
Abstract
We investigated whether pharmacological increase of “M-type” (KCNQ, Kv7) K + channel currents by the M-channel opener, retigabine (RTG), acutely after repetitive traumatic brain injuries (rTBIs), prevents or reduces their long-term detrimental effects. rTBIs were studied using a blast shock air wave mouse model. Animals were monitored by video and electroencephalogram (EEG) records for nine months after the last injury to assess the occurrence of post-traumatic seizures (PTS), post-traumatic epilepsy (PTE), sleep–wake cycle architecture alterations, and the power of the EEG signals. We evaluated the development of long-term changes in the brain associated with various neurodegenerative diseases in mice by examining transactive response DNA-binding protein 43 (TDP-43) expression and nerve fiber damage ~ 2 years after the rTBIs. We observed acute RTG treatment to reduce the duration of PTS and impair the development of PTE. Acute RTG treatment also prevented post-injury hypersomnia, nerve fiber damage, and cortical TDP-43 accumulation and translocation from the nucleus to the cytoplasm. Mice that developed PTE displayed impaired rapid eye movement (REM) sleep, and there were significant correlations between seizure duration and time spent in the different stages of the sleep–wake cycle. We observed acute RTG treatment to impair injury-induced reduction of age-related increase in gamma frequency power of the EGG, which has been suggested to be necessary for a healthy aged brain. The data show that RTG, administered acutely post-TBI, is a promising, novel therapeutic option to blunt/prevent several long-term effects of rTBIs. Furthermore, our results show a direct relationship between sleep architecture and PTE.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Traumatic brain injury (TBI) can result in post-traumatic seizures (PTS) and the development of post-traumatic epilepsy (PTE), which in humans can have a latent period of up to ten years [1, 2]. Approximately 6% of all epilepsy cases are caused by head trauma [3]. Compounding the problem is that PTS facilitates the process of epileptogenesis. However, whereas all anticonvulsants tested previously have been effective in reducing PTS, they have not been effective in preventing PTE development [1, 4].
Sleep disorders are also common following TBIs. Short-term sleep disorders affect 46–50% of TBI patients and include symptoms such as apnea, insomnia, or, conversely, hypersomnia [5, 6]. Sleep disorders are also common comorbidities in many epilepsy patients [7]. Using a rat TBI model, others have shown that most generalized PTE seizures occur at the transition from slow-wave sleep (SWS) to rapid eye movement (REM) sleep and that alterations in sleep electrographic characteristics can even be used as a biomarker of PTE [8].
The occurrence of mild repetitive traumatic brain injuries (rTBIs) is common in athletes, victims of domestic violence, and military personnel. rTBIs produce a cumulative and sometimes synergistic effect that increases the risk of multiple brain disorders [9]. A history of rTBIs can result in the development of various chronic neurodegenerative diseases such as chronic traumatic encephalopathy (CTE) [10, 11], Alzheimer’s disease (AD) [12], amyotrophic lateral sclerosis (ALS) [12], and frontotemporal dementia (FTD) [13]. Damage of nerve fibers is commonly seen in patients with CTE [14], AD [15], ALS [16], and FTD [17]. Accumulation of transactive response DNA-binding protein 43 (TDP-43) is also observed in patients with FTD [18, 19], ALS [19, 20], AD [21], and CTE [22, 23]. Additionally, both ALS and FTD induce increased levels of TDP-43 phosphorylation and translocation to the cytoplasm from the nucleus [24, 25]. TDP-43 regulates gene expression and in pathological conditions accumulates outside the nucleus forming neurotoxic plaques [26].
Of particular interest to military personnel, blast‐induced TBI is a result of exposure to shockwaves of high intensity, usually induced by explosive devices or the discharge of large-caliber weapons. It can lead to cell death, intracranial hemorrhages, edema, axonal injury, and inflammation [27, 28]. Using a mouse model of shock air wave-induced brain injury, we have reported that mild repetitive blast TBIs (rbTBIs), but not a single mild blast TBI, induced the occurrence of PTS [29]. We also observed that three rbTBIs resulted in the development of PTE in 46.1% of the mice (determined by 72 h of video/electroencephalogram (EEG) recording one month after rbTBIs). rbTBIs also induced a hyperexcitability phenotype in hippocampal neurons (shortened latency to the first spike and decreased action potential threshold) [29]. Hence, we decided to test the therapeutic effects of acute pharmacological M-current increase using the same rbTBI mouse model.
M-type (KCNQ, Kv7) channels are voltage-gated K+ channels that underlie the “M current” and play a crucial role in regulating neuronal excitability via acute or long-term up/down-regulation of activity [30]. M-type channels are named after the inhibition of its currents by activation of muscarinic receptors, and they can be composed of homo- or heterotetrameric assembly of different subunits (Kv7.1-Kv7.5). However, only Kv7.2-Kv7.5 are expressed in the nervous system [30, 31]. We have recently shown that acute pharmacological M-current increase with RTG blocks/reduces the development of various short-term deleterious effects of one TBI [32]. RTG has also been shown to reduce short-term deleterious effects in a zebrafish TBI model [33]. Here, we present evidence that acute pharmacological M-current increase after rbTBIs could be a possible novel therapeutic intervention to diminish the development of PTE, TBI-induced sleep disorders, and long-term neuropathology.
Methods
Animals
Adult 12-week-old male C57BL/6 J mice (Jackson Labs, Bar Harbor, ME) were group-housed with food and water ad libitum in a 12:12-h light: dark cycle. Only male mice were used in this study. Nonetheless, we have previously shown that pharmacological M-current increase is also beneficial for occluding the deleterious effects of a TBI in female mice as well [32]. Experimenters were blinded to group allocation during data collection and analysis. The experiments followed the National Institutes of Health Guide for Care and Use of Laboratory Animals and are reported here according to the Animal Research: Reporting in Vivo Experiments guidelines. The Institutional Animal Care and Use Committee at University of Texas Health San Antonio approved all experiments.
Repetitive Blast Traumatic Brain Injury
The rbTBI protocol used was the same described previously [29]. The shock tube (Applied Research Associates) had a 43 cm inner diameter and a 91.5 cm end opening. The blast was generated by a 61 cm driver with a 244 cm long driven section and a 122 cm expansion cone with a 7.5-degree angle of expansion. Before reaching the mice, the air blast wave passed through a 4-mm thick aluminum membrane of the alloy AL-2200. Pencil probe gages (PCB Piezotronics) were placed beside the mice to monitor the pressure of the air-driven blast wave. The airwaves presented the desired Friedlander waveform, with an average maximum peak incident overpressure of 14.6 ± 0.5 psi and a positive phase of 2.8 ± 0.5 ms. During the procedure, mice were anesthetized using ketamine (25–75 mg/kg) and dexmedetomidine (0.25 mg/kg). The animals were subjected to three directed head-on mild blast TBIs with 24 h intervals between each occurrence. On the first day of blast exposure, animals were given a subcutaneous injection of buprenorphine SR (1.2 mg/kg) in the interscapular region. Sham animals were anesthetized and placed in the shock tube but were not subjected to the rbTBIs. After each blast exposure, mice were given one subcutaneous injection of atipamezole (1 mg/kg) to reverse the anesthesia and were placed into a recovery chamber with a controlled temperature. In the following days, mice were monitored for signs of pain and distress.
Retigabine Administration
Retigabine (RTG) (AdooQ Bioscience) dilution and administration were performed as previously described [32]. RTG was diluted to a concentration of 330 µM in sterile 0.9% (w/v) saline solution with 0.5% (w/v) methylcellulose. Each animal from the “Blast + RTG” group received one i.p. injection of RTG (1.2 mg/kg) 30 min after each blast exposure. Mice from the “Blast” group were injected with the same volume of only vehicle solution. Mice from the Sham group did not receive any treatment besides what was necessary for the anesthesia procedure.
