reported in the Lmna−/− mice, muscular hypotonia observed in our patient may also be attributed to the abnormal gene expression and functioning of LMNA. In addition, psychomotor retardation manifested by our proband may result not only from hypotonia and muscular weakness, but also from the impediment of the growth of the patient’s brain. This is often seen in complex forms of CS, even in cases where there is no overt evidence for increased intracranial pressure.
Interestingly, LMNA gene dosage may be also implicated in the pathogenesis of CS, as its overexpression correlates with an elevated level of osteogenic factors including RUNX2 and OCN (Bermeo et al. 2015; Tsukune et al. 2017). Consistently, we demonstrated that LMNA expression measured in peripheral blood of the index patient was elevated by 1.4-fold in comparison with healthy controls. This slight overexpression may contribute to craniosynostosis but is rather inconsistent with muscular hypotonia, as this feature is related to decreased level of LMNA (Tong et al. 2011). Considering a possibility that dosage imbalance of other than LMNA and BGLAP genes could contribute to the observed phenotype, we analyzed the predicted effect of haploinsufficiency (HI) of all protein-coding genes included in the duplication. Out of seven genes with a high (< 10%) or moderate (< 25%) HI score (NES, CCT3, MEF2D, LAMTOR2, ETV3, SSR2, NTRK1), none of them have been linked to craniosynostosis (Huang et al. 2010). A partly overlapping deletion in the 1q22-q23.1 region has been recently described in a patient presenting with intellectual disability and multiple congenital anomalies, and two of the HI genes, NES and MAF2D, were shown to be necessary for normal neuron development in mice (Ikeshima et al. 1995; Park et al. 2010; Aleksiuniene et al. 2018). Therefore, dosage imbalance could possibly contribute to mental retardation observed in the index patient.
In conclusion, we hypothesize that the complex phenotype of our proband, although caused be a novel 1q22-q23.1 duplication, does not result directly from the increased gene dosage of LMNA and BGLAP, but additionally from the change of a regulatory landscape within the duplicated region. As a consequence, misexpression of LMNA and BGLAP in a specific developmental time point during embryogenesis would give rise to the complex CS and muscular hypotonia observed in our patient, although further studies are needed to support this hypothesis.
Change history
25 August 2018
This article was originally published electronically on 29 May 2018 with incorrect copyright line in the Publisher’s internet portal (currently SpringerLink). The copyright line of the article should be “© The Author (s) 2018”.
References
Aebi U, Cohn J, Buhle L, Gerace L (1986) The nuclear lamina is a meshwork of intermediate-type filaments. Nature 323:560–564. https://doi.org/10.1038/323560a0
Akter R, Rivas D, Geneau G et al (2009) Effect of Lamin A/C knockdown on osteoblast differentiation and function. J Bone Min Res 24:283–293. https://doi.org/10.1359/jbmr.081010
Aleksiuniene B, Preiksaitiene E, Morkuniene A et al (2018) A de novo 1q22q23.1 interstitial microdeletion in a girl with intellectual disability and multiple congenital anomalies including congenital heart defect. Cytogenet Genome Res 154:6–11. https://doi.org/10.1159/000486947
Bermeo S, Vidal C, Zhou H, Duque G (2015) Lamin A/C acts as an essential factor in mesenchymal stem cell differentiation through the regulation of the dynamics of the Wnt/β-catenin pathway. J Cell Biochem 116:2344–2353. https://doi.org/10.1002/jcb.25185
