
Summary
Narcolepsy and other syndromes associated with excessive daytime sleepiness can be challenging to treat. New classifications now distinguish narcolepsy/hypocretin deficiency (also called type 1 narcolepsy), a lifelong disorder with well-established diagnostic procedures and etiology, from other syndromes with hypersomnolence of unknown causes. Klein-Levin Syndrome, a periodic hypersomnia associated with cognitive and behavioral abnormalities, is also considered a separate entity with separate therapeutic protocols. Non hypocretin-related hypersomnia syndromes are diagnoses of exclusion. These diagnoses are only made after eliminating sleep deprivation, sleep apnea, disturbed nocturnal sleep, and psychiatric comorbidities as the primary cause of daytime sleepiness. The treatment of narcolepsy/hypocretin deficiency is well-codified, and involves pharmacotherapies using sodium oxybate, stimulants, and/or antidepressants, plus behavioral modifications. These therapies are almost always needed, and the risk-to-benefit ratio is clear, notably in children. Detailed knowledge of the pharmacological profile of each compound is needed to optimize use. Treatment for other syndromes with hypersomnolence is more challenging and less codified. Preferably, therapy should be conservative (such as modafinil, atomoxetine, behavioral modifications), but it may have to be more aggressive (high-dose stimulants, sodium oxybate, etc.) on a case-by-case, empirical trial basis. As cause and evolution are unknown in these conditions, it is important to challenge diagnosis and therapy over time, keeping in mind the possibility of tolerance and the development of stimulant addiction. Kleine-Levin Syndrome is usually best left untreated, although lithium can be considered in severe cases with frequent episodes. Guidelines are provided based on the literature and personal experience of the author.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
According to the Diagnostic and Statistical Manual of Mental Disorders, 5th edition (DSM-5) currently being finalized, syndromes with primary hypersomnolence can be practically divided into 3 groups: 1) narcolepsy caused by hypocretin (orexin) deficiency, a disorder associated with Human Leukocyte Antigen (HLA) marker DQB1*06:02 and believed to be autoimmune (almost all cases with cataplexy), 2) Kleine-Levin Syndrome (KLS), and 3) syndromes with hypersomnolence unexplained by hypocretin abnormalities (generally without cataplexy) [1]. This last group is the most challenging and the most frequent diagnosis.
A similar revised International Sleep Disorder Classification, 3rd edition (ICSD3) is also being finalized and will use a similar framework to that of the DSM-5 with minor differences. In the ICSD3, narcolepsy caused by hypocretin deficiency is called “type 1 narcolepsy” while other hypersomnias (not likely due to hypocretin abnormalities) will stay subdivided into “type 2 narcolepsy” in the presence of a positive Multiple Sleep Latency Test (MSLT) with multiple Sleep Onset REM periods (SOREMPs) vs idiopathic hypersomnia otherwise (for more detail see American Psychiatric Association [1]).
In this publication, we will use the DSM-5 classification as a guideline because there is no evidence that the pathophysiology or therapeutic response is substantially different for hypersomnia with or without SOREMPs on the MSLT. First we will review the most commonly used medications across all conditions. Treatment specificities in children and for each of the three conditions will be discussed last.
Amphetamines
Amphetamines are simple derivatives of catecholamines (dopamine, norepinephrine, epinephrine) that are made more lipophilic so that they enter the central nervous system easily (Fig. 1). These are very old chemical entities, first made available in 1935 [2]. These compounds mimic many of the catecholaminergic actions in the brain, primarily substituting for monoamines in presynaptic synapses and producing monoaminergic release [3] (Table 1).

Chemical structures of amphetamine-like stimulants, modafinil, and caffeine (a xanthine derivative), as compared to dopamine and norepinephrine
Amphetamine derivatives not only affect adrenergic and dopaminergic synapses [4], but also to a lesser extent serotoninergic synapses; specificity for each monoamine varies if further modifications are made (3,4-methylenedioxy-N-methylamphetamine [MDMA], a drug of abuse also known as Ecstasy, for example, has more of a serotonin effect).
As a rule, amphetamine isomers of the D-type are more active than isomers of the L-type, and have more effects on dopaminergic synapses than on other monoaminergic synapses [5]. L and D isomers of any derivatives may also have different pharmacokinetic properties. The addition of another methyl group to amphetamine creates methamphetamine, further increasing brain penetration and potency (Fig. 1). Effective doses are listed in Table 2, keeping in mind that there is great variation in effectiveness across formulations, and that it is unusual and probably not as safe to have to go beyond 60 mg/day.
Amphetamines have complex pharmacological effects that are dose dependent and not fully characterized [6]. Amphetamines substitute for monoamines at the level of the vesicular monoamine transporter (VMAT), disrupting granular storage of monoamine through a reserpine-like effect and increasing cytosolic dopamine [7]. Next, they substitute for monoamines at the level of monoaminergic transporters (notably the dopamine reuptake site [DAT]), and not only inhibit reuptake, but also enter the presynaptic synapse while creating a paradoxal reverse efflux of monoamines through the same transporter [3, 6, 7]. Finally, amphetamines can also inhibit the monoamine oxidase (MAO) enzymes at very high doses. These complex actions are not without consequences, as high doses of amphetamines are cytotoxic for monoaminergic neurons in animals [6, 7], an effect mostly documented with MDMA and serotoninergic synapses, but also present with regular amphetamines and dopaminergic transmission [7, 8]. The stepwise recruitment of additional effects on monoamine transmission likely explains the high efficacy of these compounds. Amphetamines are exceptionally wake-promoting, and at high doses also reduce cataplexy in narcoleptic patients [9], an effect best explained by its action on adrenergic and serotoninergic synapses.
Amphetamines are water-soluble (thus risk of diversion to intravenous administration) and very potent wake-promoting compounds. The pharmacokinetics for each isomer is different, with a primarily urinary elimination that is favored at high urinary pH, decreasing half-life. As for other stimulants (i.e., methylphenidate and modafinil), the formulation and type of salt used in each preparation is almost as important as the milligram dosage and isomer composition. Notably, due to its use in attention deficit hyperactivity disorder (ADHD), a countless number of slow or extended-release formulations are available [10, 11], increasing duration of action from a few hours (3-6 for D-amphetamine) to 12 hours or more. Slow-release formulations also slow down absorption, reducing peak concentration after administration, a property that reduces the “rush” or “quick” effects of nonextended-release formulation. The rapid rise in concentration, rush effects, and rapid on–off changes in dopamine released are likely important contributors to the addictive potential of amphetamines [12]. Amphetamines can be addictive, leading patients to feel unreasonably anxious or unwilling to stop, also seeking the drug at ever-increasing doses.
Numerous studies have shown that increased dopamine release is the main property explaining wake-promotion [3, 13], although norepinephrine effects also contribute. Side effects of amphetamines include peripheral release of norepinephrine, resulting in cardiac stimulation and vasoconstriction. Increased heart rate and blood pressure, palpitations, and sweating are common [14]. Increased anxiety can occur in predisposed patients. Mood may be temporarily enhanced, but the effect is generally not sustainable alone in patients with depression. At a high dose, an amphetamine may precipitate psychosis [15], although most typically delusions are of the persecution subtype and are reversible with cessation of the medication. As with all psychotropic medications, however, more severe and irreversible psychiatric complications can emerge.
Co-administration of an amphetamine with MAO inhibitors is contraindicated, as it can potentialize its effects, notably on blood pressure. Many MAO inhibitors are irreversible “suicide substrates” at MAO A and B enzymes that have an extremely long duration of action (weeks), and have minimal effects on wakefulness, making their use difficult to control. However, it is notable to remark that Selegiline (also called L-Desprenyl), a competitive MAO-B inhibitor that has been shown to be effective in narcolepsy, is primarily a metabolic precursor of amphetamine and exerts most of its therapeutic effects through amphetamine metabolism [16].