Electroencephalogram
To study the occurrence of seizures and the sleep–wake cycle architecture, EEG electrodes were implanted in the mice 24 h after the last blast injury. Sham mice were also implanted with EEG electrodes. We performed simultaneous video and EEG monitoring as in our previous work [29, 32, 34]. Prefabricated head mounts with screw electrodes (Pinnacle Technologies) were used. Two electrocorticogram (ECOG) signals were obtained using four depths electrodes: a ground screw (left frontal lobe; -2 mm rostra-caudal and 1.5 mm medial–lateral from the bregma), a reference screw electrode (left parietal lobe; 4 mm rostra-caudal and 1.5 mm medial–lateral from the bregma), a right frontal lobe recording electrode (-2 mm rostra-caudal and 1.5 mm medial–lateral from the bregma), and a right parietal lobe recording electrode (4 mm rostra-caudal and 1.5 mm medial–lateral from the bregma). The two ECOG signals obtained from these electrodes are presented as the two top lines of the EEG records shown in Figs. 1 and 2. Furthermore, one EEG signal was obtained from surface bipolar recording electrodes that were in contact with the animal’s skull in the parietal region of both hemispheres (3 mm rostra-caudal and 0.5 mm medial–lateral from the bregma). The signal obtained with these surface electrodes is presented in Figs. 1 and 2 as the bottom line of the EEG records shown. Starting one day after EEG surgery (2 days after the last blast injury), mice were video/EEG monitored for 72 h. Afterward, mice were monitored for 48 h every month for 8 months (starting one month after the last blast injury). For EEG recording, the head mount was connected to a pre-amplifier (Gain 25X; Pinnacle Technologies) connected to extracellular amplifiers of the Stellate Harmonie acquisition hardware. During all the record sessions, mice were under 12:12-h light: dark cycle with food and water ad libitum. The EEG signal was filtered during acquisition with a 60 Hz notch noise filter. During seizure analysis, the EEG signal was filtered with a 0.1 to 45 Hz bandpass filter.

Retigabine (RTG) treatment reduced the duration of post-traumatic seizures (PTS). a Bars summarize data of animals from “Sham,” “Blast,” and “Blast + RTG” groups that displayed spontaneous seizures on days 2–4 after the third blast exposure. The number of animals that displayed seizures in each group and the total number of animals per group are indicated above each bar. The lines in graphs (b), (c), and (d) show the average and standard error of the mean for each group. b Each dot in the graph represents the average seizure duration of each mouse. Acute post-rbTBIs RTG treatment significantly reduced the duration of the PTS. c Graphical representation of the total number of PTS recorded in each mouse (represented by each dot in the graph) from “Blast” and “Blast + RTG” groups. d The frequency of seizures recorded per hour in the first video/electroencephalogram (EEG) session (2–4 days post-injury) is shown for each mouse (represented by each dot) from Blast and “Blast + RTG” groups. e Examples of EEG recordings of seizures in animals from “Blast” and “Blast + RTG” groups of mice. Red bars indicate the seizure events, and the voltage and time scales can be seen in the bottom left corner. The same data are presented in two different voltage and time scales. f Summarized data of the percentage of PTS from “Blast” and “Blast + RTG” groups that happened in awake mice (white background and horizontal lines) or when mice were asleep (dark background and small dots). “Sham” n = 5, “Blast” n = 6, and “Blast + RTG” n = 12; *p < 0.05; Pearson chi-square test (a) and (f), one-way ANOVA (b)–(d)

Retigabine (RTG) treatment impaired post-traumatic epilepsy (PTE) development. a Bars summarize the percentage of mice from “Sham,” “Blast,” and “Blast + RTG” groups that had seizures 1–9 months after the third blast exposure. The occurrence of spontaneous seizures during this time window was considered as a sign of PTE. The number of animals that displayed seizures in each group and the total number of animals per group are indicated above each bar. Eight “Sham” mice were also video/electroencephalogram (EEG) monitored, and no seizures were observed. Acute RTG treatment after each blast-induced injury significantly reduced the development of PTE. Each dot in graph (b) represents the average seizure duration obtained for each mouse. Each dot of graph (c) shows the total number of PTE seizures recorded in each mouse. The lines in graphs (b) and (c) show each group’s average and standard error of the mean. d Examples of EEG recording of a seizure in an animal in the “Blast” group and a phenotypically normal EEG recording in a mouse in the “Blast + RTG” group. The red bar marks the seizure event and voltage, and time scale bars can be seen in the bottom left corner of the panel. The EEG recording of a representative PTE seizure is shown in two different time and voltage scales. e The graph shows the percentage of PTE seizures recorded from “Blast” and “Blast + RTG” mice that happened when the animals were awake (white background and horizontal lines) and the percentage that occurred during sleep (dark background and small dots). “Sham” n = 8, “Blast” n = 12, and “Blast + RTG” n = 16; *p < 0.05; Pearson chi-square test (a) and (e), one-way ANOVA (b) and (c)
Seizure Analysis
Seizures were identified visually by the investigator’s analysis of the video/EEG records. Only seizure events that included both seizure-like EEG signals and behavioral changes concomitantly were considered seizures. For EEG signal analysis, seizures were defined as an abnormal increase in the frequency combined with an increase in the amplitude of one or more signals from the EEG electrodes. To be counted as such, increases in EEG signal frequency and amplitude must vary in intensity during the seizure event and occur concomitantly with a seizure-like behavioral change. Seizure epileptiform activity included spikes, polyspikes, slow waves, and spike-and-wave discharges. Interictal epileptiform spiking activity before the onset of the seizure and postictal depression (decrease of EEG signal below background after seizure) was typically observed. Seizure-like behavioral changes were determined following the parameters defined by Shimada and Yamagata [35], which include frequent jerking/twitching of the head, limbs, and tail (typical of myoclonic seizures) or tail suspended stiff in an upward position combined with unusually prolonged stiffening of the front and/or back limbs and whole-body shaking that immobilized the animal causing him to fall on its side or back (commonly observed in clonic and tonic–clonic seizures). These behavioral changes were only considered a seizure if they occurred in combination with the previously described changes in EEG signal. The total number of seizures, frequency of the seizures, average seizure duration, and percentage of mice from each group that had seizures were quantified. Seizure duration was calculated for each mouse independently by averaging the duration of all the seizures observed in this animal. Seizures presented in the first 72 h of record (2–4 days after rbTBIs) were considered PTS, whereas recurrent seizures that occurred one month or more after the last blast injury were considered indications of PTE. The frequency of PTS per hour was calculated by dividing the total number of PTS observed by the 72 h of recording. The frequency of PTE seizures was not calculated as it is unlikely to be accrued due to the limitations imposed by the sparse recording protocol applied to identify mice that developed PTE over such a long time period. The percentage of recorded seizures that occurred during sleep and awake states was quantified for PTS and PTE. No seizure events were observed in mice from the Sham group. We did observe the occurrence of abnormal EEG signals that could be classified as “seizure-like” but without a corresponding behavioral change. These events were observed in mice from the three groups and were not counted as seizure events in this study.
The EEG experiments started with 16 mice in each group as determined by power analysis using results from our pilot experiments (not shown). Unfortunately, we were unable to record the first session of 72 h (PTS) for 8 mice from the “Sham” group, 10 mice from the “Blast” group, and 4 mice from the “Blast + RTG” group because of a quarantine imposed to our animal facility due to an infection of mouse mites. Although our animals were not affected by this infection, the facility was isolated for a month. Hence, there is a lower number of mice in the PTS experiments (Fig. 1; Sham = 5, Blast = 6, and Blast + RTG = 12) compared to the PTE experiments (Fig. 2; Sham = 8, Blast = 12, and Blast + RTG = 16). Additionally, for the PTE experiments, only mice from which we collected data in all 8 months of PTE recording were considered. As a result, 8 mice from the “Sham” group and 4 mice from the “Blast” group were excluded since their head mount fell off before the 8 months of PTE recording sessions were completed. Therefore, from the 16 original mice per group, the numbers of mice in the Sham, Blast, and Blast + RTG groups were 8, 12, and 16 mice, respectively, in the PTE data set (Fig. 2a).
Sleep–Wake Cycle Architecture Analysis
To evaluate the sleep–wake cycle, video/EEG records from the first 72 h long session (starting 2 days after the last injury), the second last (48 h long, 8 months after the last injury), and the last session (48 h long, 9 months after the last injury) were analyzed. To select one EEG channel from each animal/session for analysis, we performed visual inspections of the raw local field potentials and their time–frequency decompositions (spectrograms) recorded from the two cortical electrodes. EEG channels with signal saturation and background noise or artifacts were discarded. Then, the EEG channel with the highest percentage power at the 5–12 Hz frequency band was used to classify the sleep–wake cycle (Fig. S1). Awake state (WK), SWS, REM sleep, and transition/undetermined states (US) were classified based on two power spectral ratios, as described by Gervasoni et al. [36]. In brief, this analysis first measures the relative power within fast (0.5–20/0.5–50 Hz) and slow (0.5–4.5/0.5–9 Hz) frequency bands and then generates a 2D state-space on which clusters are manually selected and associated with each state of the sleep–wake cycle.