Cohen MM (1979) Craniosynostosis and syndromes with craniosynostosis: incidence, genetics, penetrance, variability, and new syndrome updating. Birth Defects Orig Artic Ser 15:13–63
Cohen MM (2002) Malformations of the craniofacial region: evolutionary, embryonic, genetic, and clinical perspectives. Am J Med Genet 115:245–268. https://doi.org/10.1002/ajmg.10982
De Sandre-Giovannoli A, Bernard R, Cau P et al (2003) Lamin a truncation in Hutchinson-Gilford progeria. Science (80-) 300:2055. https://doi.org/10.1126/science.1084125
Eshel G, Lahat E, Reish O, Barr J (2002) Neurodevelopmental and behavioral abnormalities associated with deletion of chromosome 9p. J Child Neurol 17:50–51. https://doi.org/10.1177/088307380201700113
French LR, Jackson IT, Melton LJ (1990) A population-based study of craniosynostosis. J Clin Epidemiol 43:69–73
Greenwald JA, Mehrara BJ, Spector JA et al (2000) Regional differentiation of cranial suture-associated dura mater in vivo and in vitro: implications for suture fusion and patency. J Bone Min Res 15:2413–2430. https://doi.org/10.1359/jbmr.2000.15.12.2413
Hassan MQ, Javed A, Morasso MI et al (2004) Dlx3 transcriptional regulation of osteoblast differentiation: temporal recruitment of Msx2, Dlx3, and Dlx5 homeodomain proteins to chromatin of the osteocalcin gene. Mol Cell Biol 24:9248–9261. https://doi.org/10.1128/MCB.24.20.9248-9261.2004
Huang N, Lee I, Marcotte EM, Hurles ME (2010) Characterising and predicting haploinsufficiency in the human genome. PLoS Genet. https://doi.org/10.1371/journal.pgen.1001154
Hurst JA, Jenkins D, Vasudevan PC et al (2011) Metopic and sagittal synostosis in Greig cephalopolysyndactyly syndrome: five cases with intragenic mutations or complete deletions of GLI3. Eur J Hum Genet 19:757–762. https://doi.org/10.1038/ejhg.2011.13
Ikeshima H, Imai S, Shimoda K et al (1995) Expression of a MADS box gene, MEF2D, in neurons of the mouse central nervous system: implication of its binary function in myogenic and neurogenic cell lineages. Neurosci Lett 200:117–120
Jabs EW, Müller U, Li X et al (1993) A mutation in the homeodomain of the human MSX2 gene in a family affected with autosomal dominant craniosynostosis. Cell 75:443–450
Kariminejad A, Kariminejad R, Tzschach A et al (2009) Craniosynostosis in a patient with 2q37.3 deletion 5q34 duplication: association of extra copy of MSX2 with craniosynostosis. Am J Med Genet A 149A:1544–1549. https://doi.org/10.1002/ajmg.a.32949
Katsianou MA, Adamopoulos C, Vastardis H, Basdra EK (2016) Signaling mechanisms implicated in cranial sutures pathophysiology: Craniosynostosis. BBA Clin 6:165–176. https://doi.org/10.1016/j.bbacli.2016.04.006
Keupp K, Li Y, Vargel I et al (2013) Mutations in the interleukin receptor IL11RA cause autosomal recessive Crouzon-like craniosynostosis. Mol Genet Genomic Med 1:223–237. https://doi.org/10.1002/mgg3.28
Kim HK, Lee JY, Bae EJ et al (2011) Hutchinson-Gilford progeria syndrome with G608G LMNA mutation. J Korean Med Sci 26:1642–1645. https://doi.org/10.3346/jkms.2011.26.12.1642
Klopocki E, Lohan S, Brancati F et al (2011) Copy-number variations involving the IHH locus are associated with syndactyly and craniosynostosis. Am J Hum Genet 88:70–75. https://doi.org/10.1016/j.ajhg.2010.11.006
Kruse K, Kracht U (1986) Evaluation of serum osteocalcin as an index of altered bone metabolism. Eur J Pediatr 145:27–33
Kutkowska-Kazmierczak A, Gos M, Obersztyn E (2018) Craniosynostosis as a clinical and diagnostic problem: molecular pathology and genetic counseling. J Appl Genet 1:1–15. https://doi.org/10.1007/s13353-018-0438-5
Lajeunie E, Le Merrer M, Bonaïti-Pellie C et al (1995) Genetic study of nonsyndromic coronal craniosynostosis. Am J Med Genet 55:500–504. https://doi.org/10.1002/ajmg.1320550422