Methylphenidate
Methylphenidate shares many of the actions of amphetamines, although it does not fully substitute for catecholamines at the level of the dopamine transporter. The compound became popular for use in narcolepsy in the 1950s [17]. It is primarily a DAT reuptake inhibitor, but it has also been shown to increase dopamine release [18, 19]. As an amphetamine, methylphenidate is also not fully specific for dopaminergic transmission, also affecting more mildly other monoamines. Furthermore, D-isomers have different pharmacokinetics and are more specific for dopaminergic synapses than the L-isomer. D-isomers are also more potent [20].
Methylphenidate has a short half-life and is water-soluble [21]. It is readily absorbed and enters the brain effectively, also producing rapidly increasing concentration in the brain and a sensation of rush. As for amphetamine, methylphenidate is addictive and can lead to drug-seeking behaviors and tolerance [18, 22]. Onset of effects after oral administration is 20 minutes and duration of action is only approximately three hours.
The addictive potential of methylphenidate is highly dependent on formulation, with extended-release formulations having less potential for addiction. Because it is soluble, prescribed methylphenidate can be reformulated differently and has a street value for stimulant abusers. The large number of formulations available for methylphenidate (D- (dextro), L- (levo), both, and various release formulations) as a treatment for ADHD has also increased choice for hypersomnia therapies [10, 11].
Methylphenidate has a similar side effect profile to amphetamines (e.g. cardiovascular effects, psychosis) [14], although as it is primarily a DAT inhibitor, it has not been shown to be as cytotoxic in animal studies. As for amphetamines, methylphenidate suppresses primarily non-REM sleep, promoting wakefulness, but also REM sleep. It is mostly effective on wakefulness and has minimal effects on cataplexy and other narcolepsy symptoms. Effective doses are listed in Table 1, keeping in mind that as for the amphetamines, there is great variation in effectiveness across preparations, and it is unusual to have to go beyond 60 mg/day, preferably using long-acting formulations.
Modafinil
Modafinil was developed and first used for the treatment of narcolepsy in France in the 1980s. It has been studied in double-blind, placebo-controlled fashion in a large number of narcoleptic patients [23, 24] and is approved by the Food and Drug Administration (FDA) for narcolepsy, shift work disorder [25], and residual sleepiness in treated sleep apnea [26]. It was recognized as an active metabolite of adrafinil, a compound developed as a cognitive enhancer for the elderly. The compound is highly insoluble in water and has a low potency, meaning that active doses are in the range of hundreds of milligrams vs tens of milligrams for doses of amphetamine or methylphenidate that are needed to produce similar wake-promoting effects (equipotency). As a chemical entity, it is poorly and slowly absorbed due to the insolubility in water. Formulation is key, and when formulated as Provigil (racemic mixture of D- and L-modafinil), it is compounded into small beads that increase absorption and consistency of effects.
Because it was rapidly shown that modafinil is pharmacologically distinct from amphetamine (but less so from methylphenidate) in some animal tests, and that abuse potential for the compound was low, the mode of action of modafinil was touted as totally different from other stimulants, and did not involve dopamine [27, 28]. Indeed, after more than 20 years of experience with the compound, abuse with dependence remains rare, although there is no doubt that the compound is misused to increase performance rather than to treat disease in some cases [28]. In the late 1980s, studies at Stanford showed that the compound had very similar properties to very selective DAT reuptake inhibitors in a canine model of narcolepsy [29]. In particular, as for other DAT compounds, it was shown to reduce wakefulness without affecting REM sleep or reducing cataplexy, a symptom that in canine narcolepsy is mostly sensitive to adrenergic reuptake inhibition [3, 29].
This led us to test whether modafinil also binds the DAT transporter in vitro at concentrations consistent with its potent wake-promoting effects, something we demonstrated in 1994 [30, 31]. The debate on whether or not modafinil effects were due to Dopamine reuptake inhibition continued for many years, with most other scientists and the drug companies involved being unconvinced by this hypothesis. Of note, selective and high-affinity DAT inhibitors are not available for human use, as animal studies generally indicate high risk of abuse, and cocaine being a relatively specific DAT reuptake inhibitor. In 2001, together with Jonathan Wisor and Dale Edgar, we found that the wake-promoting effects of modafinil were completely abolished in DAT knockout mice [13], as are those of amphetamine-like stimulants and DAT reuptake inhibitors. In spite of these findings, most of those in the scientific community remained unconvinced. It was only when Nora Wolkov, in a series of experiments, found that modafinil administered at wake-promoting doses can displace DAT positron emission tomographic ligands in vivo, that the tide finally turned, with most investigators now agreeing with a primary dopaminergic mediation of modafinil wake-promoting effects [32, 33].
The fact that modafinil increases wakefulness through dopamine reuptake inhibition does not reduce its therapeutic value as a unique compound in the treatment of excessive daytime sleepiness. Indeed, positron emission tomography studies previously mentioned are increasingly showing that DAT occupation alone may not be sufficient for addiction [12]. Most notably, the speed at which DAT inhibitors enter the brain, leading to high brain peak concentrations with subsequent rapid decreases, may be most important, explaining why mode of administration is critical for abuse potential, with intravenous and smoking being the worst, followed by snorting. Oral administration of stimulants, especially as slow-release formulations, has lower abuse potential, even for amphetamine and methylphenidate. One likely hypothesis for the low risk of abuse with modafinil may thus be its very poor solubility and low potency, making it impossible to reformulate for adequate, high-speed brain delivery. As for any pharmaceutical entity, it is also possible that modafinil does not bind the DAT transporter in the same way as other DAT inhibitors [34], or that other unknown effects are involved, keeping with the pharmaceutical adage that any drug has effects that are known, and many that are unknown.
Modafinil is available as a racemic mixture, or as the R-isomer only. As L-modafinil has a lower affinity for the dopamine transporter and a threefold shorter half-life than R-modafinil, the effects of the racemic mixture are more similar to those of R-modafinil once steady state concentrations are reached, although small differences have been reported [35, 36]. Patients may benefit from one daily administration in the morning or from two administrations at morning and noon. A common side effect is headache (as with other stimulants), but this side effect generally subsides when doses are more slowly increased. Increased anxiety or blood pressure can also be observed. As for many other medications, modafinil can induce an allergic reaction, an effect that can escalate to a Stevens Johnson Syndrome (SJS) life-threatening situation. Because such an effect was suspected in a few cases during a pediatric trial of racemic modafinil, the compound is not approved for pediatric use [37]. The risk of a possible Stevens Johnson Syndrome with modafinil is to be balanced with that of an increased risk of addiction with more classic stimulants.
Modafinil is one of the few treatments that has been subjected to rigorous, double-blind, placebo-controlled studies in narcolepsy [24] and is approved by the Food and Drug Administration. Modafinil has also been approved for treating sleepiness in the context of shift work sleep disorder [38] and residual sleepiness in pressure airway therapy (PAP) or otherwise treated sleep apnea [39]. Because of the relatively low risk of addiction, modafinil can be more easily prescribed in patients without a clear, biochemically defined central hypersomnia syndrome, and is also easier to stop, if needed. It is also a schedule IV compound.
A typical treatment with modafinil (racemic mixture) in a typical adult hypersomnia/narcolepsy patient may start with 100 mg in the morning, and generally a second 100 mg dose will be added at noon. Then, the total dose may be increased to as much as 400 mg/day. For R-modafinil, as the potency is approximately twice, the initial dose may be 50 mg or 150 mg in the morning to increase to as much as 250 mg in the morning (equivalent to ~500 mg of the racemic mixture). Experience suggests that modafinil is effective for at least half of the patients, whereas in others it is simply too weak a stimulant. Slightly higher doses can be helpful in some patients, but are typically not covered by insurance. In such cases, we either would switch to other stimulants (methylphenidate) or use sodium oxybate and other treatment strategies. Of note, occassionally, it is helpful to add a dose of short-acting stimulant, such as methylphenidate (not slow-release) in addition to modafinil at times when alertness is particularly challenging (early afternoon), or as needed in case of an emergency (driving, etc.).