EEG Signal Coherence and Power Analysis
The power of the EEG signal in the different frequencies was calculated using the “pwelch” function of Matlab. In brief, the power of each frequency was decomposed in a distribution histogram (Fig. 4b). The power of the EEG signal in the gamma (30–50 Hz) and the delta (2–9 Hz) frequencies was calculated as the average of the power in these ranges of frequencies. The average power obtained was then divided by the average power of the EEG signal for all the frequencies. Therefore, the power of the gamma or the delta frequencies is presented as a percentage of the total power of the EEG signal. The power of the EEG signals was calculated during the SWS state. For coherence analysis, the Matlab function “mscohere” was used. For this analysis, in brief, the difference in the phase of the wave between two EEG signals of the same mouse during the SWS stage was calculated. The EEG signals compared were the ones obtained in the right frontal lobe electrode and the right parietal lobe electrode. These two EEG signals were obtained with intracranial depth electrodes, whereas the third EEG signal was collected with a subcutaneous bipolar surface electrode. Whereas the power analysis reflected the amplitude of a wave in a specific frequency, the coherence analysis reflected the synchrony of two EEG signals in a particular frequency.
Sample Collection from Aged Mice
Cortical and hippocampal samples used for immunohistochemistry, immunoblotting, and Luxol fast blue were collected from mice at 26–28 months of age. During the aging period, many mice died due to complications from the EEG headmount and other unknown reasons, although such mice were not distinguished in any way evident to the experimenters. Therefore, an independent cohort of mice was aged in parallel with the mice used for the EEG recordings. These mice were subjected to the same rbTBIs and RTG/vehicle treatment procedures, or sham, but not to the EEG surgery and recording. Those two cohorts of mice were combined to compensate for the loss of mice during aging and allowed us to study neuronal molecular and structural changes observed in age-related neurodegenerative diseases. Hence, samples used in immunohistochemistry, immunoblotting, and Luxol fast blue analyses were from both the EEG-subjected mice and from this parallel independent cohort of mice. Samples used for immunoblotting were from the left hemisphere, whereas the right hemisphere was used for immunohistochemistry and Luxol fast blue.
Immunoblotting
Expression of TDP-43 was first analyzed by immunoblotting performed as previously described [32]. Samples for immunoblotting were dissected in ice-cold Ringer’s solution, snap-frozen in dry ice, and stored at -80 °C until analysis. Samples were homogenized in RIPA buffer (Thermo Scientific) using a tissue homogenizer (Ultra EZgrind—Denville Scientific) at 5000 rpm. Proteins were separated in 4–20% gels (Bio-Rad) and transferred to a polyvinylidene difluoride (PVDF) membrane (Bio-Rad). Primary antibodies used were TDP-43 (1:1000, Proteintech 12,892–1-AP), pTDP-43 (1:1000, Cosmo Bio TIP-PTD-M01), Tau (1:1500, MilliporeSigma MAB361), and pTau (1:200, PHF-1 provided by Dr. Peter Davis from Albert Einstein Institute). Immunoblot membranes were stripped (Santa Cruz Biotechnology) and re-probed to stain glyceraldehyde 3-phosphate dehydrogenase (GAPDH) (1:25,000, Abcam ab125247) or β-actin (1:20,000, Sigma A5316) as loading controls. Striping was also performed to stain total and phosphorylated Tau or TDP-43. Exemplary images of entire immunoblot films can be seen in the Supplementary Fig. S2. Results were standardized to the average of the results from Sham mice.
Immunohistochemistry
Complementary to the immunoblot experiments, immunohistochemistry of TDP-43 was performed as previously described [32]. Left-hemisphere samples were removed and fixed overnight in 4% paraformaldehyde (Electron Microscopy Sciences). Subsequently, samples were transferred into 30% (w/v) sucrose in phosphate-buffered saline (PBS) and kept for 3 days at 4 °C before being frozen in Tissue-Plus O.C.T. Compound (Fisher Scientific) using 2-methylbutane immersed in dry ice. Samples were stored at -80 °C until cryosectioning. Twenty-µm-thick coronal sections of the prefrontal cortex (PFC) were mounted on Superfrost Plus microscope slides (Fisher Scientific) previously coated with gelatin (Sigma). Before staining, slices were blocked with 8% (v/v) donkey serum with triton-x (0.1% v/v) and Tween (0.1% v/v). Primary antibody against TDP-43 (1:200, Proteintech 12,892–1-AP) was used. The NeuroTrace 500/525 Green Fluorescent Nissl Stain (Thermo Fisher Scientific—N21480) was used to stain neurons. Slides were mounted using an anti-fade mounting medium (Vectashield) containing 4′,6-diamidino-2-phenylindole (DAPI) (Vector Laboratories) for nuclear staining. Images were taken from slices of the PFC layers II/III at 60X magnification using a Zeiss LSM 710 confocal microscope. The range indicator option was used to confirm that there was no camera saturation by the fluorescent signal in every image in the three channels. Images were obtained by combining four sweeps in each focal plane, and Z stack images were obtained with 0.5 µm intervals. Fluorescence settings and parameters were held constant for all images (DAPI: laser at 2% power, gain 725, digital offset 140, digital gain 1; NeuroTrace: laser at 3.5% power, gain 770, digital offset 150, digital gain 1; TDP-43: laser at 6% power, gain 850, digital offset 225, digital gain 1.5). The mean and maximum fluorescence signals were obtained by automatic quantification with ImageJ (National Institutes of Health, USA). Regions of interest were determined automatically using the “analyze particles” function of ImageJ (size 0.1–2000 pixel2; circularity 0.00–5.00). For colocalization analyses, the Mander’s overlap coefficients (M1) were obtained using the ImageJ plugin JACoP. The percentage of neurons per image in which TDP-43 staining was visualized was quantified manually.
Luxol Fast Blue
Tissues collected for immunohistochemistry experiments were also used for Luxol fast blue staining. Twenty-µm-thick coronal sections of the striatum and corpus callosum were stained to study nerve fiber integrity. These brain areas were selected due to the high content of myelinated fibers. To stain myelin, slices were incubated in Luxol fast blue solution (Sigma) overnight at 60 °C. Eosin Y stain (Ricca chemical company) and Cresyl Violet (0.1% w/v, Abcam) were used for counterstaining. Photos were taken in an Olympus microscope (BX 60) with an Olympus camera (DP-71) and a 10X objective. Images were analyzed using ImageJ. The diameter or the area of each nerve fiber was measured in the corpus callosum and the striatum, respectively. All nerve fibers with boundaries that could be determined clearly by the complete lack of Luxol fast blue staining around it were measured in all images.
Statistical Analysis
The percentage of mice in each group that displayed seizures and the percentage of seizures that occurred during sleep were analyzed using the Pearson chi-square test since those are dichotomous nominal variables. All other data were checked for normal distributions using the Shapiro–Wilk normality test and Q-Q plot analysis. If the data showed a non-normal distribution, outliers were excluded from the analysis as a normalization strategy [37, 38]. Outliers were defined as values that fall more than 1.5 times the interquartile range below the first quartile or above the third quartile [38]. If the dataset had no outliers, it was transformed to Log10 [39]. If these two normalization strategies were ineffective, outliers were returned to the dataset, and the data was considered to have a non-normal distribution. Non-normally distributed data were analyzed using the Kruskal–Wallis test followed by Mann–Whitney U (MWU) test with Bonferroni correction as post hoc analysis. These data are presented using box and whisker plots in which minimum, first quartile, median, third quartile, and maximum values are displayed. ANOVA and Student–Newman–Keuls (SNK) post hoc tests were used for normally distributed data. Normally distributed data are presented using scatter plot graphs with mean and standard error of the mean (SEM). The relationship between seizure-related parameters and time spent in different sleep–wake cycle states was tested by linear regression analysis since these variables were collected in the same cohort of mice.
Results
Acute RTG Treatment Reduced the Duration of rbTBI-Induced PTS
Mice subjected to three rbTBIs were video/EEG monitored for 3 consecutive days (starting 48 h after the last blast TBI), and PTS was evaluated. We did not observe a significant difference in the percentage of mice that displayed PTS between animals injected with RTG after each blast (Blast + RTG) and those that received vehicle only (Blast) (χ(1) = 0.4; p = 0.50) (Fig. 1a). All mice from which we were able to record seizures were included in the analysis of different seizure-related parameters. RTG administration caused a significant decrease in the average seizure duration (F1,8 = 5.4; p = 0.04) (Fig. 1b, e). There was no significant difference between Blast and Blast + RTG groups of mice in number of seizures observed (F1,8 = 0.5; p = 0.51) and of the frequency of seizures per hour (F1,8 = 0.5; p = 0.51) (Fig. 1c, d). During PTS recording, 49.6% of seizures in the Blast group occurred while the mice were sleeping, and 51.0% of seizures occurred during sleep in the Blast + RTG group (Fig. 1f). There was no significant difference between the groups (χ(1) = 0.04; p = 0.83). Five sham mice were also video/EEG monitored, for which no seizures were observed.
