
Abstract
The objective of this study was to analyse the genetic variability and phylogenetic analysis of six strains of rabbit haemorrhagic disease virus (RHDV), including four Czech strains (CAMPV-351, CAMPV-561, CAMPV-562, CAMPV-558) and two French strains (Fr-1, Fr-2), on the basis of a fragment of the VP60 capsid structural protein-coding gene N-terminal region. The results of our own studies were compared to 26 RHDV strains obtained from GenBank. The analysis of the genetic variability of six RHDV strains indicated that the CAMPV-561 strain is the most genetically variable. Less variable were the Fr-1 and Fr-2 strains, while the least variable was CAMPV-351. In turn, the genetic distance among the six analysed strains and 26 strains obtained from GenBank was the greatest for CAMPV-351 and Whn/China [11.3 % according to the observed divergence (OD) method and 12.2 % according to the maximum likelihood (ML) method], while it was the lowest for CAMPV-351 and FRG (0.8 % in both the OD and ML methods). In turn, the scale of the genetic distances among the six analysed strains and five RHDVa strains (99-05, NY-01, Whn/China, Triptis, Iowa2000) ranged from 9.3–10.3 % in the OD method to 10.3–13.7 % in the ML method. The image of phylogenetic dependencies generated for the strains analysed and those obtained from GenBank revealed their distribution to be in five genetic groups (G1–G5), whereas the analysed strains were included in genetic groups 2 and 3.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The rabbit haemorrhagic disease virus (RHDV) is an aetiological factor of rabbit haemorrhagic disease and belongs to the Caliciviridae family. The disease caused by the virus was first recorded in 1984 in China, in Jiangsu Province, in Angora rabbits imported from Germany (Hukowska-Szematowicz 2006). At present, it is known that RHDV occurs on all continents and is still a very significant threat to wild animals, meat races and Angora rabbits, as the latter are bred principally for their meat and fur (Abrantes et al. 2012). The course of the disease caused by RHDV can vary, from the hyperacute and acute forms with high mortality rate, to the subacute form with lower mortality rate. The incubation period after experimental infection ranges from 4 to 12–26 h, with death most frequently occurring after 12–36 h from the first symptoms (Tokarz-Deptuła 2009).
The first reports on the disease, and the first hypothesis in regards to the pathogenesis of the disease, suggested that the mechanism of pathogenic impact of RHDV onto rabbit organism was related to its affinity to blood vessels, where it causes damage to the endothelium and disseminated intravascular coagulations (DIC syndrome), leading to ecchymosis and haemorrhages (Hukowska-Szematowicz 2006). With time and the growing interest of researchers in the virus and the pathogenesis of the disease caused by it, the participation of other factors in the pathogenesis of the disease was considered. Further studies focused on the liver, where the virus causes the greatest anatomic and pathological lesions, and it turned out that, in this organ, intensified cell death processes occur by way of necrosis (Park et al. 1995) and apoptosis (Alonso et al. 1998); the latter of these processes is also related to peripheral blood leukocytes from rabbits infected with RHDV (Niedźwiedzka-Rystwej and Deptuła 2012a, b). Moreover, other examinations showed that, during rabbit infection with RHDV, there is overexpression of pro-inflammatory cytokines in the liver, which is probably one of the causes of pathological lesions in this organ (Tuñón et al. 2011a, b). Tokarz-Deptuła’s team (Tokarz-Deptuła 2009) has clearly contributed to explanation of the pathogenesis of the disease by proving that, in the course of infection with RHDV, many changes occur to the immune system of the infected rabbits, which are manifested with changes of immunity parameters, imaging phagocytary activity and metabolism of neutrophil granulocytes, as well as qualitative changes to peripheral blood regarding lymphocytes and their subpopulations. Further new facts in the area of the pathogenesis of the disease were presented in 2011, when it was revealed that histo-blood group antigens (HBGA) can act as factors facilitating infection with RHDV, while polymorphism of genes coding such antigens can contribute to the emergence of genetic immunity to RHDV at the population level (Nyström et al. 2011).
In the aspect of its structure, RHDV is a small virus, of the size 28–40 nm and density 1.310–1.365 g/cm3, features cubic symmetry and occurs in the form of a regular icosahedron, with 32 capsomeres. It contains linear one-strand RNA with positive polarity, comprising 7,437 nucleotides. There are two reading frames in the RHDV genome: a longer—ORF1 (7,034 nucleotides), coding non-structural proteins of the virus—p16, p23, p37, p30, VPg and structural capsid protein VP60, and a shorter—ORF2 (353 nucleotides), coding a VP12 protein (Meyers et al. 1991; Wirblich et al. 1996). The main component of RHDV capsid with the structure of a regular icosahedron is VP60 polypeptide, with a molecular weight of 60 kDa (Meyers et al. 1991; Wirblich et al. 1996). VP60 protein is an antigen recognised by the immune system of the rabbit host, and, thus, plays an important role in the immune response against the infection with RHDV (Viaplana et al. 1997). The capsid features three functional domains: outer surface, inner surface and the hinge region linking them. The outer surface is formed by the C-terminal part of the VP60 protein, and the domain contains two regions C and E among the six regions differentiated for this protein (A, B, C, D, E, F), and, in these regions, antigen determinants of the virus are located. In turn, the inner surface is formed by the N-terminal part of the VP60 protein, and it directly protects the genome of the virus, and, also, as suggested by Viaplana et al. (1997), this region of the protein is also immunogenic, can contain antigen determinants to evoke the immune response, probably of humoral type, yet, such suggestions have not yet been experimentally confirmed. Due to the key role of the VP60 protein in the biology of RHDV, this item is the most frequently selected molecular target to analyse the variability and phylogenesis of RHDV strains originating from various parts of the world. Such data can serve to determine variable and constant regions, and, therefore, to develop a detailed antigen map of the virus, which may, in turn, find application in the works on vaccines against RHD.
Currently, in the GenBank database, there are 35 RHDV strains with completely sequenced genomes, sequences of the structural capsid protein VP60 of 48 RHDV strains and 200 various fragments of the genome sequenced for 50 RHDV strains. Apart from the above sequences placed in GenBank, many publications (Fitzner and Kęsy 2003; Chrobocińska and Mizak 2007; Niedźwiedzka-Rystwej et al. 2009; Oem et al. 2009; Pawlikowska et al. 2009, 2010; Fitzner et al. 2012) describe the sequences of 65 other RHDV strains. The above data on RHDV sequences prove that there is high interest in the virus, because, on the basis of biological sequences, one can trace the genetic variability of the virus and perform phylogenetic analyses, providing information on the relationship of the strains and evolutionary dependencies. Studies and analyses performed on the basis of the sequences have allowed for the identification of 55 specific strains among the RHDV, referred to as antigen variants RHDVa, one Chinese strain referred to as a new RHDVa variant and one French strain referred to as a new RHDV variant (Table 1).