Lemonnier J, Delannoy P, Hott M et al (2000) The Ser252Trp fibroblast growth factor receptor-2 (FGFR-2) mutation induces PKC-independent downregulation of FGFR-2 associated with premature calvaria osteoblast differentiation. Exp Cell Res 256:158–167. https://doi.org/10.1006/excr.2000.4820
Lemonnier J, Haÿ E, Delannoy P et al (2001) Role of N-cadherin and protein kinase C in osteoblast gene activation induced by the S252W fibroblast growth factor receptor 2 mutation in Apert craniosynostosis. J Bone Min Res 16:832–845. https://doi.org/10.1359/jbmr.2001.16.5.832
Manilal S, Sewry CA, Pereboev A et al (1999) Distribution of emerin and lamins in the heart and implications for Emery-Dreifuss muscular dystrophy. Hum Mol Genet 8:353–359
Maraldi NM, Lattanzi G, Capanni C et al (2006) Laminopathies: a chromatin affair. Adv Enzym Regul 46:33–49. https://doi.org/10.1016/j.advenzreg.2006.01.001
Massalska D, Bijok J, Kucińska-Chahwan A et al (2014) Prenatal diagnosis of craniosynostosis (compound Saethre-Chotzen syndrome phenotype) caused by a de novo complex chromosomal rearrangement (1; 4; 7) with a microdeletion of 7p21.3-7p15.3, including TWIST1 gene--a case report. Ginekol Pol 85:541–544
McGuigan F, Kumar J, Ivaska KK et al (2010) Osteocalcin gene polymorphisms influence concentration of serum osteocalcin and enhance fracture identification. J Bone Min Res 25:1392–1399. https://doi.org/10.1002/jbmr.32
Mefford HC, Shafer N, Antonacci F et al (2010) Copy number variation analysis in single-suture craniosynostosis: multiple rare variants including RUNX2 duplication in two cousins with metopic craniosynostosis. Am J Med Genet A 152A:2203–2210. https://doi.org/10.1002/ajmg.a.33557
Morriss-Kay GM, Wilkie AO (2005) Growth of the normal skull vault and its alteration in craniosynostosis: insights from human genetics and experimental studies. J Anat 207:637–653. https://doi.org/10.1111/j.1469-7580.2005.00475.x
Novelli G, Muchir A, Sangiuolo F et al (2002) Mandibuloacral dysplasia is caused by a mutation in LMNA-encoding Lamin A/C. Am J Hum Genet 71:426–431. https://doi.org/10.1086/341908
Odgren PR, Pratt CH, Mackay CA et al (2010) Disheveled hair and ear (Dhe), a spontaneous mouse Lmna mutation modeling human laminopathies. PLoS One 5:e9959. https://doi.org/10.1371/journal.pone.0009959
Park D, Xiang AP, Mao FF et al (2010) Nestin is required for the proper self-renewal of neural stem cells. Stem Cells 28:2162–2171. https://doi.org/10.1002/stem.541
Rauner M, Sipos W, Goettsch C et al (2009) Inhibition of Lamin A/C attenuates osteoblast differentiation and enhances RANKL-dependent osteoclastogenesis. J Bone Min Res 24:78–86. https://doi.org/10.1359/jbmr.080902
Renier D, Sainte-Rose C, Marchac D, Hirsch JF (1982) Intracranial pressure in craniostenosis. J Neurosurg 57:370–377. https://doi.org/10.3171/jns.1982.57.3.0370
Sandberg MM, Aro HT, Vuorio EI (1993) Gene expression during bone repair. Clin Orthop Relat Res:292–312
Shannon P, Markiel A, Ozier O et al (2003) Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res 13:2498–2504. https://doi.org/10.1101/gr.1239303
Sharma VP, Fenwick AL, Brockop MS et al (2013) Mutations in TCF12, encoding a basic helix-loop-helix partner of TWIST1, are a frequent cause of coronal craniosynostosis. Nat Genet 45:304–307. https://doi.org/10.1038/ng.2531
Shen JJ, Brown CA, Lupski JR, Potocki L (2003) Mandibuloacral dysplasia caused by homozygosity for the R527H mutation in Lamin A/C. J Med Genet 40:854–857
Stratton RF, Dobyns WB, Greenberg F et al (1986) Interstitial deletion of (17)(p11.2p11.2): report of six additional patients with a new chromosome deletion syndrome. Am J Med Genet 24:421–432. https://doi.org/10.1002/ajmg.1320240305