Other Stimulants or Wake-Promoting Compounds
Unfortunately, most other DAT compounds available are either rarely used or have been withdrawn. These include pemoline (a low potency DAT inhibitor that was withdrawn due to dose-dependent hepatotoxicity) and mazindol (a high potency DAT inhibitor that also has additional properties making it difficult to increase the dose without experiencing side effects) [9, 31]. The antiviral amantadine and benztropine, an anticholinergic compound with DAT inhibitory effects used in Parkinson’s disease, are also notable. Higher affinity and more specific compounds, such as nomifensine (dual norepinephrine and dopamine reuptake inhibitor) and amineptine [29] have generally been withdrawn because of reports of abuse or misuse. Bupropion also deserves a brief mention, as it is a low potency nonspecific monoamine reuptake inhibitor that also has DAT inhibitory effects, probably explaining its stimulatory effects as an antidepressant. Selegiline was also previously mentioned.
Nondopaminergic wake-promoting compounds that are safe to use and efficacious are few. Caffeine, an adenosinergic antagonist, remains widely used, but has intolerable side effects at high doses (including cardiovascular), and it is generally not efficient enough for patients with hypersomnia or narcolepsy. Adrenergic reuptake inhibitors, such as atomoxetine [40, 41] or reboxetine (in Europe),which are compounds that have been developed for depression or ADHD, have a clear use in the therapeutic arsenal against narcolepsy and hypersomnia although undocumented by clinical trials (Table 1). These compounds increase wakefulness (but generally less strongly than DAT inhibitors, although there is great inter-individual variation) and they reduce cataplexy. We found these compounds to be very helpful in addition to (or instead of) modafinil in cases in which abuse could be an issue. Side effects include tachycardia and urinary retention, and increased anxiety in some individuals. Other modes of actions, such as histamine receptor H3 antagonists and hypocretin agonists, are being developed and will be discussed in the conclusion.
Antidepressants as Anti-Cataplectic Agents
Reuptake inhibitor antidepressants generally do not have strong wake-promoting effects, although selective reuptake inhibitors with dual adrenergic serotoninergic effects can have some mild stimulant effects [42]. The use of these compounds is mostly reserved to treat cataplexy and other ancillary symptoms in narcolepsy. In animals and humans, reuptake inhibitors reduce REM sleep very potently after acute administration, likely explaining effects on cataplexy, a symptom with similarities to REM sleep atonia. Similarly, antidepressants also improve ancillary symptoms related to abnormal REM sleep, such as sleep paralysis and hypnagogic hallucinations.
A large number of antidepressants are active on cataplexy, including older compounds, such as tricyclic antidepressants [42]. Clomipramine, imipramine, desipramine, or protriptyline may all be used, although these compounds not only inhibit monoamine reuptake, they also have anticholinergic properties responsible for additional side effects (increased constipation, blurred vision, dry mouth, cardiac conduction impact). Other tricyclic antidepressants with antihistaminic and alpha-1 adrenergic blocking effects are also effective, but as they also induce sedation and orthostatic hypotension, they are rarely the best choice in narcolepsy.
New generation, selective serotoninergic (SSRI) or adrenergic reuptake inhibitors are generally preferred and are very effective. In narcoleptic canines, adrenergic reuptake inhibition has been shown to be the key property mediating anti-cataplectic effects for these compounds [29]. In humans, very selective serotoninergic reuptake inhibitors (e.g., escitalopram) are generally not as effective as dual serotoninergic-adrenergic reuptake compounds. For this reason, venlafaxine [43], or more rarely duloxetine or desmethylvenlafaxine, are commonly used and are probably the best first choice [41]. Unfortunately however, venlafaxine has a short duration of action so the extended-release form is preferable. With these compounds, doses inferior to those typically used for depression or anxiety treatment are generally sufficient. Venlafaxine, one of most used compounds, is typically started at 37.5 mg (extended release) in the morning, but is most often effective at doses of 75 to 150 mg extended release/day, although higher doses are needed in some patients.
Fluoxetine, a primary SSRI with some adrenergic effect via its metabolite desmethylfluoxetine, is effective, but higher doses, closer to the antidepressive effective doses (e.g., 20-60 mg/day) are needed. Fluoxetine, however, is occasionally useful when depressive or anxiety symptoms associated with narcolepsy are present, or when longer half-life is needed.
An advantage of antidepressant therapy is that it is immediately active on the symptom of cataplexy, unlike with depression or anxiety. A major problem, however, is rebound cataplexy with the cessation of treatment [44]. The effect is dramatic, it occurs the day after interruption of treatment (even with long-acting medication), and it may last for several weeks. For this reason, when patients are reporting the treatment to be ineffective, it is essential to check for compliance (or question the diagnosis of narcolepsy/cataplexy).
Sodium Oxybate: A Compound with Beneficial Effects on All Narcolepsy Symptoms
Sodium oxybate, also called gamma hydroxybutyric acid (GHB), was first developed as an anesthetic agent. Unlike other anesthetic agents and sleep inducers, GHB was found to induce slow wave sleep and REM sleep, suggesting a very distinct mode of action and pharmacological profile. Based on this observation and considering the fact that many patients with narcolepsy/hypocretin deficiency have disturbed nocturnal sleep, the compound was tried with the hypothesis that increased sleep, notably REM sleep, would reduce sleep pressure during the day and allow narcoleptic patients to be more awake [45]. Numerous double-blind studies have now demonstrated that sodium oxybate is effective on many narcolepsy symptoms [46–50] and the compound is FDA approved for the treatment of cataplexy and excessive daytime sleepiness in narcolepsy. The compound also has additional documented effects on all other symptoms of narcolepsy (disturbed nocturnal sleep, sleep paralysis, hypnagogic hallucinations).
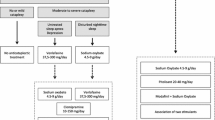
Because of its short half-life (30 minutes) [51] and a duration of action of only two to four hours, however, sodium oxybate typically needs to be administered twice during the night to fully consolidate a six to eight hour night. It is also a very low potency compound, requiring 6-9 g in adults per night to be effective (Table 1). Practically, in adults, we start with a 4.5 g dose split into two (one at bedtime and another in the middle of the night). We explain that the sedative effect is initially very strong, and that it is good to have someone else watch over the patient the first night, noting anything, such as snoring or gasping. Dizziness may occur, thus it is important to take the compound while in bed. We also explain that the unresponsiveness decreases somewhat as the patient becomes used to the medication. Patients are asked to call us back after the first day to give their impression, mostly for reassurance purposes. The dose is then increased to a total dose of 6 to 9 g, as 4.5 g is generally only partially effective, generally by 1.5 g steps every three to seven days, with weekly phone consultations.
Based on each individual’s response in terms of side effects (see as follows), and most importantly, their response in terms of duration of action on sleep and other symptoms, the dose is then adjusted not only quantitatively but also in its mode of administration. For example, we find that in many cases, the duration of action is not long enough to cover the entire night, such that a patient may take a 3 g dose at 10 PM to wake up at midnight, having to wait until 2 to 3 AM to take the take the second 3 g dose to fully sleep until 6 AM. In such patients, if we determine they have no difficulties falling asleep at 10 PM (exceptions would be in teenagers), we may suggest to first sleep without sodium oxybate for 1.5 h, then take the first dose at 11:30 PM to sleep until 2 AM, and then take the second dose and wake up at 6 AM. In rare cases, doses may even be split into three administrations, and may also be unequal in size. Our goal is to maintain close contact with the patient for the first few months of treatment to optimize the regimen so that nocturnal sleep is as good as possible, while maintaining the total amount required nightly within the active range (4.5 to 9 g), and also minimizing side effects.