Acute RTG Treatment Impaired the Development of PTE
Starting one month after the rbTBIs, the same cohort of mice was recorded for 8 months to assay for PTE, defined as the occurrence of recurrent spontaneous seizures one month or more after rbTBIs. Acute RTG treatment after each TBI significantly decreased the percentage of mice that developed PTE (χ(1) = 4.9; p = 0.02). Whereas 67% of mice that received vehicle only developed PTE, only 25% of “Blast + RTG” mice displayed PTE (Fig. 2a, d). All mice from which we were able to record seizures 1–9 months post-rbTBIs were included in the analysis of different seizure-related parameters. There was no significant difference in the duration of the seizures (F1,10 = 1.3; p = 0.27) (Fig. 2b) or in the number of recorded seizures (F1,10 = 0.5; p = 0.50) (Fig. 2c) between “Blast” and “Blast + RTG” mice. During PTE recording, 41.2% of seizures recorded in mice from the Blast group and 41.7% of seizures in mice from the Blast + RTG group occurred during sleep (Fig. 2e). There was no significant difference between the groups (χ(1) = 0.0007; p = 0.98). Eight sham mice were also video/EEG monitored, for which no seizures were observed.
Acute RTG Treatment Blocked rbTBI-Induced Hypersomnia
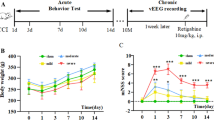
To study short and long-term changes in sleep–wake cycle architecture, the first (2–5 days after rbTBIs), the second last (8 months after rbTBIs), and the last (9 months after rbTBIs) video/EEG recording sessions were analyzed. In the first session, mice from the “Blast” group presented a hypersomnia phenotype with a lower percentage of time spent awake than “Blast + RTG” animals (one-way ANOVA: F2,17 = 5.9; p = 0.01 // SNK: q = 4.4; p < 0.01) and “Sham” animals (q = 4.4; p = 0.01) (Fig. 3b). There was no difference between the mice of the different groups in the percentage of time spent in SWS (F2,20 = 0.6; p = 0.53) (Fig. 3c), REM (F2,20 = 0.8; p = 0.47) (Fig. 3d), or US (F2,20 = 1.5; p = 0.24) (Fig. 3e) states. In the second last and last video/EEG record sessions, we did not observe any significant difference between any of the groups of mice (Figs. S3 and S4).

Retigabine (RTG) treatment impaired repetitive blast traumatic brain injuries (rbTBIs)-induced hypersomnia. a Three examples of 2D state maps obtained from “Sham,” “Blast,” and “Blast + RTG” animals. Black points represent one-second intervals from ratio 1 and ratio 2 of the local field potentials. Each of these examples shows three clusters scattered through different regions of the graph, in which each cluster can be considered a sleep stage. Red circles represent slow-wave sleep (SWS), blue circles represent rapid eye movement (REM) sleep, and green circles represent awake state (WK). Note that points placed out of these clusters were considered as transition/undetermined states (US). b A significant decrease in the percentage of time spent in the WK state was observed in mice from the “Blast” group but not in mice treated with RTG during the first session of video/electroencephalogram (EEG) recording. c–e No other significant difference was observed between the three groups of mice in the first recording session. Data are displayed as the mean and standard error of the mean; “Sham” n = 5; “Blast” n = 6, and “Blast + RTG” n = 12; *p < 0.05, and.**p < 0.01
To evaluate changes in the sleep–wake cycle architecture of mice with PTE, animals from all the groups (Sham, Blast, and Blast + RTG) were combined and divided between “Epileptic” and “Non-epileptic” mice, independent of treatment. Epileptic mice displayed impaired REM sleep, and various seizure-related parameters correlated significantly with the percentage of time spent in different states of the sleep–wake cycle (Fig. S5).
RTG Treatment Preserved Physiological Age-Induced Increase in the Power of the Gamma Frequency That Was Otherwise Impaired by rbTBIs
Age-related neurodegenerative diseases like AD induce alterations in the EEG signal (e.g., impaired gamma frequency), and healthy brain aging seems to have the opposite effect (e.g., increased power of the gamma frequency) [40,41,42,43,44,45]. To study the age-related effects of the rbTBIs and of RTG treatment, the power of the gamma and the delta frequencies was calculated in the first (2–5 days after rbTBIs) and last (9 months after rbTBIs) EEG sessions for the two intracranial electrodes. When comparing the power of the gamma frequency in the first EEG recording session against the last session, both recorded in the right parietal lobe electrode, we observed a significant increase in mice from the “Sham” (two-way ANOVA: effect of group: F2,56 = 3.8; p = 0.02 / effect of sessions: F1,56 = 12.4; p < 0.001 / interaction effect: F2,56 = 0.3; p = 0.74 // SNK: q = 3.3; p = 0.02) and the “Blast + RTG” groups (q = 3.3; p = 0.02), but not in mice from the “Blast” group (q = 2.1; p = 0.14) (Fig. 4c). Nevertheless, the power of the gamma frequency in the last EEG record session was significantly higher in “Sham” mice compared to both “Blast” (q = 3.6; p = 0.03) and “Blast + RTG” (q = 3.4; p = 0.01) mice. There was no significant difference in the power of the gamma frequency in the last video/EEG session between mice from the “Blast” and “Blast + RTG” groups (q = 0.6; p = 0.67). There was also no significant difference between mice from the three groups in the first EEG session (Sham X Blast: q = 1.5; p = 0.52 // Sham X Blast + RTG: q = 1.6; p = 0.23 // Blast X Blast + RTG: q = 0.1; p = 0.95). Still in the right parietal lobe electrode, a two-way ANOVA test of the power of the delta frequency revealed a significant effect of group (F2,56 = 3.6; p = 0.03), a significant effect of the sessions (F1,56 = 4.8; p = 0.03), and no interaction effect (F2,56 = 0.5; p = 0.58). However, post hoc analysis with the SNK test did not show any significant difference between the sessions within the groups or between the groups within both sessions studied (Fig. 4d) (Supplementary Table S1).

Physiological age-induced increase in the power of the gamma frequency is impaired by traumatic brain injury (TBI), but preserved by acute post-repetitive blast traumatic brain injuries (rbTBIs) retigabine (RTG) treatment. a Shown are representative exemplary raw electroencephalogram (EEG) signals for each group of mice during the first (black) (2–5 days after rbTBIs) and the last (red) (9 months after rbTBIs) EEG sessions. b Shown are the relative power of the EEG signals in the different frequencies for each group of mice in the first (S1 – black) and last (S9 – red) EEG sessions. The relative power of the gamma (c) and delta (d) frequencies recorded in the right parietal lobe electrode was calculated in the first and the last EEG sessions. Animals from the “Sham” and “Blast + RTG” groups displayed an increase in the power of the gamma frequency when comparing the first to the last EEG sessions. This increase in the power of the gamma frequency is a physiological age-induced phenomenon previously observed in mice and is believed to be important for healthy brain aging [45]. No such increase was observed in “Blast” mice injected with vehicle only. Additionally, “Sham” mice had significantly higher power of the gamma frequency signal in the last EEG session compared to “Blast” and “Blast + RTG” mice. Hence, although RTG treatment reduced the deleterious effects of rbTBIs, it did not completely prevent the impairment of the gamma frequency in the last session. Data are displayed as the mean and standard error of the mean; “Sham” n = 5–8; “Blast” n = 6–12, and “Blast + RTG” n = 12–16; *p < 0.05
Analysis of the power of the gamma and delta frequencies in the right frontal lobe electrode showed no significant differences between the groups within the sessions or between the sessions within any of the groups (Fig. S6).
We also analyzed the power of the gamma and delta frequencies in the second last EEG session (8 months after rbTBIs). Once more, when analyzing the data from the right frontal lobe electrode, there was no difference in the power of the gamma or delta frequencies compared to the first or the last EEG sessions (Fig. S7). In signals from the right parietal lobe electrode, there was also no significant difference in the delta frequency comparing the second last session to the first or the last EEG session (Fig. S8). In the right parietal lobe electrode, there was no significant difference in the gamma frequency comparing the second last session to the last EEG session (Fig. S8). However, in the right parietal lobe electrode, there was a significant increase in the gamma frequency of the Blast + RTG group comparing the second last session to the first EEG session (Fig. S8), similar to what was observed in the comparison between the first and the last EEG sessions in the same EEG electrode (Fig. 4c). Additionally, we analyzed the coherence of the EEG signal between the two electrodes in the gamma and the delta frequencies during the first and the last video/EEG record sessions. No significant difference was observed between the groups within a session or between the sessions within a group, neither in the delta nor in the gamma frequencies (Fig. S9).