The study of the genetic variability of RHDV (Milton et al. 1992; Boga et al. 1994; Rasschaert et al. 1995; Gould et al. 1997; Nowotny et al. 1997; Le Gall et al. 1998; Asgari et al. 1999; Moss et al. 2002; Le Gall-Reculé et al. 2003; Matiz et al. 2006; McIntosh et al. 2007; Tian et al. 2007; Forrester et al. 2008; Abrantes et al. 2008; Yang et al. 2008; Muller et al. 2009; Oem et al. 2009; Wang et al. 2012) referred to about 200 strains (among which 55 are RHDVa strains), comprising fragmentary sequences of VP60 coding gene, the entire VP60 gene or sequences of p30 protein-coding gene. The above studies indicate that the variability of RHDV strains oscillates at the level of 14 %, while variability among RHDVa strains amounts to a value from 9.9 to 10.3 % (Milton et al. 1992; Boga et al. 1994; Rasschaert et al. 1995; Gould et al. 1997; Nowotny et al. 1997; Le Gall et al. 1998; Asgari et al. 1999; Moss et al. 2002; Le Gall-Reculé et al. 2003; Matiz et al. 2006; McIntosh et al. 2007; Tian et al. 2007; Forrester et al. 2008; Yang et al. 2008; Muller et al. 2009; Oem et al. 2009; Wang et al. 2012). Furthermore, observations by Le Gall-Reculé et al. (2011) indicate that new RHDV strains are beginning to appear which differ greatly from the original RHDV subtype and the new RHDVa subtype, which suggests the isolation of a new group of RHDV strains, while the first of them, the French RHDV variant (Le Gall-Reculé et al. 2011), showed variability at the level of 14.3 %. In turn, in the case of 37 domestic RHDV strains, variability amounted to from 7.5 % in the case of ŻD to 10.3 % in the case of Polish RHDVa strains (Fitzner and Kęsy 2003; Chrobocińska and Mizak 2007; Fitzner et al. 2012). Phylogenetic analysis and investigation of the phylodynamics of RHDV strains was performed by many centres worldwide (Nowotny et al. 1997; Le Gall et al. 1998; Moss et al. 2002; Le Gall-Reculé et al. 2003; Fitzner and Kęsy 2003; Forrester et al. 2003, 2006a, b; Hukowska-Szematowicz 2006; Matiz et al. 2006; Chrobocińska and Mizak 2007; McIntosh et al. 2007; Hukowska-Szematowicz et al. 2009; Muller et al. 2009; Kerr et al. 2009; Niedźwiedzka-Rystwej et al. 2009; Pawlikowska et al. 2009, 2010; Alda et al. 2010; Hukowska-Szematowicz and Deptuła 2010; Kinnear and Linde 2010; Fitzner et al. 2012; Wang et al. 2012). The analyses performed indicate that RHDV strains are grouped depending on the time of strain isolation and on their geographic location, and that the strains referred to as antigen variants RHDVa are genetically distanced from the strains referred to as original RHDV subtype, which is manifested by the formation of completely separate genetic groups. Furthermore, it must be noticed that, due to the appearance of an increasing number of RHDV strains, the analyses performed are no longer limited to the specification of affinity among RHDV strains, but also owing to phylodynamics (Kerr et al. 2009), analysing evolutionary relations in the strain group, which allows for reconstruction of the changes to the virus sequence divergence matrix, which, in turn, allows for templates of virus migration.
Purpose
The purpose of this study was to analyse the genetic variability and to perform phylogenetic analysis of six strains of RHDV, including four Czech strains (CAMPV-351, CAMPV-561, CAMPV-562, CAMPV-558) and two French strains (Fr-1, Fr-2), on the basis of a fragment of the VP60 capsid structural protein-coding gene N-terminal region. The results of our own studies were compared with 26 RHDV strains obtained from GenBank.
Materials and methods
RHDV strains
The study involved four Czech strains [CAMPV-351 from 1987 (passage from 2000), CAMPV-561 from 1996, CAMPV-562 from 1992, CAMPV-558 from 1988] and two French strains (Fr-1 from 1994, Fr-2 from 1992) (Table 2), prepared in the form of lyophilisate according to the procedure previously described by Fitzner et al. (1996). The Czech strains originated from the Collection of Animal Pathogenic Microorganisms (CAPM), Veterinary Research Institute in Brno, Czech Republic, while the French strains were obtained from Centre National d’Etudes Vétérinaires et Alimentaires (CNEVA), Laboratoire Central de Recherches Avicole et Porcine, Ploufragan, France, from Dr. J. P. Morisse.
Isolation of viral RNA
Complete RNA of RHDV was isolated from lyophilisates using the RNA set Total RNA (A&A Biotechnology, Poland), according to the provided protocol.
Reverse transcription (RT) reaction—cDNA synthesis
Complementary cDNA strands were obtained on a matrix of viral RNA, using reverse transcriptase enzyme (M-MLV Reverse Transcriptase, Invitrogen, USA). 25 μl of the reaction mixture contained the following: 1.0 μl of specific antisense starter (P2) at 100 mM concentration (Metabion GmbH, Niemcy), 1.0 μl of dNTPs nucleotide blend at 25 mM concentration (Promega, USA), 0.5 μl of reverse transcriptase enzyme M-MLV RT (Invitrogen, USA), 2.0 μl of 5-fold concentrated RT-PCR buffer (Invitrogen, USA), 0.5 μl of DTT 0.1 M (Invitrogen, USA), 1.0 μl of RNase inhibitor RNase OUT (Invitrogen, USA), 14 μl of water for molecular biology (Eppendorf, Niemcy) and 5.0 μl of RNA of an appropriate RHDV strain. Before the reaction mixture was prepared, the RNA of the six tested RHDV strains was heated for 5 min at 65 °C and then stored on ice until the mixture was prepared. RT-PCR reaction was conducted in a T-gradient Thermocycler (Biometra, Germany) using the following temperature–time profile: 25 °C for 10 min, 37 °C for 60 min, 95 °C for 5 min and 4 °C for 1 min. The resulting cDNA was stored at a temperature of 2–8 °C for further analyses.