Sodium oxybate is without any doubt one of the most effective medications available for narcolepsy/hypocretin deficiency. Not only does it have some effects on all symptoms, but a comparison of clinical trial effects on sleepiness, as measured subjectively (Epworth sleepiness scale) or objectively (MSLT or the Maintenance Test of Wakefulness (MWT), a variant of the MSLT most useful to assess whether patients can fight sleepiness) suggests that sodium oxybate is more effective than modafinil alone (400 mg) [24, 46]. The mode of action is unknown, but sodium oxybate is known to activate the Gamma-aminobutyric acid receptor type B (GABA-B receptor), an effect that has been shown to be essential in rodents for the mediation of sedation [52]. Other animal studies, however, suggest that the sleep effects of GHB may be different from those of baclofen [53]. In our opinion, because the compound has a very low affinity/low potency on the GABA-B receptor and has differential effects across species, it has been very difficult to demonstrate whether or not the GABA-B effect is sufficient for the therapeutic effects in narcolepsy. Baclofen, a prototypical GABA-B agonist, was used in narcolepsy and was found to be ineffective, suggesting that GABA-B alone may not be the entire story [54]. Unfortunately, however, baclofen has a longer half-life so it could still be that it is the combination of a very short-acting compound and GABA-B agonism that is responsible for the therapeutic effect.
A necessary area of investigation is whether or not optimizing nocturnal sleep with sodium oxybate, as previously described, is important for the full therapeutic effects in narcolepsy [55]. In one scenario, it is not, and then the effects on cataplexy and sleepiness are pharmacological and independent of sleep debt restoration. In the other scenario, sodium oxybate could act by reducing sleep debt so that when patients are waking up in the morning, they are so refreshed that they can stay awake much longer. Such a restorative effect would fit with the observation that the full therapeutic effect of sodium oxybate often takes weeks to months to manifest, not unlike the way sleep debt needs multiple days, if not weeks, to be fully recovered. One study also found correlations between increased slow wave sleep (SWS) activity and restorative effects after sodium oxybate in healthy subjects [56]. Because this is still unknown, and disturbed nocturnal sleep a clinical complaint on its own, our strategy always to try to optimize nocturnal sleep using sodium oxybate while also looking at the response of the other symptoms.
The use of sodium oxybate is limited by its side effect profile and the fact that it is difficult to prescribe. Regarding availability, prescription of sodium oxybate requires registration and training, and distribution to the patient is all made through a central pharmacy. The rationale for this decision was threefold. First, the compound has a very poor (but not necessarily deserved) public reputation. It is easily synthesized and has been used recreationally (e.g. to “cool down” after taking abused stimulants or MDMA) [57]. Second, as it is a strong sedative, it has also been used as a date rape drug [57]. Finally, under certain circumstances, such as continuous use at high dose, cessation can create severe withdrawal symptoms [58] and overdoses can be fatal, although in almost all cases fatal overdosing occurs in the context of polypharmacy.
In spite of this, however, in our experience that sodium oxybate is safe and efficacious when prescribed within the active dose range and when administration is limited to nighttime hours. When correctly prescribed, abrupt cessation does not lead to significant rebound or withdrawal effects, although a return to disturbed nocturnal sleep obviously occurs [59]. We certainly consider the compound first line for any patient with disturbed nocturnal sleep, cataplexy and obesity, and use it often in other cases. The most common side effects are nausea and weight loss [60]. Most of the time, these effects can be managed either by lowering the dose or by more progressively increasing the dose, or in very rare cases by adding anti-nausea medications, such as cyproheptadine or ondansetron. Rarely, however, nausea is so severe that administration is impossible.
Similarly, administration of sodium oxybate frequently leads to significantly decreased weight [60]. As narcolepsy/hypocretin deficiency is usually associated with weight gain (especially in children when the onset has been abrupt), weight loss is actually a very useful effect that allows a return to baseline. Weight loss, however, can be occasionally problemsome in patients who have not had weight gain and who have pre-existing anorectic tendencies. Another common side effect is enuresis, notably in younger patients, but this side effect is usually dose- and time-dependent, so that increasing the dose more slowly usually improves the situation. More rarely, parasomnias, such as sleep-walking, night eating (also common in untreated patients) or expiratory groaning [61, 62] can occur, and these side effects are also dose-dependent.
Other concerns have been raised and are not as well-established. First, sodium oxybate is a strong sedative, and as such there is the theoretical risk of increasing sleep-disordered breathing or hypoventilation (a common occurrence in obese narcoleptic subjects). Studies of the compound in sleep apnea patients have not found dramatic changes in apnea and hypopnea index, a measure of sleep apnea severity [63, 64], although the distribution of events by sleep stage was impaired. Others have raised the possibility that the effect is extremely variable and unpredictable, so that even some patients with mild sleep apnea could see their condition be exacerbated [65]. A controversy has even arisen on whether or not a prescription of sodium oxybate could be associated with a slightly increased death rate, a phenomenon that could not be assessed due to underreporting by the central pharmacy [66, 67]. A reanalysis of the data, however, does not suggest significantly increased mortality in subjects taking sodium oxybate [68].
To address this issue, we generally have a conservative common sense attitude so that patients with a high risk of sleep apnea or hypoventilation are monitored with polysmnography and CO2 monitoring while taking the drug. If there is significant disease, they are then treated with positive airway pressure (PAP) therapy first prior to the introduction of sodium oxybate, which paradoxically helps tolerate the PAP therapy in narcoleptic patients who can often have disturbed nocturnal sleep masked by sleep apnea [69]. Not uncommonly, we conduct titration of PAP under the influence of sodium oxybate, or restudy patients using polysomnography once a stable dose has been established. This is also systematically done if the response to sodium oxybate therapy on narcolepsy symptoms has not been as good as anticipated.
Second, clinical trials have shown that administration of sodium oxybate slightly increases anxiety ratings in many trials [70], although not always significantly, or in the pathological range. Other investigators have noted that in some rare cases reported as case reports, psychiatric complications may emerge, ranging from increased anxiety, to depression or even psychosis. Although not established, it is our experience that these effects are likely the result of increased daytime anxiety on a premorbid personality. For example, increased anxiety, depression, or a paradoxical feeling that the drug makes the sleepiness even worse can be the result of increased anxiety in a subject already pathologically anxious at baseline. Similarly, an individual with a paranoid personality may be biased toward having persecution delusions (similar to when stimulants are added). These effects are typically reversible once sodium oxybate is stopped, although it is not uncommon for us to then first add a course of SSRI to reduce anxiety and then reintroduce sodium oxybate with good results. Although these side effects are rare, this is an area in need of more research, as proper psychiatric screening is likely helpful to avoid potential problems if there is suspicion of psychiatric comorbidity. The situation is often complex in children and subjects close to onset, as in our experience a number of patients may develop narcolepsy together with behavioral disturbances secondary to sleepiness or a bona fide psychiatric disorder, such as schizophrenia, possibly the result of a larger autoimmune process.
Therapeutic Approaches to KLS (Recurrent Hypersomnia, KLS Type)
No treatment has been shown to be clearly efficacious in KLS [71–73]. During episodes, patients are not only sleepy but cognitively impaired. Giving a stimulant can produce a paradoxical agitation, and thus is not indicated unless the episodes are mild (e.g., at a later phase of the disease when KLS is “burning out,” typically after 30 years of age).
In most cases, notably when episodes are not too frequent (e.g., 1-3 times/year) the best approach is to do no harm and to let the patient sleep through the episodes undisturbed, asking for accommodation at school, etc. Although depression is not a core feature of KLS, some patients can become extremely upset during episodes, and it is important to monitor mood. It is also important to make sure the patient does not leave the house unattended, driving or putting themselves in a dangerous situation during episodes. Reassurance regarding the long-term evolution of the condition is key.