Acute RTG Treatment Prevented rbTBI-Induced Increases in Cortical Levels of TDP-43 in Aged Mice
TDP-43 is a protein that accumulates in many age-related neurodegenerative diseases, for which TBI is a risk factor [18,19,20,21,22,23]. TDP-43 translocates to the cytoplasm from the nucleus in pathological conditions and its accumulation there is neurotoxic [24,25,26]. Immunoblotting analysis of brain samples from aged (26–28 months old; 23–25 months post-rbTBIs) mice revealed a significant increase in the cortical levels of TDP-43 in animals from the “Blast” group compared to mice from both “Sham” (one-way ANOVA: F2,29 = 3.6; p = 0.04 // SNK: q = 3.1; p = 0.03) and “Blast + RTG” (q = 3.5; p = 0.04) groups (Fig. 5a). There was no significant difference in the cortical levels of TDP-43 between mice from “Sham” and “Blast + RTG” groups (q = 0.3; p = 0.82). There was no significant difference in the ratio of TDP-43 phosphorylation in the cortex of the animals (Fig. 5b) (F2,36 = 1.24; p = 0.30). There was also no significant difference in the levels of TDP-43 between any of the three groups of mice in the hippocampus (Fig. S10). Additionally, we analyzed the expression of Tau and the ratio of Tau phosphorylation in the cortex of the mice. No significant difference between the three groups of mice was observed (Fig. S11).

Acute retigabine (RTG) treatment impaired repetitive blast traumatic brain injuries (rbTBIs)-induced transactive response DNA-binding protein 43 (TDP-43) accumulation in the cytoplasm from the nucleus. a Immunoblot analysis of cortical expression of TDP-43. TDP-43 levels were normalized to the levels of beta-actin. Each panel shows the graphical representation of the data on the top and representative exemplary bands on the bottom. The average of all the groups was normalized by the average of “Sham.” rbTBIs resulted in increased levels of TDP-43, which was blocked by acute post-rbTBIs RTG treatment. b Shown is the ratio of cortical TDP-43 phosphorylation measured by immunoblot. No significant difference between the three groups of mice was observed. Data are displayed as mean and SEM. (a–b) “Sham” n = 11–14, “Blast” n = 9–11, and “Blast + RTG” n = 12. Panel c shows representative images of immunohistochemistry staining of TPD-43, Neurotrace (Niss), and DAPI within layers II/III of the PFC in “Sham,” “Blast,” and “Blast + RTG” samples. A scale bar of 10 µm is shown in the top right corner of each image. The overlapping of the signals from the three channels is shown in the last column of images in (c). In the center is the image of one focal plane (XY). The combination of all the focal planes was obtained with Z-stack imaging. An XZ focal plane (image below XY image) and a YZ focal plane (image on the right of XY image) are shown. No significant difference between the groups was observed in the mean (d) and maximum (e) intensity of the immunofluorescence signal. f rbTBIs induced a significant increase in the number of neurons per image that expressed TDP-43. Such an increase was prevented by acute-RTG treatment. Each dot on graph (f) represents the percentage of neurons expressing TDP-43 observed in one image. We observed a significant decrease in the colocalization of DAPI and TDP-43 staining in “Blast” images compared to “Sham” and “Blast + RTG” images in the XZ (h) and the YZ (i) focal planes but not in the XY focal plane (g). Data are displayed in box and whisker plots or mean and standard error of the mean. “Sham” = 72 images from 9 mice; “Blast” = 30 images from 5 mice; “Blast + RTG” = 64 images from 7 mice; *p < 0.05, **p < 0.01, and.***p < 0.001
Acute RTG Treatment Impaired the rbTBI-Induced Increase in Neuronal TDP-43 Expression and Dislocation to Outside the Nucleus in Aged Mice
To better localize the rbTBI-induced increase in TDP-43 levels, we performed immunofluorescence staining of TDP-43, NeuroTrace (Nissl), and DAPI (nucleus) in slices from the PFC (layers II/III) of the three groups of mice (Fig. 5c). Samples were collected when mice were 26–28 months of age. No significant difference between the three groups was observed in the mean (Fig. 5d) (Kruskal–Wallis: p = 0.04 // MWU: Blast x Sham: p = 1.00 // Blast x Blast + RTG: p = 0.13 // Sham x Blast + RTG: p = 0.09) and maximum (Fig. 5e) (Kruskal–Wallis: p = 0.02 // MWU: Blast x Sham: p = 1.00 // Blast x Blast + RTG: p = 0.12 // Sham x Blast + RTG: p = 0.06) values of the fluorescent signal obtained with TDP-43 immunostaining. Nonetheless, the rbTBIs induced a significant increase in the percentage of neurons expressing TDP-43 that was prevented by acute RTG treatment (Fig. 5f) (one-way ANOVA: F2,164 = 6.6; p = 0.002; // SNK: Blast x Sham: q = 5.1; p < 0.001 // Blast x Blast + RTG: q = 3.9; p = 0.005 // Sham x Blast + RTG: q = 1.4; p = 0.32).
We also performed colocalization analysis to investigate if rbTBIs could induce changes in TDP-43 cellular localization. Colocalization of TDP-43 immunostaining with the nuclear staining DAPI was quantified using the Mander’s overlap coefficient (Fig. 5g–i). No significant changes in TDP-43 colocalization with DAPI were observed in the XY focal plane (Fig. 5g) (p = 0.38). The Mander’s overlap coefficient M1 revealed that ~ 4–2% of all TDP-43 was localized within the nuclei of neurons, independent of the animal’s group when observing in the XY focal plane. XZ and YZ focal planes were also obtained by combining images of the XY focal plane taken in series with the Z-stack imaging protocol. rbTBIs induced a reduction of TDP-43 colocalization with DAPI that was impaired by acute RTG treatment in both the XZ (Fig. 5h) (Kruskal–Wallis: p < 0.001 // MWU: Blast x Sham: p = 0.02 // Blast x Blast + RTG: p < 0.001 // Sham x Blast + RTG: p = 0.10) and YZ (Fig. 5i) (Kruskal–Wallis: p = 0.004 // MWU: Blast x Sham: p = 0.01 // Blast x Blast + RTG: p = 0.005 // Sham x Blast + RTG: p > 0.99) focal planes. In XZ and YZ focal planes, approximately 10–14% of all TDP-43 was localized within the nucleus in Sham and Blast + RTG mice, but only ~ 2% of the TDP-43 was localized in the nucleus in neurons from mice of the Blast group.
Acute RTG Treatment Prevented the rbTBI-Induced Damage of Nerve Fibers in Aged Mice
Finally, long-term damage to nerve fibers was studied. Mice from the “Blast” group had significantly thinner nerve fibers in the corpus callosum than “Sham” (Kruskal–Wallis: p < 0.001 // MWU: p < 0.001) and “Blast + RTG” (p < 0.001) mice (Fig. 6a–c). Nerve fibers in the striatum of mice from the “Blast” group were also thinner than nerve fibers of mice from the “Sham” (Kruskal–Wallis: p < 0.001 // MWU: p < 0.001) and the “Blast + RTG” (p < 0.001) groups (Fig. 6d–f). No significant difference between mice from the “Sham” and “Blast + RTG” groups was observed neither in the corpus callosum (p = 0.61) nor the striatum (p = 0.20).