Starters
Starters suggested by Guittré et al. (1996) were used, based on the complete sequence of the RHDV-FRG virus genome, developed by Meyers et al. (1991) and allowing for amplification of the fragment of the VP60 structural protein-coding gene N-terminal region (position in the genome at 5182–5692). Using starters P1 (sense) 5′gagctcgagcgacaacaggc3′ and P2 (antisense) 5′caaacacctgacccggcaac3′, a 510-bp fragment was amplified. Starter synthesis was performed by Metabion GmbH (Germany).
PCR
50 μl of the reaction mixture contained the following: 2.0 μl of starters (1.0 μl of each P1 and P2) at 10 mM concentration each (Metabion GmbH, Germany), 1.0 μl of dNTPs blend at 10 mM concentration (Promega, USA), 5.0 μl of 10-fold concentrated Taq Plus buffer (GenoPlast, Poland), 1.0 μl of Taq Plus DNA polymerase (GenoPlast, Poland), 1.0 μl of 10-fold concentrated Mg buffer (Promega, USA), 38.0 μl of water for molecular biology (Eppendorf, Germany) and 2.0 μl of cDNA of an appropriate RHDV strain (added to the reaction at the end). PCR reaction was conducted in a T-gradient Thermocycler (Biometra, Germany). The following temperature–time profile was used: preliminary denaturation 94 °C for 2 min, 35 cycles involving: denaturations (94 °C for 30 s), starter affixing (50 °C for 1 min), chain elongation (72° for 2 min), final elongation (72° for 5 min) and cooling the reaction mixture to 4 °C. Reaction products were stored at a temperature of 2–8 °C for further analyses.
Electrophoresis of PCR products in agarose gel
For the purpose of visualisation of PCR products, electrophoresis was performed in 1.5 % agarose gel (Prona, USA) dyed with ethidium bromide (Fermentas, Lithuania). Molecular mass marker GeneRuler 100 and 50 (Fermentas, Lithuania) was used for the evaluation of the size of products. Electrophoretic separation was conducted in 1.0-fold concentrated TBE buffer, at room temperature, with a current voltage of 100 V/cm of gel for 45 min, using a set for electrophoresis (Bio-Rad, Germany). The storage and interpretation of results was completed using a UV visualisation set (Vilber Lourmat, France).
Preparative amplification, purification and preparation of analysed fragments of the RHDV genome for sequencing
Following PCR results visualisation, mass PCR was performed along with electrophoretic separation, using conditions identical to those described above. Next, DNA isolation from gel was performed using a Gel OUT set (A&A Biotechnology, Poland), according to the manufacturer’s recommended procedure. Such prepared samples were sent for automatic sequencing to Metabion GmbH, Germany.
Comparative analysis of nucleotide sequences, and creation of distance matrices and phylogenetic trees
The obtained nucleotide sequences of a VP60 protein-coding gene N-terminal region fragment from the six tested RHDV strains (CAMPV-351, CAMPV-561, CAMPV-562, CAMPV-558, Fr-1, Fr-2) were compared to each other (alignment) and to 26 homologous sequences of RHDV obtained from GenBank (Table 2), and the analysis was performed in DNAMAN software version 5.2.10 (Lynnon BioSoft, Canada). Next, on the basis of the alignment created in DNAMAN software, distance matrices were developed using the observed divergence (OD) method and the maximum likelihood (ML) method. The distance matrix shows the genetic distances for all sequence pairs in the set of analysed strains within the range of values from 0.000 to 1.000. The genetic distance between the sequence pair corresponded to the number of nucleotide substitutions (expressed as a percentage), which occurred in such sequences from their divergence from the input sequence. The values of the resulting matrices were then graphically transformed into phylogenetic trees constructed using the ML method. The generated phylogenetic tree was a rooted tree, and the outgroup was formed by the sequence of the RCV virus (X96868). The correctness of the constructed tree was verified using the bootstrap method with formation of 1,000 comparisons of the analogical variability level. For each new node, bootstrap values were estimated and expressed as a percentage, illustrating the frequency of identical node occurrence in each thousand newly constructed trees.
Results
PCR yielded the amplification of a 510-bp-long genome fragment, which was subsequently sequenced. The obtained sequences of five analysed strains (CAMPV-561, CAMPV-562, CAMPV-558, Fr-1, Fr-2) of RHDV were submitted to GenBank, while the CAMPV-351 sequence is under preparation for submission (Table 2). The obtained nucleotide sequences of the six strains (CAMPV-351, CAMPV-561, CAMPV-562, CAMPV-558, Fr-1, Fr-2) were compared to each other and to 26 homologous sequences of RHDV (Table 2). Variability of the nucleotide sequence in the tested VP60 structural protein-coding gene N-terminal region fragment for the six analysed Czech and French strains amounted to 3.5 %. Genetic divergence of the tested Czech and French strains was manifested by the occurrence of 40 polymorphic loci with repeated local mutations of the nature of transitions A↔G and C↔T and transversions A↔T and A↔C. The comparative analysis of the amino acid sequence in the strains analysed, designed to test the importance of mutations in the nucleotide sequence, showed that most of them are silent mutations and the differences in the amino acid sequence were only observed in three positions.
The largest genetic distance (Table 3) among the six analysed strains was recorded between CAMPV-561 and Fr-1 (0.068 in the OD method and 0.071 in the ML method, which corresponds to 6.8 and 7.1 % nucleotide substitutions, respectively). Similar but slightly lower values were obtained in the case of strains CAMPV-561 and Fr-2 (0.060 OD method/0.062 ML methods, which corresponds to 6.0 and 6.2 % nucleotide substitutions, respectively), CAMPV-351 and CAMPV-561 (0.055 OD method/0.057 ML method, which corresponds to 5.5 and 5.7 % nucleotide substitutions, respectively), CAMPV-561 and CAMPV-558 (0.055 OD method/0.057 ML method, which corresponds to 5.5 and 5.7 % nucleotide substitutions, respectively) and between CAMPV-561 and CAMPV-562 (0.055 OD method/0.056 ML method, which corresponds to 5.5 and 5.6 % nucleotide substitutions, respectively). Smaller genetic distances in the generated matrix were recorded for the strain pairs CAMPV-351 and Fr-1 (0.034 OD method/0.035 ML method, corresponding to 3.4 and 3.5 % nucleotide substitutions, respectively), CAMPV-562 and Fr-1 (0.034 OD method/0.035 ML method, corresponding to 3.4 and 3.5 % nucleotide substitutions, respectively), CAMPV-562 and Fr-2 (0.026 in both models, which yields 2.6 % nucleotide substitutions), CAMPV-558 and Fr-1 (0.029 in both models, which yields 2.9 % nucleotide substitutions), CAMPV-558 and Fr-2 (0.026 in both models, which corresponds to 2.6 % nucleotide substitutions) and between Fr-1 and Fr-2 (0.024 in both models, which corresponds to 2.4 % nucleotide substitutions). In turn, the genetic distance range was the smallest for strain pairs CAMPV-351 and CAMPV-562, as well as CAMPV-351 and CAMPV-558 (0.016 in both models, which corresponds to 1.6 % substitutions) and CAMPV-562 and CAMPV-558 (0.016 in both models, which corresponds to 1.6 % substitutions).