In some cases, in which the episodes are severe and frequent (e.g., two weeks long, every 1-2 months), the only therapy that has been suggested to be efficacious in some cases is lithium, although there is controversy regarding its efficacy, with meta-analysis suggesting effects in 20 to 40 % of cases in preventing episodes and reducing severity [71, 72]. When lithium is introduced, it is important to raise the dose until adequate blood levels (0.8-1.2 mEq/ml) are attained [73]. In a few cases, lithium was added and removed, and clear effects on and relapses of lithium were observed, suggesting that the effect is real. The effects of other mood stabilizers, such as valproic acid or carbamazepine are less well-documented. Antidepressants are not efficacious. Working with a psychiatrist with experience using lithium is advised.
Therapeutic Approaches in Narcolepsy/Hypocretin Deficiency vs Other Hypersomnias
At the pathophysiological level, it is now clear that most narcolepsy cases with cataplexy, and a minority of cases (5–30 %) without cataplexy or with atypical cataplexy-like symptoms, are caused by a lack of hypocretin (orexin) of likely an autoimmune origin. In these cases, once the disease is established, the majority of the 70,000 hypocretin-producing cells have been destroyed, and the disorder is irreversible. The cause is known and etiologically homogenous, and typically lifelong treatment will be needed, with the goal of optimizing life for these patients according to their goals. In our experience, as with any other disease entity, there is a range of symptom severity and variability in treatment response and side effects, but in approximately 80 % of cases, a return close to normal functioning is possible. A combination of lifestyle changes and pharmaceutical treatment tailored to each individual is typically needed.
Patients with narcolepsy hypocretin deficiency benefit best from combined drug therapy and behavioral modifications. Scheduled napping one to three times a day (with school or work accommodation often being needed) is very helpful to reduce the need for high dose stimulant. Driving and dangerous occupations must be discussed. In terms of drug therapy, we found that the majority of these patients do well on one to three of the following drugs: sodium oxybate, modafinil, and/or venlafaxine extended release. We tend to favor sodium oxybate as a first-line therapy, as in our experience this drug is at times sufficient by itself (especially in prepubertal children), whereas it is rare that a patient with narcolepsy/hypocretin deficiency will do optimally on modafinil or venlafaxine alone. Next line therapies may involve amphetamine-like stimulants, atomoxetine, or other antidepressants (added or in replacement, trying to limit the number of drugs).
For patients unlikely to have hypocretin deficiency, for example those negative for Human Leukocyte Antigen (HLA) DQB1*06:02 or/and without cataplexy, we are more cautious in terms of drug therapy for several reasons. First, the diagnosis of hypersomnia is difficult as it is a diagnosis of exclusion, made after other causes of daytime sleepiness (depression, sleep apnea, sedatives, disturbed nocturnal sleep, sleep deprivation, abnormal sleep cycle or shift work) have been ruled out. In clinical practice however, it is rare not to have some mild sleep apnea or some degree of disturbed sleep or mood disturbances in association with hypersomnia. It is thus hard to conclude whether the hypersomnia is purely primary and central. Second, the MSLT is known to have false positive results, even in “healthy” subjects who do not complain of daytime sleepiness [74, 75]. Third, whereas it is known that narcolepsy/hypocretin deficiency is a lifelong condition, little is known regarding the evolution of other hypersomnias of presumed central origin. Because of this, it is useful to challenge the notion of a lifelong therapy, and it is important to do no harm (e.g., establishing a stimulant addiction). Finally, clinical trials with modafinil and sodium oxybate have been performed in samples containing a majority of cases with cataplexy and presumably hypocretin deficiency, thus the response of these non hypocretin deficient cases is not as well-established.
In spite of these issues, it is unacceptable to dismiss these patients without providing treatment. Anemia, hypothyroidism, low iron status, infection (viral or Lyme disease), and general causes of fatigue must have been excluded. A careful evaluation of psychiatric status, sleep habits (during work and when on vacation), and sleep phase is essential, as is a sleep study to assess sleep apnea or other factors. The possibility that multiple factors could contribute should also be examined. The documentation of excessive sleep through logs or actigraphy is important. The MSLT is mostly useful to document objective daytime sleepiness, notably in the differential diagnosis of psychiatric issues, although it may be normal in severely affected patients who have extremely extended daytime sleep or cannot stay awake between naps.
Once the diagnosis is firm, it is essential to explain to the patient that the cause and evolution of the problem is unknown, and thus it is best to proceed cautiously. We generally advocate using low addiction potential medications, such as modafinil or atomoxetine first, and we have found that a portion of subjects react favorably. Regularizing sleep and wakefulness through behavioral interventions, such as mild sleep restriction and scheduled naps (as opposed to irregular 24-h sleep wake), can be helpful if the patient complies. Treating empirically with antidepressant is also worthwhile in some cases, especially when the MSLT is normal. Finally, recent findings suggest that some cases of unexplained hypersomnia are due to the accumulation of a GABA-enhancing natural compound in the brain, a pathology that could be reversed by flumazenil (briefly considering the short half-life) or the antibiotic clarithromycin (500 mg twice a day, with a maximum of 1 g/day) [76]. In this case, the positive effect of clarithromycin is secondary to a benzodiazepine antagonist-like effect, not its antibiotic effects, and treatment must be maintained.
Stronger drugs can also be tried, but if not helpful, should be rapidly withdrawn. Sodium oxybate can help significantly, notably if sleep difficulties are present, but special attention to psychiatric status is advised. Similarly, rare cases benefit from amphetamine-like stimulants, even at high doses, but in these cases we advise use of longlasting formulations to reduce the potential for addiction, and we recommend monitoring of the dose to a maximum of 60 mg/day, with a clear agreement by the patient that a higher dose will not be prescribed.
Treatment of Children
The treatment of children with narcolepsy and hypersomnia is similar to that of adults with a few caveats. First, in children stimulants and antidepressants with adrenergic effects can reduce growth slightly [77]. Second, modafinil has not been FDA approved for use in children after reports of a few rashes, including one possible Stevens Johnson Syndrome in 1,500 cases [37]. Whether or not this report warrants no prescription vs surveillance is uncertain, and varies across countries.
In younger children (pre or peripubertal) with hypocretin deficiency, we have found that sodium oxybate can be uniquely effective (often at a dose close to the adult dose, even in younger children) reducing weight and normalizing sleep, wake, and cataplexy by itself [78–80]. The addition of one or two scheduled naps (or in a few cases adding low-dose stimulant and/or venlafaxine) is often sufficient to complement the therapy, although napping is often resisted by prepubertal children and needs gentle convincing. Failures to respond in this population are typically due to psychiatric side effects or when a psychiatric comorbidity has been developing concomitantly. Parental support is critical to proper adjustment of medication, with specificities due to age. For example, we occasionally use three lower doses of sodium oxybate in very young children so that the child can still sleep ten hours. In adolescent or older children, difficulties are often academic, requiring stronger treatment. Poor compliance due to behavioral issues can also be a problem.
Pregnancy and Breast Feeding
This area is fraught by the absence of good human data, weak correspondence between animal and human teratogenicity, and the fact that classifications reflect length of exposure so that the longer a drug has been on the market, the more likely an issue may have been reported in humans. With this caveat in mind, amphetamines, methylphenidate, venlafaxine, and modafinil are category C (studies on animals show adverse effect and toxicity on fetus; no adequate and well-controlled studies done on pregnant women; drugs should be given only if the potential benefit outweighs the potential risk to the fetus). It should also be mentioned that modafinil has been suggested to interact with low-dose contraceptives, potentially reducing efficacy, although the scientific data supporting this claim is week and rests on poorly documented anecdotes. Sodium oxybate is category B (animal-reproduction studies have not demonstrated a fetal risk, but there are no controlled studies in pregnant women). These ratings suggest that there is no obvious teratogenicity, although it is impossible to exclude small developmental effects that could manifest later in life. As an example, animal studies have shown that rodents treated in pregnancy with antidepressants gave birth to animals with a behavioral profile consistent with depression. It is also clear that most of the drugs access the fetal brain, and in many cases, neonates may experience mild symptoms consistent with withdrawal.