Repetitive blast traumatic brain injuries (rbTBIs)-induced damage of nerve fibers in the corpus callosum and striatum was blocked by acute retigabine (RTG) treatment. Panel a shows representative images of Luxol fast blue staining of the myelin in the nerve fibers of the corpus callosum from “Sham,” “Blast,” and “Blast + RTG” samples. A scale bar of 100 µm is shown in the bottom right corner of each image in panels (a) and (d). Panel b shows expanded images of the areas pointed out by the red arrows in panel (a). The green lines in the images of the panel (b) show how fiber diameter was measured. A scale bar of 10 µm is shown in the top right corner of each image in panel (b). The diameter of the nerve fibers in the corpus callosum was reduced in “Blast” mice, but this reduction was prevented by RTG treatment (c). Similar results were observed in the nerve fibers of the striatum (d–f). The red arrows point to exemplary nerve fibers that were measured (d). In panel e, these fibers are shown more closely, and the magenta circles demarcate the areas of the nerve fibers that were measured (e–f). A scale bar of 10 µm is shown in the top right corner of each image in panel (e). Data are displayed in box and whisker plots. “Sham” = 9 mice, 10–15 images per mice, 1444 and 1588 nerve fibers measured in the corpus callosum and striatum, respectively; “Blast” = 5 mice, 10–15 images per mice, 1083 (corpus callosum) and 824 (striatum) nerve fibers measured; “Blast + RTG” = 7 mice, 10–15 images per mice, 1440 (corpus callosum) and 1005 (striatum) nerve fibers measured;.***p < 0.001
Discussion
The results presented here and previously [29] indicate our rbTBIs mouse model to be well-suited to study the development of short-term and long-term pathologies after TBI in mice. The rbTBI model used here revealed increases in Tau phosphorylation [29], induced seizures, increased cytoplasmatic levels of TDP-43, nerve fibers damage, and TBI-induced sleep disorder. More studies are necessary to determine which chronic neurodegenerative diseases are best modeled by our mouse rbTBI protocol (e.g., CTE, AD, ALS, and FTD). Previously, we showed Tau phosphorylation to be upregulated shortly (6 days) after rbTBIs [29]. However, previous experiments using different mice models of repetitive TBI have shown that significant changes in Tau phosphorylation can be observed up to 6 months after the last injury [46,47,48] but not at later time points (8–12 months) [49, 50]. The results presented here corroborate these observations. Additionally, we would like to investigate further the levels and intracellular localization of TDP-43 in different brain regions and at other time points. In both mice and fly models, TBI upregulates the expression of nuclear pore complex and nucleocytoplasmic transport proteins, increasing TDP-43 translocation to outside the nucleus and accumulation in the cytoplasm [51]. Accumulation of TDP-43 outside the nucleus is also observed in various chronic neurodegenerative diseases [24, 25]. Up to this point, there are no known direct interactions between TDP-43 and M-type K+ channels.
More studies are necessary to understand by what mechanism acute post-TBI RTG treatment prevented the rbTBI-induced nerve fiber damage and TDP-43 accumulation outside the nucleus almost two years after the last mTBI. Our current hypothesis is presented in more detail by Vigil et al. [52]. We believe the long-term therapeutic effects of RTG after TBI could be explained by blocking the cascade of events that starts with neuronal hyperexcitability and, if untreated, often progresses to a deleterious cycle of cell death and a maladaptive inflammatory response. In this hypothesis, maintaining nuclear TDP-43 and nerve fiber integrity to normal levels would be a long-term downstream effect of the acute reduction of neuronal hyperexcitability by M-current increase after TBI. The hypothesis that the reduction of neuronal activity is a key player for the therapeutic effects of acute post-TBI RTG injection is corroborated by Alyenbaawi et al. [33], who observed that induction of neuronal activity with kainic acid reduces short-term therapeutic effects of RTG post-TBI. Nonetheless, in the same experiments, the synergetic increase in cell death markers induced by the combination of TBI and kainic acid injection was significantly reduced by RTG treatment [33]. The impairment of the therapeutic effects of RTG treatment by kainic acid injection does not prove that occlusion of neuronal hyper-activity is sufficient to explain RTG’s therapeutic effects.
More acute mechanisms of action beyond the decrease of neuronal hyperexcitability are likely to be related to the therapeutic effects of post-TBI RTG injection. A different possible mechanism of action of post-TBI RTG treatment is the alteration of blood supply by relaxation of blood vessel smooth muscle. RTG more weakly augments currents from Kv7.4 and Kv7.5 of smooth muscle than those of Kv7.2 and Kv7.3 of neurons, yet still decreases contraction and increases blood vessel caliber [53, 54]. Such action on cerebral blood vessels may counteract TBI-induced hypoperfusion. Another possible acute therapeutic effect of RTG treatment could be the regulation of microglial response post-TBI. Microglia cells express Kv7.2, Kv7.3, and Kv7.5. Blocking these channels reduces the migration of resting/non-inflammatory microglia [55]. Therefore, an increased microglial M current could result in a less intense inflammatory response to multiple TBIs. These hypotheses remain to be tested.
Blocking epileptogenesis in our model may also be a downstream effect of the acute post-TBI M-current increase. We observed that one dose of RTG after each blast TBI was sufficient to reduce the duration of PTS but did not completely block its occurrence. Interestingly, acute M-current increase occluded PTE development. This suggests that reducing the duration of the PTS may prevent the initial steps (priming), leading to the development of PTE in our model. This hypothesis remains to be tested. However, we observed here that not all mice that developed PTE displayed PTS. Hence, other mechanisms besides priming by PTS must play a role in PTE development in our model. For example, intense inflammatory response and blood–brain barrier (BBB) breakdown have both been shown to induce the occurrence of seizures [56, 57] and are both reduced by acute RTG treatment after a TBI [32]. Reduction of TBI-induced death of neurons, especially inhibitory neurons [58, 59], is also a possible explanation for the impairment of PTE development reported here. A combination of all these factors and the reduction of additional TBI-induced changes in the brain not yet elucidated are likely to account for the RTG-induced reduction of PTE development reported in this study.
A limitation of our study is the exclusion of seizures not detected by our electrodes (e.g., subcortical seizures) and of seizures that did not induce behavioral changes included in our seizure parameters (e.g., absence seizures). Additionally, the frequency of video/EEG recordings performed is admittedly not sufficient to identify all epileptic mice, since seizure occurrence is highly intermittent in mice with PTE [60]. Nonetheless, the percentage of vehicle-treated mice that developed PTE in our experiments (66.7%) is approximate to the frequency of animals that developed PTE in other mouse TBI models (58%) [60, 61]. Therefore, it is unlikely that our protocol resulted in a significant undercount of the percentage of mice that developed PTE.
Hypersomnia affects around 20% of TBI patients and commonly occurs days to months after a TBI, but usually, it is not a sustained post-TBI effect [5, 6, 62]. In our model, this phenotype was prevented by acute M-current increase. Although the physiological mechanisms of hypersomnia after TBI are not entirely understood, one possibility is that TBI decreases the levels of the wake-promoting neurotransmitters, hypocretin and histamine [6]. How RTG treatment might prevent a TBI-induced decrease in the levels of these neurotransmitters remains to be determined. Another possible explanation for the observed prevention of TBI-induced hypersomnia by acute M-current increase could be the reduction of the post-TBI inflammatory response. Pro-inflammatory cytokines such as interleukin-6 and tumor necrosis factor-alpha play crucial roles in sleep regulation [63]. By reducing the inflammatory response after TBI [32], RTG treatment might maintain normal sleep regulation. Our analysis of alterations in the sleep–wake cycle architecture in epileptic animals revealed an impairment of REM sleep and an intimate relationship between the sleep–wake cycle and epilepsy (Fig. S5). This is in accord with previous reports in the literature [7, 8, 64].
Our EEG data analysis also revealed that mice from the “Sham” and the “Blast + RTG” groups had an increase in the power of the cortical EEG signal in the gamma frequency when we compared the first to the last sessions. This was not the case in mice from the “Blast” group. Leparulo et al. [65] observed a similar aged-induced increase in the power of the gamma frequency in control mice but not in AD mouse model animals. In both human AD patients and AD mouse models, the gamma frequency signal is impaired and increased gamma frequency power results in better performance in memory tasks [40,41,42,43,44]. The power of the gamma frequency is increased in humans when performing working memory-associated tasks [66, 67]. Moreover, healthy aging leads to an increase in the power of the gamma frequency, which is critical to support normal working memory function [45]. It is important to remember that our experiments calculated the power of the EEG signal during the SWS state. An increase in the power of the gamma frequency during SWS has been associated with increased information processing [68]. Thus, our results corroborate with a rbTBI-induced impairment of healthy brain aging that was reduced by acute RTG treatment after each TBI. More experiments are necessary to test the effects of the observed changes in the gamma frequency on the working memory of the mice.
RTG is an FDA-approved anti-convulsive drug, and it may represent an underexplored option for treatment to prevent PTE and long-term neuropathology after TBI. After prolonged use, RTG has side effects like urinary incontinence and blue skin discoloration [69, 70]. Such effects of prolonged use of RTG have led it to be discontinued as a treatment for epilepsy. However, imporantly, the single-dose post-TBI treatment tested in our experiments avoids the side effects that occur with prolonged RTG treatment. Newer, more potent, and more specific compounds that also augment M current have been developed [71, 72] and could represent more efficient and safer options. Currently, no FDA-approved treatments are available to impair the development of long-term TBI-induced brain dysfunction. Taken together, the data presented here and previously [32], using two different TBI mouse models, show that acute RTG treatment reduces TBI-induced cellular metabolic stress, inflammation, cell death, BBB breakdown, sleep disorder, PTS, PTE, long-term neuropathology, and impairment of age-related increase in the power of the gamma frequency in mice. In a zebrafish TBI model, RTG treatment was also shown to reduce PTS, tauopathy, and cell death [33]. M-current increase acutely after one or multiple TBIs might be a new treatment option to reduce PTE development and various deleterious effects of brain trauma. More preclinical and clinical tests are necessary. Translation of a new treatment option from animal models to humans fails too often and must be carefully pursued. Nevertheless, our results are promising for this novel therapeutic approach.