In turn, comparative analysis (alignment) of all the analysed 32 RHDV strains revealed variability at the level of 4.6 %; 105 polymorphic loci were located with the dominant C↔T and G↔A transitions over T↔A and A↔C transversions. The distance matrix constructed (Table 3) showed that the CAMPV-351 strain recorded the greatest genetic distance with NY-01 (0.0108 OD method/0.115 ML method, which corresponds to 10.8 and 11.5 % nucleotide substitutions, respectively), Triptis (0.970 OD method/0.103 ML method, corresponding to 9.7 and 10.3 % substitutions, respectively) and Whn/China (0.113 OD method/0.122 ML method, corresponding to 11.3 and 12.2 % substitutions, respectively), whereas the smallest distance was shown with FRG (0.008 in both methods, which corresponds to 0.8 % nucleotide substitutions), Italy90 (0.013 in both methods, which yields 1.3 % nucleotide substitutions), WX/China (0.016 in both methods and 1.6 % substitutions) and Haute Saone (0.026 in both methods, which corresponds to 2.6 % nucleotide substitutions). The Czech strain CAMPV-561 revealed the largest genetic distance from NY-01 (0.100 OD method/0.106 ML method, corresponding to 10.0 and 10.6 % substitutions, respectively) and Whn/China (0.110 OD method/0.119 ML method, corresponding to 11 and 11.9 % nucleotide substitutions, respectively), while the smallest was with the Jena (0.016 in both methods and 1.6 % nucleotide substitutions), Bahrain (0.029 in both models and 2.9 % substitutions), BS89 (0.029 in both models and 2.9 % substitutions) and Frankfurt strains (0.029 in both models and 2.9 % nucleotide substitutions). In turn, CAMPV-562 revealed the greatest genetic distance with Whn/China (0.108 OD method/0.115 ML method, corresponding to 10.8 and 11.5 % nucleotide substitutions, respectively) and NY-01 (0.102 OD method/0.109 ML method, corresponding to 10.2 and 10.9 % substitutions, respectively), whereas the smallest was shown with strains FRG (0.008 in both models, which corresponds to 0.8 % substitutions), Italy90 (0.013 in both methods and 1.3 % substitutions) and WX/China (0.016 in both methods and 1.6 % substitutions). The fourth analysed Czech strain, CAMPV-558, was the most genetically distant from Whn/China (0.102 OD method/0.110 ML method, corresponding to 10.2 and 11.0 % substitutions, respectively) and NY-01 (0.097 OD method/0.103 ML method, corresponding to 9.3 and 10.3 % substitutions, respectively), while the least distance was shown from FRG (0.08 in both methods and 0.8 % substitutions), Italy90 (0.013 in both methods and 1.3 % substitutions), WX/China (0.016 in both methods and 1.6 % substitutions) and Haute Saone (0.016 in both methods and 1.6 % substitutions).
In turn, Fr-1 proved to be the most genetically distant from Whn/China (0.126 OD method/0.137 ML method, corresponding to 12.6 and 13.7 % nucleotide substitutions, respectively), NY-01 (0.121 OD method/0.131 ML method, corresponding to 12.1 and 13.1 % substitutions, respectively), 99-05 (0.105 OD method/0.113 ML method, corresponding to 10.5 and 11.3 % substitutions, respectively), Iowa (0.105 OD method/0.113 ML method, corresponding to 10.5 and 11.3 % substitutions, respectively) and Triptis (0.110 OD method/0.119 ML method, corresponding to 11.0 and 11.9 % substitutions). It revealed small genetic distances from strains WX/China (0.024 in both methods and 2.4 % nucleotide substitutions), FRG (0.026 OD method/0.027 ML method, corresponding to 2.6 and 2.7 % substitutions, respectively) and Mexico89 (0.029 in both methods and 2.9 % substitutions). Fr-2 presented evident genetic distance from strains Whn/China (0.123 OD method/0.134 ML method, corresponding to 12.3 and 13.4 % substitutions, respectively), NY-01 (0.118 OD method/0.127 ML method, corresponding to 11.8 and 12.7 % substitutions, respectively), Triptis (0.108 OD method/0.115 ML method, corresponding to 10.8 and 11.5 % substitutions, respectively), 99-05 (0.102 OD method/0.109 ML method, corresponding to 10.2 and 10.9 % substitutions, respectively) and Iowa (0.102 OD method/0.109 ML method, corresponding to 10.2 and 10.9 % substitutions, respectively). In turn, it had small genetic distance from WX/China (0.016 in both methods and 1.6 % substitutions), FRG (0.018 in both methods and 1.8 % substitutions) and Mexico (0.021 in both methods and 2.1 % substitutions). It must be noticed that slightly higher values of the matrix (Table 3) were obtained using the ML method, which conforms to the assumption of the method based on the evaluation of the phylogenetic distance considering the level of mutation estimated with sequence comparison and is set by the differences in sequences. In turn, the observed divergence method uses unchanged values obtained directly from sequence comparison by calculation of the divergence factor for each sequence pair (Hukowska-Szematowicz 2006).
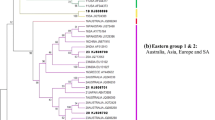
The image of phylogenetic dependencies among 32 RHDV strains (Fig. 1), generated on the basis of the VP60 structural protein-coding gene N-terminal region fragment, points to their evident distribution to five genetic groups (G1–G5), whereas the analysed strains were included in genetic groups 2 and 3. The first genetic group (G1) included strains identified during the during the first years of the plague development in Europe—1989 (AST89, Eisenhuttenstadt, SD). The second genetic group (G2) comprised five of the tested strains (CAMPV-351, CAMPV-562, CAMPV-558, Fr-1, Fr-2) and eight other strains from Europe, Mexico, Asia and Australia from the years 1987–2005 (FRG, Italy90, Haute Saone, Mexico89, WX84, Nyngan05, Saudi Arabia, 95-05). The third genetic group (G3) included the tested Czech strain CAMPV-561 and strains originating from Europe from the years 1989–2001 (Wriezen, Jena, 95-10, Rainham, Meiningen, Frankfurt, BS, Hagenow) and a strain from Asia (Bahrain). The fourth genetic group (G4) was individually formed by strain 00-13 identified in 2000 in France. The fifth genetic group (G5) gathered European, Asian and American RHDVa strains from 1996 to 2005 (French 99-05, American Iowa2000 and NY-01, Chinese Whn/China and German Triptis96). Bootstrap values estimated for particular nodes on the tree remained within the range from 70 to 100 %, confirming very high reliability of the tree.