The potential impact of any given drug varies depending on the pregnancy timing. For example, organogenesis begins three weeks after conception, so any problem before this timepoint would typically lead to a spontaneous abortion. Major teratogenic effects leading to birth defects and a viable child are likely to have occurred due to a problem occurring during the first trimester but after three weeks. Another critical period to consider is late pregnancy and delivery. As previously mentioned, mother and child may both be affected by psychotropic agents, and this may reduce vitality of the newborn and affect the ease of delivery.
At the practical level in one scenario a women may realize she is pregnant while still taking medications. In this case, reassurance is needed, as studies have shown that a large number, if not a majority, of pregnant women also report having taken some medication prior to realizing they were pregnant without any subsequent problem. In addition, as previously mentioned, there is no evidence that the drugs used in narcolepsy are notably teratogenic.
In a second scenario, a patient asks for advice for a planned pregnancy. In this case, we believe it is important to assess the potential guilt that could result if the patient kept using medications and a problem occurred. Work and family situations are also key. We generally suggest a conservative attitude and advise stopping the medications. Alternatively, careful high sensitivity monitoring of a potential pregnancy can be performed so that treatment is stopped as early as possible after conception, before organogenesis is initiated.
Later in pregnancy, it is also important to discuss delivery and the potential effects of the drugs on labor and on the immediate status of the newborn (see above), possibly stopping some medications prior to a planned delivery. Finally, when breastfeeding is initiated, it should be mentioned that all these psychotropic compounds can go into the breast milk. For these reasons, we advise using formula for a newborn if the patient wants to continue treatment.
Conclusion and Emerging Therapies
Tremendous progress has been made in the treatment of narcolepsy with hypocretin deficiency (a pathology affecting 0.03 % of the population), although treatment remains symptomatically based. It is our opinion that in the majority of cases, functioning can be restored to approximately 80 % of normal, with the caveat that narcoleptic patients have to learn to adjust to their disability in the areas of wake and sleep, and be diligent with their medication and regularity in sleep/wake patterns. Difficulties are then most notable when an excessive amount of work is being required (e.g., at the end of high school or during stressful work situations).
In this area, it is likely that progress will come from two fronts. First, it is increasingly evident that narcolepsy is autoimmune and that an upper airway infection, maybe related to H1N1 influenza, triggers narcolepsy [81]. Furthermore, patients are diagnosed closer and closer to disease onset. Thus, it is now possible to explore whether immune modulation near disease onset could rescue the disorder if caught early enough. Trials using intravenous immunoglobulins in recent onset cases had mixed results, and not once was the disease fully reversed [82]. Interestingly, recent results suggest a T-cell rather than B-cell/antibody mediation [83]. Thus, it may be that trials with newer medications targeting T-cells (e.g., alpha-4 integrin inhibitors blocking T-cell entry to the brain, such as natalizumab) would have more beneficial effects. Progress in our therapeutic arsenal in this area, together with improved diagnostic methods as the autoimmune basis of narcolepsy becomes more understood, are likely to lead to preventive or therapeutic protocols in special cases.
Second, for established narcolepsy/hypocretin deficiency cases, the most logical intervention would be to use a hypocretin receptor agonist (dual, or selective for receptor 2) during the day. Central administration of hypocretin-1 reverses narcolepsy in animal models [84, 85], but unfortunately the hypocretin peptide does not cross the blood brain barrier, thus a centrally penetrating agonist is needed to be usable. Companies have successfully synthesized numerous molecules with hypocretin receptor antagonist properties [86], one of which is awaiting FDA approval for the treatment of insomnia; it is also likely that an agonist will be found and developed. Based on the fact that central administration of hypocretin-1 is strongly wake-promoting in rodents and dogs, it is likely that such compounds will be effective in narcolepsy and other hypersomnias. Whether or not they will have other desirable (e.g., antidepressant effects) or undesirable (addictive potential) effects will likely define their future therapeutic usefulness.
Finally, whereas a large number of safe hypnotics are available, clinicians have very few options for wake-promotion beside dopamine-acting compounds, such as modafinil and amphetamine-like stimulants. This is especially problematic for hypersomnia patients without hypocretin deficiency. Adrenergic reuptake inhibitors and caffeine can be used, but these only have mild stimulant effects. Benzodiazepine antagonist-like compounds were previously mentioned as potential therapies for a subset of hypersomnia patients, but they await confirmatory data. Companies have been developing H3 antagonists (i.e., compounds that promote the release of the wake-promoting amine histamine [87]), but whether or not these compounds will be particularly useful as wake-promoting agents in this population remain to be seen [88]. As approximately 1.5 % of the general population complains of excessive daytime sleepiness or excessive sleep amounts consistent with a hypersomnia disorder [89], we believe there is a strong therapeutic need for safe and effective compounds in this area.
References
American Psychiatric Association. DSM-5 Development. In: DSM-5: The Future of Psychiatric Diagnosis. Available at: http://www.dsm5.org/.
Mignot E. A hundred years of narcolepsy research. Arch Ital Biol 2001;139:207–220.
Nishino S, Mignot E. Pharmacological aspects of human and canine narcolepsy. Prog Neurobiol 1997;52:27–78.
Robertson SD, Matthies HJ, Galli A. A closer look at amphetamine-induced reverse transport and trafficking of the dopamine and norepinephrine transporters. Mol Neurobiol 2009;39:73–80.
Kanbayashi T, Honda K, Kodama T, Mignot E, Nishino S. Implication of dopaminergic mechanisms in the wake-promoting effects of amphetamine: a study of D- and L-derivatives in canine narcolepsy. Neuroscience 2000;99:651-659.
Leviel V. Dopamine release mediated by the dopamine transporter, facts and consequences. J Neurochem 2011;118:475–489.
Riddle EL, Fleckenstein AE, Hanson GR. Role of monoamine transporters in mediating psychostimulant effects. AAPS J 2005;7:E847–E851.
Steinkellner T, Freissmuth M, Sitte HH, Montgomery T. The ugly side of amphetamines: short- and long-term toxicity of 3,4-methylenedioxymethamphetamine (MDMA, “Ecstasy”), methamphetamine and D-amphetamine. Biol Chem 2011;392:103-115.
Mitler MM, Hajdukovic R. Relative efficacy of drugs for the treatment of sleepiness in narcolepsy. Sleep 1991;14:218–220.
Connor DF, Steingard RJ. New formulations of stimulants for attention-deficit hyperactivity disorder: therapeutic potential. CNS Drugs 2004;18:1011–1030.
Chavez B, Sopko MA Jr, Ehret MJ, et al. An update on central nervous system stimulant formulations in children and adolescents with attention-deficit/hyperactivity disorder. Ann Pharmacother 2009;43:1084–1095.
Volkow ND, Fowler JS, Wang GJ, Baler R, Telang F. Imaging dopamine’s role in drug abuse and addiction. Neuropharmacology 2009;56:(suppl 1):3–8.
Wisor JP, Nishino S, Sora I, Uhl GH, Mignot E, Edgar DM. Dopaminergic role in stimulant-induced wakefulness. J Neurosci 2001;21:1787–1794.
Stiefel G, Besag FM. Cardiovascular effects of methylphenidate, amphetamines and atomoxetine in the treatment of attention-deficit hyperactivity disorder. Drug Saf 2010;33:821–842.
Guilleminault C. Amphetamines and narcolepsy: use of the Stanford database. Sleep 1993;16:199–201.
Magyar K. The pharmacology of selegiline. Int Rev Neurobiol 2011;100:65–84.
Daly DD, Yoss RE. The treatment of narcolepsy with methyl phenylpiperidylacetate: a preliminary report. Proc Staff Meet Mayo Clin 1956;31:620–625.
Leonard BE, McCartan D, White J, King DJ. Methylphenidate: a review of its neuropharmacological, neuropsychological and adverse clinical effects. Hum Psychopharmacol 2004;19:151–180.