Data Availability
Supplementary data files include raw dataset analysis of seizures and seizure-related parameters. Any additional datasets generated during and/or analyzed during the current study are available from the corresponding author upon reasonable request.
References
Verellen RM, Cavazos JE. Post-traumatic epilepsy: an overview. Therapy. 2010;7(5):527–31.
Lowenstein DH. Epilepsy after head injury: an overview. Epilepsia. 2009;50(Suppl 2):4–9.
Laskowitz D, Grant G, editors. Translational research in traumatic brain injury. Boca Raton (FL). 2016.
Kirmani BF, Robinson DM, Fonkem E, Graf K, Huang JH. Role of anticonvulsants in the management of posttraumatic epilepsy. Front Neurol. 2016;7:32.
Mathias JL, Alvaro PK. Prevalence of sleep disturbances, disorders, and problems following traumatic brain injury: a meta-analysis. Sleep Med. 2012;13(7):898–905.
Aoun R, Rawal H, Attarian H, Sahni A. Impact of traumatic brain injury on sleep: an overview. Nat Sci Sleep. 2019;11:131–40.
Jin B, Aung T, Geng Y, Wang S. Epilepsy and its interaction with sleep and circadian rhythm. Front Neurol. 2020;11:327.
Andrade P, Nissinen J, Pitkanen A. Generalized seizures after experimental traumatic brain injury occur at the transition from slow-wave to rapid eye movement sleep. J Neurotrauma. 2017;34(7):1482–7.
Bailes JE, Dashnaw ML, Petraglia AL, Turner RC. Cumulative effects of repetitive mild traumatic brain injury. Prog Neurol Surg. 2014;28:50–62.
Ledreux A, Pryhoda MK, Gorgens K, Shelburne K, Gilmore A, Linseman DA, et al. Assessment of long-term effects of sports-related concussions: biological mechanisms and exosomal biomarkers. Front Neurosci. 2020;14:761.
Breen PW, Krishnan V. Recent preclinical insights into the treatment of chronic traumatic encephalopathy. Front Neurosci. 2020;14:616.
Gardner RC, Yaffe K. Epidemiology of mild traumatic brain injury and neurodegenerative disease. Mol Cell Neurosci. 2015;66(Pt B):75–80.
Rosso SM, Landweer EJ, Houterman M, Donker Kaat L, van Duijn CM, van Swieten JC. Medical and environmental risk factors for sporadic frontotemporal dementia: a retrospective case-control study. J Neurol Neurosurg Psychiatry. 2003;74(11):1574–6.
Bi B, Choi HP, Hyeon SJ, Sun S, Su N, Liu Y, et al. Quantitative proteomic analysis reveals impaired axonal guidance signaling in human postmortem brain tissues of chronic traumatic encephalopathy. Exp Neurobiol. 2019;28(3):362–75.
Nasrabady SE, Rizvi B, Goldman JE, Brickman AM. White matter changes in Alzheimer’s disease: a focus on myelin and oligodendrocytes. Acta Neuropathol Commun. 2018;6(1):22.
Cheng L, Tang X, Luo C, Liu D, Zhang Y, Zhang J. Fiber-specific white matter reductions in amyotrophic lateral sclerosis. Neuroimage Clin. 2020;28: 102516.
Savard M, Pascoal TA, Servaes S, Dhollander T, Iturria-Medina Y, Kang MS, et al. Impact of long- and short-range fibre depletion on the cognitive deficits of fronto-temporal dementia. Elife. 2022;11.
Hu WT, Grossman M. TDP-43 and frontotemporal dementia. Curr Neurol Neurosci Rep. 2009;9(5):353–8.
Liscic RM, Grinberg LT, Zidar J, Gitcho MA, Cairns NJ. ALS and FTLD: two faces of TDP-43 proteinopathy. Eur J Neurol. 2008;15(8):772–80.
Suk TR, Rousseaux MWC. The role of TDP-43 mislocalization in amyotrophic lateral sclerosis. Mol Neurodegener. 2020;15(1):45.
Meneses A, Koga S, O’Leary J, Dickson DW, Bu G, Zhao N. TDP-43 pathology in Alzheimer’s disease. Mol Neurodegener. 2021;16(1):84.
Tribett T, Erskine B, Bailey K, Brown T, Castellani RJ. Chronic traumatic encephalopathy pathology after shotgun injury to the brain. J Forensic Sci. 2019;64(4):1248–52.
Mez J, Daneshvar DH, Kiernan PT, Abdolmohammadi B, Alvarez VE, Huber BR, et al. Clinicopathological evaluation of chronic traumatic encephalopathy in players of American football. JAMA. 2017;318(4):360–70.
Archbold HC, Jackson KL, Arora A, Weskamp K, Tank EM, Li X, et al. TDP43 nuclear export and neurodegeneration in models of amyotrophic lateral sclerosis and frontotemporal dementia. Sci Rep. 2018;8(1):4606.
Ederle H, Dormann D. TDP-43 and FUS en route from the nucleus to the cytoplasm. FEBS Lett. 2017;591(11):1489–507.
Gao J, Wang L, Huntley ML, Perry G, Wang X. Pathomechanisms of TDP-43 in neurodegeneration. J Neurochem. 2018.
Skotak M, Townsend MT, Ramarao KV, Chandra N. A comprehensive review of experimental rodent models of repeated blast TBI. Front Neurol. 2019;10:1015.
Kovacs SK, Leonessa F, Ling GS. Blast TBI models, neuropathology, and implications for seizure risk. Front Neurol. 2014;5:47.
Bugay V, Bozdemir E, Vigil FA, Chun SH, Holstein DM, Elliott WR, et al. A mouse model of repetitive blast traumatic brain injury reveals post-trauma seizures and increased neuronal excitability. J Neurotrauma. 2020;37(2):248–61.
Brown DA, Passmore GM. Neural KCNQ (Kv7) channels. Br J Pharmacol. 2009;156(8):1185–95.
Jentsch TJ. Neuronal KCNQ potassium channels: physiology and role in disease. Nat Rev Neurosci. 2000;1(1):21–30.
Vigil FA, Bozdemir E, Bugay V, Chun SH, Hobbs M, Sanchez I, et al. Prevention of brain damage after traumatic brain injury by pharmacological enhancement of KCNQ (Kv7, “M-type”) K(+) currents in neurons. J Cereb Blood Flow Metab. 2020;40(6):1256–73.
Alyenbaawi H, Kanyo R, Locskai LF, Kamali-Jamil R, DuVal MG, Bai Q, et al. Seizures are a druggable mechanistic link between TBI and subsequent tauopathy. Elife. 2021;10.
Bugay V, Gregory SR, Belanger-Coast MG, Zhao R, Brenner R. Effects of sublethal organophosphate toxicity and anti-cholinergics on electroencephalogram and respiratory mechanics in mice. Front Neurosci. 2022;16:866899.
Shimada T, Yamagata K. Pentylenetetrazole-induced kindling mouse model. J Vis Exp. 2018;(136).
Gervasoni D, Lin SC, Ribeiro S, Soares ES, Pantoja J, Nicolelis MA. Global forebrain dynamics predict rat behavioral states and their transitions. J Neurosci. 2004;24(49):11137–47.
Aguinis H, Gottfredson R, Joo H. Best-practice recommendations for defining, identifying, and handling outliers. Organ Res Methods. 2013;16:270–301.
Leys C, Ley C, Klein O, Bernard P, Licata L. Detecting outliers: do not use standard deviation around the mean, use absolute deviation around the median. J Exp Soc Psychol. 2013;49:764–6.
Bland JM, Altman DG, Rohlf FJ. In defence of logarithmic transformations. Stat Med. 2013;32(21):3766–8.
Etter G, van der Veldt S, Manseau F, Zarrinkoub I, Trillaud-Doppia E, Williams S. Optogenetic gamma stimulation rescues memory impairments in an Alzheimer’s disease mouse model. Nat Commun. 2019;10(1):5322.