Phylogenetic tree for 32 strains of rabbit haemorrhagic disease virus (RHDV) constructed using the maximum likelihood (ML) method on the basis of the comparison of nucleotide sequences of the fragment of the gene coding the N-terminal region of a structural protein VP60 capsid
Discussion
The variability of nucleotide sequences obtained in our own study (4.6 %) for 32 analysed RHDV sequences (alignment), identified within the period 1987–2005, is comparable to the variability recorded by other researchers (0–14.3 %) when analysing sequence variability within the entire RHDV genome or its fragments (Milton et al. 1992; Boga et al. 1994; Rasschaert et al. 1995; Gould et al. 1997; Nowotny et al. 1997; Le Gall et al. 1998; Asgari et al. 1999; Moss et al. 2002; Le Gall-Reculé et al. 2003, 2011; Matiz et al. 2006; McIntosh et al. 2007; Tian et al. 2007; Yang et al. 2008; Muller et al. 2009; Oem et al. 2009; Wang et al. 2012).
Within the group of tested Czech and French strains (CAMPV-351, CAMPV-561, CAMPV-562, CAMPV-558, Fr-1, Fr-2), the most genetically distanced was CAMPV-561, which revealed a maximum distance of 5.5 % according to the OD method and 7.1 % according to the ML method from all the analysed Czech and French strains. In view of the result obtained, one can suggest that CAMPV-561 is the most variable strain among the strains tested. Contrary to CAMPV-561, a smaller scale of genetic distances was presented by strains Fr-1 (2.4 %/3.5 %) and Fr-2 (2.6 %), while the smallest was shown by CAMPV-351 (1.6 %). It is worth recording that three of the analysed Czech strains (CAMPV-351, CAMPV-562 and CAMPV-558) revealed the smallest genetic distance from the reference German strain FRG, which amounted to 0.8 % in the OD and ML models, while the result obtained is confirmed with previous observations by Gould et al. (1997). The results obtained can be referred to analogical studies on nine Polish RHDV strains (SGM, KGM, PD, MAŁ, BLA, GSK, ŻD, ŻD1, LUB) (Fitzner and Kęsy 2003). In the studies, nucleotide variability was obtained at the level of 0.2–1.2 %, while detailed analysis pointed to BLA and ŻD strains as being the most variable, at the level of 3.8–4.0 %.
In turn, when considering the six strains tested and 26 obtained from GenBank, 32 RHDV strains in total, it can be noticed that a clear genetic distance was recorded for all six strains tested (CAMPV-351, CAMPV-561, CAMPV-562, CAMPV-558, Fr-1, Fr-2) as compared to RHDVa strains (new RHDVa subtype), in particular to NY-01, Whn/China and Iowa strains. As in the results from previous studies (Le Gall et al. 1998; Fitzner and Kęsy 2003; Le Gall-Reculé et al. 2003; Forrester et al. 2006b; Farnós et al. 2007; McIntosh et al. 2007; Kerr et al. 2009; Oem et al. 2009; Wang et al. 2012), the variability of nucleotide sequences in strains referred to as antigen variants RHDVa grows as compared to strains referred to as RHDV (original RHDV subtype); such a tendency was also recorded in our own study. In fact, the genetic distances between the six tested strains and RHDVa strains amounted to 9.3–10.3 % according to the OD method and 10.3–13.7 % according to the ML method, and these were the largest for the strain pair Fr-1 and Whn/China, while it was the smallest for the strain pair CAMPV-558 and NY-01. Moreover, it must be pointed out that detailed analysis of the genetic distance scale in the group of 32 strains (Table 3) revealed that the largest distance recorded amounted to 12.6 % according to the OD method and 13.7 % according to the ML method. Furthermore, the study tracks the scale of genetic distances for strains referred to in the literature as “reference” strains (AST89, SD89, CAMPV-351, FRG, WX84, BS89), namely, such that appeared in the first years of plague development in Europe and Asia, as compared to RHDVa strains. The analyses indicate that “reference” strains show genetic distances to RHDVa strains in the range 8.4–11.8 % in the OD method and 8.8–12.8 % in the ML method. It is worth noticing that all “reference” strains showed the maximum genetic distance to strains NY-01 and Whn/China, which amounted to 10.0 % in the OD method and 11.5–12.8 % in the ML method.
When analysing the genetic distance in the group of French strains, both tested (Fr-1, Fr-2) and collected from GenBank (SD, Haute Saone, 95-05, 95-10, 00-13, 99-05), it must be stated that it amounted to 2.6 % in the OD model and 10.9–11.3 % in the ML model. It must also be pointed out that the tested strains Fr-1 and Fr-2 were the most genetically distanced from the French strain collected from GenBank, 99-05 RHDVa (10.2–10.5 % for the OD model and 10.9–11.3 % for the ML model), and other strains originating from France, namely, 00-13 (8.9–9.2 % in the OD model and 9.4–9.7 % in the ML model), 95-10 (7.1–7.9 % in the OD model and 7.4–8.2 % in the ML model) and SD (6.3–7.1 % in the OD model and 6.5–7.4 % in the ML model), 95-05 (5.8–6.0 % in the OD model and 5.9–6.2 % in the ML model). In turn, the lowest heterogeneity of nucleotide sequences in the group of French strains occurred between Fr-1 and Fr-2 strains and Haute Saone (2.6–3.4 % in the OD model and 2.6–3.5 % in the ML model).