Schenk JO. The functioning neuronal transporter for dopamine: kinetic mechanisms and effects of amphetamines, cocaine and methylphenidate. Prog Drug Res 2002;59:111–131.
Markowitz JS, Patrick KS. Differential pharmacokinetics and pharmacodynamics of methylphenidate enantiomers: does chirality matter? J Clin Psychopharmacol 2008;28:S54–S61.
Volkow ND, Fowler JS, Wang GJ, Ding YS, Gatley SJ. Role of dopamine in the therapeutic and reinforcing effects of methylphenidate in humans: results from imaging studies. Eur Neuropsychopharmacol 2002;12:557–566.
Bogle KE, Smith BH. Illicit methylphenidate use: a review of prevalence, availability, pharmacology, and consequences. Curr Drug Abuse Rev 2009;2:157–176.
Broughton RJ, Fleming JA, George CF, et al. Randomized, double-blind, placebo-controlled crossover trial of modafinil in the treatment of excessive daytime sleepiness in narcolepsy. Neurology 1997;49:444–451.
US Modafinil in Narcolepsy Multicenter Study Group. Randomized trial of modafinil as a treatment for the excessive daytime somnolence of narcolepsy. Neurology 2000;54:1166–1175.
Czeisler CA, Walsh JK, Wesnes KA, Arora S, Roth T. Armodafinil for treatment of excessive sleepiness associated with shift work disorder: a randomized controlled study. Mayo Clin Proc 2009;84:958–972.
Schwartz JR, Khan A, McCall WV, Weintraub J, Tiller J. Tolerability and efficacy of armodafinil in naive patients with excessive sleepiness associated with obstructive sleep apnea, shift work disorder, or narcolepsy: a 12-month, open-label, flexible-dose study with an extension period. J Clin Sleep Med 2010;6:450–457.
Duteil J, Rambert FA, Pessonnier J, Hermant JF, Gombert R, Assous E. Central alpha 1-adrenergic stimulation in relation to the behaviour stimulating effect of modafinil; studies with experimental animals. Eur J Pharmacol 1990;180:49–58.
Myrick H, Malcolm R, Taylor B, LaRowe S. Modafinil: preclinical, clinical, and post-marketing surveillance—a review of abuse liability issues. Ann Clin Psychiatry 2004;16:101–109.
Mignot E, Renaud A, Nishino S, Arrigoni J, Guilleminault C, Dement WC. Canine cataplexy is preferentially controlled by adrenergic mechanisms: evidence using monoamine selective uptake inhibitors and release enhancers. Psychopharmacology (Berl) 1993;113:76–82.
Mignot E, Nishino S, Guilleminault C, Dement WC. Modafinil binds to the dopamine uptake carrier site with low affinity. Sleep 1994;17:436–437.
Nishino S, Mao J, Sampathkumaran R, Shelton J. Increased dopaminergic transmission mediates the wake-promoting effects of CNS stimulants. Sleep Res Online 1998;1:49–61.
Volkow ND, Fowler JS, Logan J, et al. Effects of modafinil on dopamine and dopamine transporters in the male human brain: clinical implications. JAMA 2009;301:1148–1154.
Spencer TJ, Madras BK, Bonab AA, et al. A positron emission tomography study examining the dopaminergic activity of armodafinil in adults using [(1)(1)C]altropane and [(1)(1)C]raclopride. Biological psychiatry 2010;68:964–970.
Schmitt KC, Reith ME. The atypical stimulant and nootropic modafinil interacts with the dopamine transporter in a different manner than classical cocaine-like inhibitors. PLoS One 2011;6:e25790.
Darwish M, Kirby M, Hellriegel ET, Robertson P Jr. Armodafinil and modafinil have substantially different pharmacokinetic profiles despite having the same terminal half-lives: analysis of data from three randomized, single-dose, pharmacokinetic studies. Clin Drug Investig 2009;29:613–623.
Darwish M, Kirby M, Hellriegel ET, Yang R, Robertson P Jr. Pharmacokinetic profile of armodafinil in healthy subjects: pooled analysis of data from three randomized studies. Clin Drug Investig 2009;29:87–100.
Rugino T. A review of modafinil film-coated tablets for attention-deficit/hyperactivity disorder in children and adolescents. Neuropsychiatr Dis Treat 2007;3:293–301.
Czeisler CA, Walsh JK, Roth T, et al. Modafinil for excessive sleepiness associated with shift-work sleep disorder. N Engl J Med 2005;353:476–486.
Kingshott RN, Vennelle M, Coleman EL, Engleman HM, Mackay TW, Douglas NJ. Randomized, double-blind, placebo-controlled crossover trial of modafinil in the treatment of residual excessive daytime sleepiness in the sleep apnea/hypopnea syndrome. Am J Respir Crit Care Med 2001;163:918–923.
Niederhofer H. Atomoxetine also effective in patients suffering from narcolepsy? Sleep 2005;28:1189.
Mignot E, Nishino S. Emerging therapies in narcolepsy-cataplexy. Sleep 2005;28:754–763.
Morgenthaler TI, Kapur VK, Brown T, et al. Practice parameters for the treatment of narcolepsy and other hypersomnias of central origin. Sleep 2007;30:1705–1711.
Moller LR, Ostergaard JR. Treatment with venlafaxine in six cases of children with narcolepsy and with cataplexy and hypnagogic hallucinations. J Child Adolesc Psychopharmacol 2009;19:197–201.
Broderick M, Guilleminault C. Rebound cataplexy after withdrawal from antidepressants. Sleep Med 2009;10:403–404.
Broughton R, Mamelak M. The treatment of narcolepsy-cataplexy with nocturnal gamma-hydroxybutyrate. Can J Neurol Sci 1979;6:1–6.
Xyrem MSG. A randomized, double blind, placebo-controlled multicenter trial comparing the effects of three doses of orally administered sodium oxybate with placebo for the treatment of narcolepsy. Sleep 2002;25:42–49.
Xyrem MSG. A 12-month, open-label, multicenter extension trial of orally administered sodium oxybate for the treatment of narcolepsy. Sleep 2003;26:31–35.
Black J, Houghton WC. Sodium oxybate improves excessive daytime sleepiness in narcolepsy. Sleep 2006;29:939–946.
Black J, Pardi D, Hornfeldt CS, Inhaber N. The nightly use of sodium oxybate is associated with a reduction in nocturnal sleep disruption: a double-blind, placebo-controlled study in patients with narcolepsy. J Clin Sleep Med 2010;6:596–602.
Xyrem MSG. Sodium oxybate demonstrates long-term efficacy for the treatment of cataplexy in patients with narcolepsy. Sleep Med 2004;5:119–123.
Scharf MB, Lai AA, Branigan B, Stover R, Berkowitz DB. Pharmacokinetics of gammahydroxybutyrate (GHB) in narcoleptic patients. Sleep 1998;21:507–514.
Koek W, France CP. Cataleptic effects of gamma-hydroxybutyrate (GHB) and baclofen in mice: mediation by GABA(B) receptors, but differential enhancement by N-methyl-d-aspartate (NMDA) receptor antagonists. Psychopharmacology 2008;199:191–198.
Vienne J, Bettler B, Franken P, Tafti M. Differential effects of GABAB receptor subtypes, {gamma}-hydroxybutyric Acid, and Baclofen on EEG activity and sleep regulation. J Neurosci 2010;30:14194–14204.
Huang YS, Guilleminault C. Narcolepsy: action of two gamma-aminobutyric acid type B agonists, baclofen and sodium oxybate. Pediatr Neurol 2009;41:9–16.
Arnulf I, Mignot E. Sodium oxybate for excessive daytime sleepiness in narcolepsy-cataplexy. Sleep 2004;27:1242–1243.
Walsh JK, Hall-Porter JM, Griffin KS, et al. Enhancing slow wave sleep with sodium oxybate reduces the behavioral and physiological impact of sleep loss. Sleep 2010;33:1217–1225.