Mably AJ, Colgin LL. Gamma oscillations in cognitive disorders. Curr Opin Neurobiol. 2018;52:182–7.
Mondragon-Rodriguez S, Gu N, Manseau F, Williams S. Alzheimer’s transgenic model is characterized by very early brain network alterations and beta-CTF fragment accumulation: reversal by beta-secretase inhibition. Front Cell Neurosci. 2018;12:121.
Herrmann CS, Demiralp T. Human EEG gamma oscillations in neuropsychiatric disorders. Clin Neurophysiol. 2005;116(12):2719–33.
Benussi A, Cantoni V, Grassi M, Brechet L, Michel CM, Datta A, et al. Increasing brain gamma activity improves episodic memory and restores cholinergic dysfunction in Alzheimer’s disease. Ann Neurol. 2022;92(2):322–34.
Barr MS, Radhu N, Guglietti CL, Zomorrodi R, Rajji TK, Ritvo P, et al. Age-related differences in working memory evoked gamma oscillations. Brain Res. 2014;1576:43–51.
Albayram O, Kondo A, Mannix R, Smith C, Tsai CY, Li C, et al. Cis P-tau is induced in clinical and preclinical brain injury and contributes to post-injury sequelae. Nat Commun. 2017;8(1):1000.
Petraglia AL, Plog BA, Dayawansa S, Dashnaw ML, Czerniecka K, Walker CT, et al. The pathophysiology underlying repetitive mild traumatic brain injury in a novel mouse model of chronic traumatic encephalopathy. Surg Neurol Int. 2014;5:184.
Yu J, Zhu H, Taheri S, Mondy W, Perry S, Kindy MS. Impact of nutrition on inflammation, tauopathy, and behavioral outcomes from chronic traumatic encephalopathy. J Neuroinflammation. 2018;15(1):277.
Mouzon BC, Bachmeier C, Ferro A, Ojo JO, Crynen G, Acker CM, et al. Chronic neuropathological and neurobehavioral changes in a repetitive mild traumatic brain injury model. Ann Neurol. 2014;75(2):241–54.
Cheng WH, Martens KM, Bashir A, Cheung H, Stukas S, Gibbs E, et al. CHIMERA repetitive mild traumatic brain injury induces chronic behavioural and neuropathological phenotypes in wild-type and APP/PS1 mice. Alzheimers Res Ther. 2019;11(1):6.
Anderson EN, Morera AA, Kour S, Cherry JD, Ramesh N, Gleixner A, et al. Traumatic injury compromises nucleocytoplasmic transport and leads to TDP-43 pathology. Elife. 2021;10.
Vigil FA, Carver CM, Shapiro MS. Pharmacological manipulation of K v 7 channels as a new therapeutic tool for multiple brain disorders. Front Physiol. 2020;11:688.
Joshi S, Sedivy V, Hodyc D, Herget J, Gurney AM. KCNQ modulators reveal a key role for KCNQ potassium channels in regulating the tone of rat pulmonary artery smooth muscle. J Pharmacol Exp Ther. 2009;329(1):368–76.
Yeung S, Schwake M, Pucovsky V, Greenwood I. Bimodal effects of the Kv7 channel activator retigabine on vascular K+ currents. Br J Pharmacol. 2008;155(1):62–72.
Vay SU, Flitsch LJ, Rabenstein M, Moniere H, Jakovcevski I, Andjus P, et al. The impact of hyperpolarization-activated cyclic nucleotide-gated (HCN) and voltage-gated potassium KCNQ/Kv7 channels on primary microglia function. J Neuroinflammation. 2020;17(1):100.
Seiffert E, Dreier JP, Ivens S, Bechmann I, Tomkins O, Heinemann U, et al. Lasting blood-brain barrier disruption induces epileptic focus in the rat somatosensory cortex. J Neurosci. 2004;24(36):7829–36.
Rana A, Musto AE. The role of inflammation in the development of epilepsy. J Neuroinflammation. 2018;15(1):144.
Wang W, Zinsmaier AK, Firestone E, Lin R, Yatskievych TA, Yang S, et al. Blocking tumor necrosis factor-alpha expression prevents blast-induced excitatory/inhibitory synaptic imbalance and parvalbumin-positive interneuron loss in the hippocampus. J Neurotrauma. 2018.
Hunt RF, Scheff SW, Smith BN. Synaptic reorganization of inhibitory hilar interneuron circuitry after traumatic brain injury in mice. J Neurosci. 2011;31(18):6880–90.
Bolkvadze T, Pitkanen A. Development of post-traumatic epilepsy after controlled cortical impact and lateral fluid-percussion-induced brain injury in the mouse. J Neurotrauma. 2012;29(5):789–812.
Di Sapia R, Moro F, Montanarella M, Iori V, Micotti E, Tolomeo D, et al. In-depth characterization of a mouse model of post-traumatic epilepsy for biomarker and drug discovery. Acta Neuropathol Commun. 2021;9(1):76.
Haboubi NH, Long J, Koshy M, Ward AB. Short-term sequelae of minor head injury (6 years experience of minor head injury clinic). Disabil Rehabil. 2001;23(14):635–8.
Krueger JM, Obal FJ, Fang J, Kubota T, Taishi P. The role of cytokines in physiological sleep regulation. Ann N Y Acad Sci. 2001;933:211–21.
Konduru SS, Wallace EP, Pfammatter JA, Rodrigues PV, Jones MV, Maganti RK. Sleep-wake characteristics in a mouse model of severe traumatic brain injury: relation to posttraumatic epilepsy. Epilepsia Open. 2021;6(1):181–94.
Leparulo A, Bisio M, Redolfi N, Pozzan T, Vassanelli S, Fasolato C. Accelerated aging characterizes the early stage of Alzheimer's disease. Cells. 2022;11(2).
Barr MS, Farzan F, Rusjan PM, Chen R, Fitzgerald PB, Daskalakis ZJ. Potentiation of gamma oscillatory activity through repetitive transcranial magnetic stimulation of the dorsolateral prefrontal cortex. Neuropsychopharmacology. 2009;34(11):2359–67.
Meltzer JA, Zaveri HP, Goncharova II, Distasio MM, Papademetris X, Spencer SS, et al. Effects of working memory load on oscillatory power in human intracranial EEG. Cereb Cortex. 2008;18(8):1843–55.
Valderrama M, Crepon B, Botella-Soler V, Martinerie J, Hasboun D, Alvarado-Rojas C, et al. Human gamma oscillations during slow wave sleep. PLoS ONE. 2012;7(4):e33477.
Beacher NG, Brodie MJ, Goodall C. A case report: retigabine induced oral mucosal dyspigmentation of the hard palate. BMC Oral Health. 2015;15(1):122.
Zaugg BE, Bell JE, Taylor KY, Bernstein PS. Ezogabine (Potiga) maculopathy. Retin Cases Brief Rep. 2017;11(1):38–43.
Kumar M, Reed N, Liu R, Aizenman E, Wipf P, Tzounopoulos T. Synthesis and evaluation of potent KCNQ2/3-specific channel activators. Mol Pharmacol. 2016;89(6):667–77.
Kalappa BI, Soh H, Duignan KM, Furuya T, Edwards S, Tzingounis AV, et al. Potent KCNQ2/3-specific channel activator suppresses in vivo epileptic activity and prevents the development of tinnitus. J Neurosci. 2015;35(23):8829–42.
Acknowledgements
This study was supported by Department of Defense Congressionally Directed Medical Research Programs grants W81XWH-15-1-0284 (M.S.S. and R.B.), W81XWH-13-1-0284 (J.D.L.), and W81XWH-22-1-0195 (F.A.V.). We also thank the support of the American Heart Association (20POST35180050 to F.A.V.), of the National Institute of Health (NS114906 to R.B. and V.B.), of the Conselho Nacional de Desenvolvimento Científico e Tecnológico (D.C.D. and H.B.), and of the William and Ella Owens Medical Research Foundation (M.S.S.). We thank William R. Elliott, Cassie J. Sprague, and David O. Zamora from the United States Army Institute of Surgical Research (Fort Sam Houston), as well as Gregory Rule from Applied Research Associates, for their advice and support on the use of the blast facility.
Required Author Forms
Disclosure forms provided by the authors are available with the online version of this article.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Vigil, F.A., Belchior, H., Bugay, V. et al. Acute Treatment with the M-Channel (Kv7, KCNQ) Opener Retigabine Reduces the Long-Term Effects of Repetitive Blast Traumatic Brain Injuries. Neurotherapeutics 20, 853–869 (2023). https://doi.org/10.1007/s13311-023-01361-9
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13311-023-01361-9