Phylogenetic analysis performed for the 32 RHDV strains allowed for the differentiation of five genetic groups (G1–G5) (Fig. 1). The obtained distribution into genetic groups corresponds to the view according to which strains in the groups are linked by the time of their identification rather than their geographic origin (Le Gall et al. 1998; Le Gall-Reculé et al. 2003; Matiz et al. 2006). In this respect, antigen variants differ, as they are always located in one genetic group (Capucci et al. 1998; Le Gall et al. 1998; Fitzner and Kęsy 2003; Forrester et al. 2006b; Farnós et al. 2007; McIntosh et al. 2007; Kerr et al. 2009; Oem et al. 2009). In the first of the genetic groups (G1), one can clearly define the criterion according to which the strains formed the group. It was formed by European strains from Spain, Germany and France, identified in 1989, namely upon plague occurrence in Europe, with the genetic variability of strains in the group amounting to 1 % and a bootstrap value of 100 %. This genetic group can be referred to as a “cluster” because it gathers strains having a common identification time, regardless of their geographic origin. Such positioning of strains in G1 corresponds to the results previously obtained by Le Gall et al. (1998) and Le Gall-Reculé et al. (2003), according to which strains in genogroups follow the time of identification pattern, whereas strains isolated at a similar time are characterised by small variability, as observed in G1. The second genetic group included 13 strains identified over 21 years (1984–2005), and the situation of some strains in this group conforms to prior observations by Le Gall et al. (1998) and Le Gall-Reculé et al. (2003). This group included the tested Czech strains CAMPV-351, CAMPV-562 and CAMPV-558, as well as the two French strains Fr-1 and Fr-2, with the distance between strains in the group ranging from 0.5 to 6.8 %. Evolutionary relations presented in this group are clearly reflected in the epizootic analysis of the plague in Europe in the years 1987–1989, with indications of CAMPV-351 from 1987, CAMPV-558 from 1988, Haute Saone from 1988 and FRG from 1989. When analysing evolutionary relations on the basis of variability evaluated in the study, it must be stated that the CAMPV-351 strain showed the smallest distance to FRG and CAMPV-562, amounting to 0.8 %. The situation of CAMPV-558 on one branch with the French Haute Saone strain can be related by the time of identification of such strains, as both were identified in 1988, although considering the variability for these two strains as compared to each other (1.6 %) and the variability between CAMPV-558 and FRG (0.8 %), one can suggest that the CAMPV-558 strain is more evolutionarily related to FRG than to Haute Saone. In turn, the two tested French strains Fr-1 and Fr-2, from 1994 to 1992, respectively, turned out to be very remote in evolutionary aspects from other tested French strains classified to genetic groups 3, 4 and 5. The third genetic group (G3) included strains from the years 1989–2001, with variability ranging from 1.3 to 9.4 %. Judging by the branch length in this genogroup, it can be suspected that the evolutionary path of Czech strain CAMPV-561 from 1996 went through Germany and, perhaps, the strain is a descendant of the Jena strain from 1993, whereas variability between the two strains in the fragment analysed amounted to 1.6 %. Other strains in this group are linked by a common time of identification and, judging from the branch length, it can be considered that the time factor affects the genetic variability of such strains, as the most genetically distanced strains were Bahrain from 2001 and Hagenow from 1990 (5.0 % according to the OD method and 5.1 % according to the ML method), as well as Wriezen from 1996 and Hagenow from 1990 (5.0 % in the OD method and 5.1 % in the ML method). The situation of the French 00-13 strain in G4 is a rather unexpected result, because, in previous studies by Le Gall-Reculé et al. (2003), the strain was included in genogroup 5, together with other strains from France identified in the years 1994–2000. Strains originating from Europe, Asia and USA, being antigen variants RHDVa (99-05, NY-01, Whn/China, Triptis, Iowa) formed the fifth genetic group (G5), with variability ranging from 1.1 to 9.7 %, which corresponds to genogroup 6 described by Le Gall-Reculé et al. (2003).
Conclusion
From the moment of rabbit haemorrhagic disease virus (RHDV) identification in 1984 until the present, there have been continuous studies on its variability and its evolutionary paths are tracked. Studies indicate that the virus is a good model for investigating the variability and evolution of RNA viruses, which is facilitated by the continuous appearance of new strains. The performed analysis of the genetic variability of six RHDV strains, including four Czech strains (CAMPV-351, CAMPV-561, CAMPV-562, CAMPV-558) and two French strains (Fr-1, Fr-2), on the basis of a fragment of the VP60 capsid structural protein-coding gene N-terminal region revealed that the Czech CAMPV-561 strain is the most genetically variable strain. Fr-1 and Fr-2 strains proved to be less genetically variable, and CAMPV-351 was the least variable. In turn, the genetic distance among the six analysed strains and 26 strains obtained from GenBank was the greatest for CAMPV-351 and Whn/China [11.3 % according to the observed divergence (OD) method and 12.2 % according to the maximum likelihood (ML) method], while it was the lowest for CAMPV-351 and FRG (0.8 % in both the OD and ML methods). In turn, the scale of genetic distances among the six analysed strains and five RHDVa strains (99-05, NY-01, Whn/China, Triptis, Iowa2000) amounted to 9.3–10.3 % in the OD method and 10.3–13.7 % in ML method, and was the greatest for the strain pair Fr-1 and Whn/China, and the smallest for the strain pair CAMPV-558 and NY-01. Phylogenetic analysis performed for the strains tested and 26 strains obtained from GenBank indicates that three Czech strains tested, CAMPV-562 from 1992 and CAMPV-558 from 1988 (belonging to G2) and CAMPV-561 from 1996 (belonging to G3) derive from European strains, while in the case of the French strains, Fr-1 from 1994 and Fr-2 from 1992, one can wonder why their evolutionary path did not go through China or Mexico to France.