Gahlinger PM. Club drugs: MDMA, gamma-hydroxybutyrate (GHB), Rohypnol, and ketamine. Am Fam Physician 2004;69:2619–2626.
van Noorden MS, van Dongen LC, Zitman FG, Vergouwen TA. Gamma-hydroxybutyrate withdrawal syndrome: dangerous but not well-known. Gen Hosp Psychiatr 2009;31:394–396.
Xyrem MSG. The abrupt cessation of therapeutically administered sodium oxybate (GHB) does not cause withdrawal symptoms. J Toxicol Clin Toxicol 2003;41:131–135.
Husain AM, Ristanovic RK, Bogan RK. Weight loss in narcolepsy patients treated with sodium oxybate. Sleep Med 2009;10:661–663.
Wallace DM, Maze T, Shafazand S. Sodium oxybate-induced sleep driving and sleep-related eating disorder. J Clin Sleep Med 2011;7:310–311.
Poli F, Ricotta L, Vandi S, et al. Catathrenia under sodium oxybate in narcolepsy with cataplexy. Sleep Breath 2011;xx:xx-xx.
George CF, Feldman N, Inhaber N, et al. A safety trial of sodium oxybate in patients with obstructive sleep apnea: acute effects on sleep-disordered breathing. Sleep Med 2010;11:38–42.
George CF, Feldman N, Zheng Y, et al. A 2-week, polysomnographic, safety study of sodium oxybate in obstructive sleep apnea syndrome. Sleep Breath 2011;15:13–20.
Seeck-Hirschner M, Baier PC, von Freier A, Aldenhoff J, Goder R. Increase in sleep-related breathing disturbances after treatment with sodium oxybate in patients with narcolepsy and mild obstructive sleep apnea syndrome: two case reports. Sleep Med 2009;10:154–155.
Feldman NT. Xyrem safety: the debate continues. Sleep Med 2009;10:405–406.
Feldman NT. Sodium oxybate post-marketing surveillance. J Clin Sleep Med 2011;7:417.
Wang YG, Swick TJ, Carter LP, Thorpy MJ, Benowitz NL. Sodium oxybate: updates and correction to previously published safety data. J Clin Sleep Med 2011;7:415–416.
Kovacevic-Ristanovic R, Kuzniar TJ. Use of sodium oxybate (Xyrem in patients with dual diagnosis of narcolepsy and sleep apnea. Sleep Med 2010;11:5–6.
Russell IJ, Holman AJ, Swick TJ, Alvarez-Horine S, Wang YG, Guinta D. Sodium oxybate reduces pain, fatigue, and sleep disturbance and improves functionality in fibromyalgia: results from a 14-week, randomized, double-blind, placebo-controlled study. Pain 2011;152:1007–1017.
Arnulf I, Lin L, Gadoth N, et al. Kleine-Levin syndrome: a systematic study of 108 patients. Ann Neurolp 2008;63:482–493.
Arnulf I, Zeitzer JM, File J, Farber N, Mignot E. Kleine-Levin syndrome: a systematic review of 186 cases in the literature. Brain 2005;128:2763–2776.
Arnulf I, Rico T, Mignot E. Diagnosis, disease course, and management of patients with Kleine-Levin syndrome. Lancet Neurol 2012;11:918–928.
Singh M, Drake CL, Roth T. The prevalence of multiple sleep-onset REM periods in a population-based sample. Sleep 2006;29:890–895.
Mignot E, Lin L, Finn L, et al. Correlates of sleep-onset REM periods during the Multiple Sleep Latency Test in community adults. Brain 2006;129:1609–1623.
Rye DB, Parker K, Trotti L. Endogenous GABA(A) Receptor Enhancement Modulates Vigilance in the Primary Hypersomnias. Ann Neurol 2011; 70, Supplement 15, S23.
Vitiello B. Understanding the risk of using medications for attention deficit hyperactivity disorder with respect to physical growth and cardiovascular function. Child Adolesc Psychiatr Clin N Am 2008;17:459–474.
Aran A, Einen M, Lin L, Plazzi G, Nishino S, Mignot E. Clinical and therapeutic aspects of childhood narcolepsy-cataplexy: a retrospective study of 51 children. Sleep 2010;33:1457–1464.
Peraita-Adrados R, Garcia-Penas JJ, Ruiz-Falco L, et al. Clinical, polysomnographic and laboratory characteristics of narcolepsy-cataplexy in a sample of children and adolescents. Sleep Med 2011;12:24–27.
Lecendreux M, Poli F, Oudiette D, et al. Tolerance and efficacy of sodium oxybate in childhood narcolepsy with cataplexy: a retrospective study. Sleep 2012;35:709–711.
Han F, Lin L, Warby SC, et al. Narcolepsy onset is seasonal and increased following the 2009 H1N1 pandemic in China. Ann Neurol 2011;70:410–417.
Knudsen S, Mikkelsen JD, Bang B, Gammeltoft S, Jennum PJ. Intravenous immunoglobulin treatment and screening for hypocretin neuron-specific autoantibodies in recent onset childhood narcolepsy with cataplexy. Neuropediatrics 2010;41:217–222.
Hallmayer J, Faraco J, Lin L, et al. Narcolepsy is strongly associated with the T-cell receptor alpha locus. Nat Genet 2009;41:708–711.
Fujiki N, Yoshida Y, Ripley B, Mignot E, Nishino S. Effects of IV and ICV hypocretin-1 (orexin A) in hypocretin receptor-2 gene mutated narcoleptic dogs and IV hypocretin-1 replacement therapy in a hypocretin-ligand-deficient narcoleptic dog. Sleep 2003;26:953–959.
Mieda M, Willie JT, Hara J, Sinton CM, Sakurai T, Yanagisawa M. Orexin peptides prevent cataplexy and improve wakefulness in an orexin neuron-ablated model of narcolepsy in mice. Proc Natl Acad Sci U S A 2004;101:4649–4654.
Hoever P, de Haas SL, Dorffner G, Chiossi E, van Gerven JM, Dingemanse J. Orexin receptor antagonism: an ascending multiple-dose study with almorexant. J Psychopharmacol 2012;26:1071–1080.
Stocking EM, Letavic MA. Histamine H3 antagonists as wake-promoting and pro-cognitive agents. Curr Top Med Chem 2008;8:988–1002.
Inocente C, Arnulf I, Bastuji H, et al. Pitolisant, an inverse agonist of the histamine H3 receptor: an alternative stimulant for narcolepsy-cataplexy in teenagers with refractory sleepiness. Clin Neuropharmacol 2012;35:55–60.
Ohayon MM, Dauvilliers Y, Reynolds CF 3rd. Operational definitions and algorithms for excessive sleepiness in the general population: implications for DSM-5 nosology. Arch Gen Psychiatry 2012;69:71–79.
Acknowledgments
The salary of the author is mostly covered by the National Institutes of Health (grant no. NN23724). The Stanford Sleep Center has received gifts and contracts from Cephalon (modafinil) and Jazz Pharmaceuticals (sodium oxybate); Dr. Mignot has consulted for and continues to consult for these companies. The opinions expressed in this review are not all based on double blind placebo controlled experiments or head to head clinical trials, thus can and should be challenged. Rather they represent the best effort of the author to convey his experience in areas where rigorous data does not exist and yet clinical practice mandates practical action. Full conflict of interest disclosure is available in the electronic supplementary material for this article.
Required Author Forms
Disclosure forms provided by the authors are available with the online version of this article.
Open Access
This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
Author information
Authors and Affiliations
Corresponding author
Electronic Supplementary Material
Below is the link to the electronic supplementary material.
ESM 1
(PDF 265 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 2.0 International License (https://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Mignot, E.J.M. A Practical Guide to the Therapy of Narcolepsy and Hypersomnia Syndromes. Neurotherapeutics 9, 739–752 (2012). https://doi.org/10.1007/s13311-012-0150-9
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13311-012-0150-9