Tong J, Li W, Vidal C et al (2011) Lamin A/C deficiency is associated with fat infiltration of muscle and bone. Mech Ageing Dev 132:552–559. https://doi.org/10.1016/j.mad.2011.09.004
Tsukune N, Naito M, Kubota T et al (2017) Lamin A overexpression promotes osteoblast differentiation and calcification in the MC3T3-E1 preosteoblastic cell line. Biochem Biophys Res Commun 488:664–670. https://doi.org/10.1016/j.bbrc.2017.02.110
Twigg SR, Versnel SL, Nürnberg G et al (2009) Frontorhiny, a distinctive presentation of frontonasal dysplasia caused by recessive mutations in the ALX3 homeobox gene. Am J Hum Genet 84:698–705. https://doi.org/10.1016/j.ajhg.2009.04.009
Twigg SR, Vorgia E, McGowan SJ et al (2013) Reduced dosage of ERF causes complex craniosynostosis in humans and mice and links ERK1/2 signaling to regulation of osteogenesis. Nat Genet 45:308–313. https://doi.org/10.1038/ng.2539
Vissers LE, Cox TC, Maga AM et al (2011) Heterozygous mutations of FREM1 are associated with an increased risk of isolated metopic craniosynostosis in humans and mice. PLoS Genet 7:e1002278. https://doi.org/10.1371/journal.pgen.1002278
Wilkie AO, Bochukova EG, Hansen RM et al (2007) Clinical dividends from the molecular genetic diagnosis of craniosynostosis. Am J Med Genet A 143A:1941–1949. https://doi.org/10.1002/ajmg.a.31905
Wilkie AO, Byren JC, Hurst JA et al (2010) Prevalence and complications of single-gene and chromosomal disorders in craniosynostosis. Pediatrics 126:e391–e400. https://doi.org/10.1542/peds.2009-3491
Worman HJ, Bonne G (2007) ‘Laminopathies’: a wide spectrum of human diseases. Exp Cell Res 313:2121–2133. https://doi.org/10.1016/j.yexcr.2007.03.028
Yang SH, Meta M, Qiao X et al (2006) A farnesyltransferase inhibitor improves disease phenotypes in mice with a Hutchinson-Gilford progeria syndrome mutation. J Clin Invest 116:2115–2121. https://doi.org/10.1172/JCI28968
Zhou YX, Xu X, Chen L et al (2000) A Pro250Arg substitution in mouse Fgfr1 causes increased expression of Cbfa1 and premature fusion of calvarial sutures. Hum Mol Genet 9:2001–2008
Acknowledgements
We are grateful to the patient and family members for participating in this study. This work was supported by the grants from the Polish National Science Centre, Poland UMO-2016/21/D/NZ5/00064 to AS-S and UMO-2016/22/E/NZ5/00270 to AJ.
Author information
Authors and Affiliations
Contributions
AJ and DL recruited and clinically diagnosed the patient; AS-S wrote the manuscript and performed and analyzed the results of molecular testing; EO contributed in sample preparation and a part of experimental procedures; MS contributed in data analysis; AJ critically revised and corrected the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Informed consent
All patients agreed to participate in this study and informed consent was taken from all subjects or their legal guardians prior to genetic testing.
Ethical approval
All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki Declaration and its later amendments or comparable ethical standards. Ethics approval was granted by the Institutional Review Board of Poznan University of Medical Sciences.
Conflict of interest
The authors declare no conflict of interest.
Additional information
Communicated by: Michal Witt
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Sowińska-Seidler, A., Olech, E.M., Socha, M. et al. Novel 1q22-q23.1 duplication in a patient with lambdoid and metopic craniosynostosis, muscular hypotonia, and psychomotor retardation. J Appl Genetics 59, 281–289 (2018). https://doi.org/10.1007/s13353-018-0447-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13353-018-0447-4