References
Abrantes J, Esteves PJ, van der Loo W (2008) Evidence for recombination in the major capsid gene VP60 of the rabbit haemorrhagic disease virus (RHDV). Arch Virol 153:329–335
Abrantes J, van der Loo W, Le Pendu J, Esteves PJ (2012) Rabbit haemorrhagic disease (RHD) and rabbit haemorrhagic disease virus (RHDV): a review. Vet Res 43:12. doi:10.1186/1297-9716-43-12
Alda F, Gaitero T, Suárez M, Merchán T, Rocha G, Doadrio I (2010) Evolutionary history and molecular epidemiology of rabbit haemorrhagic disease virus in the Iberian Peninsula and Western Europe. BMC Evol Biol 10:347. doi:10.1186/1471-2148-10-347
Alonso C, Oviedo JM, Martín-Alonso JM, Díaz E, Boga JA, Parra F (1998) Programmed cell death in the pathogenesis of rabbit hemorrhagic disease. Arch Virol 143:321–332
Asgari S, Hardy JRE, Cooke BD (1999) Sequence analysis of rabbit haemorrhagic disease virus (RHDV) in Australia: alterations after its release. Arch Virol 144:135–145
Boga JA, Casais R, Marin MS, Martin-Alonso JM, Carmenes RS, Prieto M, Parra F (1994) Molecular cloning, sequencing and expression in Escherichia coli of the capsid protein gene from rabbit haemorrhagic disease virus (Spanish isolate AST/89). J Gen Virol 75:2409–2413
Capucci L, Fallacara F, Grazioli S, Lavazza A, Pacciarini ML, Brocchi E (1998) A further step in the evolution of rabbit hemorrhagic disease virus: the appearance of the first consistent antigenic variant. Virus Res 58:115–126
Chrobocińska M, Mizak B (2007) Phylogenetic analysis of partial capsid protein gene of rabbit haemorrhagic disease virus (RHDV) strains isolated between 1993 and 2005 in Poland. Bull Vet Inst Pulawy 51:189–197
Farnós O, Rodríguez D, Valdés O, Chiong M, Parra F, Toledo JR, Fernández E, Lleonart R, Suárez M (2007) Molecular and antigenic characterization of rabbit hemorrhagic disease virus isolated in Cuba indicates a distinct antigenic subtype. Arch Virol 152:1215–1221
Fitzner A, Kęsy A (2003) Variability of Polish isolates of the RHD virus (in Polish). Medycyna Wet 59:905–908
Fitzner A, Kęsy A, Niedbalski W, Paprocka G, Walkowiak B (1996) The identification of polypeptide VP60 national isolates of the RHD virus (in Polish). Medycyna Wet 52:303–305
Fitzner A, Niedbalski W, Paprocka G, Kęsy A (2012) Identification of Polish RHDVa subtype strains based on the analysis of a highly variable part of VP60 gene. Pol J Vet Sci 15:21–29
Forrester NL, Boag B, Moss SR, Turner SL, Trout RC, White PJ, Hudson PJ, Gould EA (2003) Long-term survival of New Zealand rabbit haemorrhagic disease virus RNA in wild rabbits, revealed by RT-PCR and phylogenetic analysis. J Gen Virol 84:3079–3086
Forrester NL, Abubakr MI, Abu Elzein EME, al-Afaleq AI, Housawi FMT, Moss SR, Turner SL, Gould EA (2006a) Phylogenetic analysis of rabbit haemorrhagic disease virus strains from the Arabian Peninsula: did RHDV emerge simultaneously in Europe and Asia? Virology 344:277–282
Forrester NL, Trout RC, Turner SL, Kelly D, Boag B, Moss S, Gould EA (2006b) Unravelling the paradox of rabbit haemorrhagic disease virus emergence, using phylogenetic analysis; possible implications for rabbit conservation strategies. Biol Conserv 131:296–306
Forrester NL, Moss SR, Turner SL, Schirrmeier H, Gould EA (2008) Recombination in rabbit haemorrhagic disease virus: possible impact on evolution and epidemiology. Virology 376:390–396
Gould AR, Kattenbelt JA, Lenghaus C, Morrissy C, Chamberlain T, Collins BJ, Westbury HA (1997) The complete nucleotide sequence of rabbit haemorrhagic disease virus (Czech strain V351): use of the polymerase chain reaction to detect replication in Australian vertebrates and analysis of viral population sequence variation. Virus Res 47:7–17
Guittré C, Ruvoen-Clouet N, Barraud L, Cherel Y, Baginski I, Prave M, Ganiere JP, Trépo C, Cova L (1996) Early stages of rabbit haemorrhagic disease virus infection monitored by polymerase chain reaction. J Vet Med B 43:109–118
Hukowska-Szematowicz B (2006) Immunological-genetic characteristics of chosen strains of RHD (rabbit haemorrhagic disease) virus (in Polish). Doctoral dissertation, University of Szczecin, Poland
Hukowska-Szematowicz B, Deptuła W (2010) Phylogenetic analysis of French strains of the RHD virus on the basis of a fragment of gene encoding C-terminal part of structural capsie VP60 protein. Adv Agric Sci 12:159–168
Hukowska-Szematowicz B, Pawlikowska M, Deptuła W (2009) Genetic variability of Czech and German RHD virus strains. Pol J Microbiol 58:237–245
Kerr PJ, Kitchen A, Holmes EC (2009) Origin and phylodynamics of rabbit hemorrhagic disease virus. J Virol 83:12129–12138
Kinnear M, Linde CC (2010) Capsid gene divergence in rabbit hemorrhagic disease virus. J Gen Virol 91:174–181
Le Gall G, Arnauld C, Boilletot E, Morisse JP, Rasschaert D (1998) Molecular epidemiology of rabbit haemorrhagic disease virus outbreaks in France during 1988 to 1995. J Gen Virol 79:11–16
Le Gall-Reculé G, Zwingelstein F, Laurent S, De Boisséson C, Portejoie Y, Rasschaert D (2003) Phylogenetic analysis of rabbit haemorrhagic disease virus in France between 1993 and 2000, and the characterisation of RHDV antigenic variants. Arch Virol 148:65–81
Le Gall-Reculé G, Zwingelstein F, Boucher S, Le Normand B, Plassiart G, Portejoie Y, Decors A, Bertagnoli S, Guérin JL, Marchandeau S (2011) Detection of a new variant of rabbit haemorrhagic disease virus in France. Vet Rec 168:137–138
Marchandeau S, Le Gall-Reculé G, Bertagnoli S, Aubineau J, Botti G, Lavazza A (2005) Serological evidence for a non-protective RHDV-like virus. Vet Res 36:53–62
Matiz K, Ursu K, Kecskeméti S, Bajmócy E, Kiss I (2006) Phylogenetic analysis of rabbit haemorrhagic disease virus (RHDV) strains isolated between 1988 and 2003 in eastern Hungary. Arch Virol 151:1659–1666
McIntosh MT, Behan SC, Mohamed FM, Lu Z, Moran KE, Burrage TG, Neilan JG, Ward GB, Botti G, Capucci L, Metwally SA (2007) A pandemic strain of calcivirus threatens rabbit industries in the Americas. Virol J 2:96. doi:10.1186/1743-422X-4-96
Meyers G, Wirblich C, Thiel HJ (1991) Rabbit hemorrhagic disease virus—molecular cloning and nucleotide sequencing of a calicivirus genome. Virology 184:664–676
Milton ID, Vlasak R, Nowotny N, Rodak L, Carter MJ (1992) Genomic 3′ terminal sequence comparison of three isolates of rabbit haemorrhagic disease virus. FEMS Microbiol Lett 72:37–42
Moss SR, Turner SL, Trout RC, White PJ, Hudson PJ, Desai A, Armesto M, Forrester NL, Gould EA (2002) Molecular epidemiology of rabbit haemorrhagic disease virus. J Gen Virol 83:2461–2467
Muller A, Freitas J, Silva E, Le Gall-Reculé G, Zwingelstein F, Abrantes J, Esteves PJ, Alves PC, van der Loo W, Kolodziejek J, Nowotny N, Thompson G (2009) Evolution of rabbit haemorrhagic disease virus (RHDV) in the European rabbit (Oryctolagus cuniculus) from the Iberian Peninsula. Vet Microbiol 135:368–373
Niedźwiedzka-Rystwej P, Deptuła W (2012a) Apoptosis of peripheral blood leukocytes from rabbits infected with non-haemagglutinating strains of rabbit haemorrhagic disease virus (RHDV). Vet Immunol Immunopathol 149:54–57
Niedźwiedzka-Rystwej P, Deptuła W (2012b) Lymphocyte subpopulations and apoptosis of immune cells in rabbits experimentally infected with a strain of the RHD virus having a variable haemagglutination capacity. Pol J Vet Sci 15:43–49
Niedźwiedzka-Rystwej P, Pawlikowska M, Hukowska-Szematowicz B, Tokarz-Deptuła B, Deptuła W (2009) Immunological and genetic studies of RHD (rabbit haemorrhagic disease) virus strains. Cent Eur J Immunol 34:61–67
Nowotny N, Bascuñana CR, Ballagi-Pordány A, Gavier-Widén D, Uhlén M, Belák S (1997) Phylogenetic analysis of rabbit haemorrhagic disease and European brown hare syndrome viruses by comparison of sequences from the capsid protein gene. Arch Virol 142:657–673
Nyström K, Le Gall-Reculé G, Grassi P, Abrantes J, Ruvoën-Clouet N, Le Moullac-Vaidye B, Lopes AM, Esteves PJ, Strive T, Marchandeau S, Dell A, Haslam SM, Le Pendu J (2011) Histo-blood group antigens act as attachment factors of rabbit hemorrhagic disease virus infection in a virus strain-dependent manner. PLoS Pathog 7:e1002188. doi:10.1371/journal.ppat.1002188
Oem JK, Lee KN, Roh IS, Lee KK, Kim SH, Kim HR, Park CK, Joo YS (2009) Identification and characterization of rabbit hemorrhagic disease virus genetic variants isolated in Korea. J Vet Med Sci 71:1519–1523
Park JH, Lee YS, Itakura C (1995) Pathogenesis of acute necrotic hepatitis in rabbit hemorrhagic disease. Lab Anim Sci 45:445–449
Pawlikowska M, Hukowska-Szematowicz B, Deptuła W (2009) Genetical characteristic of six strains of RHD (Rabbit haemorrhagic disease) virus originating from Europe. Cent Eur J Immunol 34:7–13
Pawlikowska M, Hukowska-Szematowicz B, Deptuła W (2010) Phylogenetic analysis of selected strains of Rabbit haemorrhagic disease virus on the basis of N-terminal fragment of the gene encoding structural protein VP60. Bull Vet Inst Pulawy 54:129–133
Rasschaert D, Huguet S, Madelaine MF, Vautherot JF (1995) Sequence and genomic organization of a rabbit hemorrhagic disease virus isolated from a wild rabbit. Virus Genes 9:121–132
Schirrmeier H, Reimann I, Köllner B, Granzow H (1999) Pathogenic, antigenic and molecular properties of rabbit haemorrhagic disease virus (RHDV) isolated from vaccinated rabbits: detection and characterization of antigenic variants. Arch Virol 144:419–735
Tian L, Liao J, Li JW, Zhou WR, Zhang XL, Wang HN (2007) Isolation and identification of a non-haemagglutinating strain of rabbit hemorrhagic disease virus from China and sequence analysis for the VP60 gene. Virus Genes 35:745–752
Tokarz-Deptuła B (2009) Immunity phenomena in rabbits infected with the RHD (rabbit haemorrhagic disease) virus. Pol J Env Stud 7:1–81
Tuñón MJ, San Miguel B, Crespo I, Jorquera F, Santamaría E, Alvarez M, Prieto J, González-Gallego J (2011a) Melatonin attenuates apoptotic liver damage in fulminant hepatic failure induced by the rabbit hemorrhagic disease virus. J Pineal Res 50:38–45
Tuñón MJ, San Miguel B, Crespo I, Riezu-Boj JI, Larrea E, Alvarez M, González I, Bustos M, González-Gallego J, Prieto J (2011b) Cardiotrophin-1 promotes a high survival rate in rabbits with lethal fulminant hepatitis of viral origin. J Virol 85:13124–13132
Van de Bildt MWG, van Bolhuis GH, van Zijderveld F, van Riel D, Drees JM, Osterhaus ADME, Kuiken T (2006) Confirmation and phylogenetic analysis of rabbit haemorrhagic disease virus in free-living rabbits from the Netherlands. J Wildlife Disease 42:808–812
Viaplana E, Plana J, Villaverde A (1997) Antigenicity of VP60 structural protein of rabbit haemorrhagic disease virus. Arch Virol 142:1843–1848
Wang X, Hao H, Qiu L, Dang R, Du E, Zhang S, Yang Z (2012) Phylogenetic analysis of rabbit hemorrhagic disease virus in China and the antigenic variation of new strains. Arch Virol 157:1523–1530. doi:10.1007/s00705-012-1340-9
Wirblich C, Thiel HJ, Meyers G (1996) Genetic map of the calicivirus rabbit hemorrhagic disease virus as deduced from in vitro translation studies. J Virol 70:7974–7983
Yang L, Wang F, Hu B, Xue J, Hu Y, Zhou B, Wang D, Xu W (2008) Development of an RT-PCR for rabbit hemorrhagic disease virus (RHDV) and the epidemiology of RHDV in three eastern provinces of China. J Virol Methods 151:24–29
Acknowledgements
This work was supported by grant no. 2 P06K 02927 from the Polish National Science Centre.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
About this article
Cite this article
Hukowska-Szematowicz, B., Tokarz-Deptuła, B. & Deptuła, W. Analysis of genetic variability and phylogenetic analysis of selected Czech and French strains of rabbit haemorrhagic disease virus (RHDV). J Appl Genetics 54, 235–248 (2013). https://doi.org/10.1007/s13353-013-0140-6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13353-013-0140-6