
Abstract
Introduction
Suboptimal glycaemic control among people with type 1 diabetes (T1D) is known to lead to long-term micro- and macrovascular complications and, unfortunately, it is still prevalent even in the most affluent societies. Although glycated haemoglobin monitoring is considered to be the gold standard for assessing glycaemic control, such monitoring is unable to reliably measure acute glycaemic excursions. Continuous glucose monitoring (CGM) has been shown to improve glucose control and reduce the incidence of hypoglycaemia, and also allow a more complete assessment of overall glycaemic control and hyper- and hypoglycaemic excursions. The use of CGM has led to time-in-range, which is the time that a patient is within the glycaemic range of 70 to 180 mg/dL, to be adopted as a treatment target. To date, only limited data comparing the second-generation insulins glargine 300 U/mL (Gla-300) and degludec 100 U/mL (IDeg-100) in people with T1D are available, and there is no CGM literature on comparisons of the use of CGM results to assess primary, secondary and tertiary endpoints. The aim of the InRange study was to address this unmet need.
Methods
InRange is a multicentre, randomised, active-controlled, parallel-group, 12-week, open-label, phase 4, comparative study. Adults with T1D will be randomised to receive once-daily Gla-300 or IDeg-100 by subcutaneous injection in the morning. Following an 8-week titration period, CGM data will be collected over 20 consecutive days.
Planned outcomes
The primary objective is to demonstrate that Gla-300 is noninferior to IDeg-100 in terms of glycaemic control [time-in-range ≥ 70 to ≤ 180 mg/dL (≥ 3.9 to ≤ 10 mmol/L)] and variability, as assessed using CGM, in adults with T1D. The results are expected to help confirm the utility of CGM in clinical practice in this population and provide insight into its application as an outcome measure in clinical practice.
Trial registration
NCT04075513.
Video Abstract
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Use of continuous glucose monitoring (CGM) allows a more complete assessment of overall glycaemic control and supports time-in-range (time a patient is within the glycaemic range of 70 to 180 mg/dL) as a treatment target. |
At present, there is no CGM literature on the use of CGM to compare the second-generation basal insulin analogues glargine 300 U/mL (Gla-300) and degludec 100 U/mL (IDeg-100). |
InRange, a multicentre, randomised, active-controlled, parallel-group, 12-week, open-label, phase 4 study will collect CGM data over 20 consecutive days from adults with T1D randomised to receive Gla-300 or IDeg-100. |
The study is designed to demonstrate that Gla-300 is noninferior to IDeg-100 in terms of glycaemic control [time in range ≥ 70 to ≤ 180 mg/dL (≥ 3.9 to ≤ 10 mmol/L)] and coefficient of variation, as assessed using CGM, in adults with T1D. |
InRange is expected to provide further insight into the utility of CGM as an outcome measure in clinical practice. |
Digital Features
This article is published with digital features, including a summary slide, slide deck and video abstract, to facilitate understanding of the article. To view digital features for this article go to https://doi.org/10.6084/m9.figshare.11777865.
Introduction
Around 10% of the total population with diabetes have type 1 diabetes (T1D), and the incidence has been rising steadily during the last 30 years. The Diabetes Control and Complications Trial showed that maintaining glycated haemoglobin (HbA1c) levels near 7% is important in reducing the risk of long-term microvascular complications of diabetes [1]. However, despite advances in technology, hypoglycaemia and the fear it causes remain major barriers to maintaining low glucose levels [2, 3], and many people with T1D struggle to achieve this recommended glycaemic target [4, 5]. While there are many reasons why it is difficult to achieve glycaemic targets, having a more stable and reliable basal insulin replacement may contribute to better glycaemic control [6].
Continuous glucose monitoring (CGM) is a blood glucose measuring technique that has become more common in recent years among people with insulin-treated diabetes [7]. CGM devices comprise a wearable sensor that uses enzymatic technology to measure glucose in interstitial fluid, a transmitter that wirelessly transmits electrical readings and a receiver that presents the information to the user [7]. CGM measures glucose levels at intervals of 5–15 min, meaning that data can be used immediately to guide lifestyle and therapeutic adjustments, to help reduce the incidence of hypoglycaemia and to improve glycaemic control overall [7,8,9]. In addition to monitoring blood glucose, a key metric obtained by CGM is the time within the target glucose range of 70 to 180 mg/dL [time-in-range (TIR)] [9]. TIR profiles can be used to determine the frequency and duration of glycaemic excursions and they can help healthcare professionals to establish personalised glycaemic targets [9]. The recent consensus report highlighted TIR as the most important actionable metric that can be obtained from CGM [9].
Insulin glargine 300 U/mL (Gla-300) and insulin degludec 100 U/mL (IDeg-100) are long-acting second-generation basal insulin analogues indicated for the treatment of both T1D and type 2 diabetes (T2D) [10, 11]. In phase 3 clinical trials, Gla-300 and IDeg-100 once daily have demonstrated better optimised pharmacokinetic (PK) and pharmacodynamic (PD) profiles than the first-generation analogue insulin glargine 100 U/mL, with durations of action lasting over 24 h [12,13,14]. In a recent noninferiority study of insulin-naïve people with uncontrolled T2D, Gla-300 demonstrated a lower incidence of anytime (24 h) confirmed hypoglycaemia [≤ 70 mg/dL (≤ 3.9 mmol/L) and < 54 mg/dL (< 3.0 mmol/L)] than those treated with IDeg-100 during the active titration period (weeks 0–12); however, overall improvements in glycaemic control and risk of hypoglycaemia were comparable between the two insulins [15]. A second head-to-head study conducted in adults with T2D already receiving insulin did not demonstrate superiority for IDeg-100 versus Gla-300 during the maintenance period [16]. To our knowledge, two small studies have been published that compare Gla-300 and IDeg-100 in people with T2D using CGM, both conducted in Japan [17, 18]. Yamabe et al. reported no statistically significant difference between the two insulins in any of the CGM metrics assessed, including mean TIR, mean time below range (TBR; both < 70 and < 54 mg/dL) and mean coefficient of variation (CV; a CV of < 36% is taken to represent stable glycaemia [18, 19]. However, Kawaguchi et al. observed significantly lower anytime (24 h) and nocturnal (0000–0600 hours) CV and TBR with Gla-300 than with IDeg-100 [17]. Direct comparisons of Gla-300 and IDeg-100 in people with T1D are currently limited to two studies, neither of which used CGM. Interestingly, the results of these two studies contrast with each other: one was a single-centre, randomised, double-blind, crossover, euglycaemic clamp study which showed that once-daily Gla-300 induces less fluctuating steady-state PD profiles and more evenly distributed PK profiles than IDeg-100 [13], and the other, with a similar crossover design, showed that IDeg-100 provided lower within-day and day-to-day variability in glucose infusion rates than Gla-300 [20].
The InRange clinical trial will be the first study to use CGM metrics as the primary endpoint to compare the second-generation basal insulin analogues Gla-300 and IDeg-100 in people with T1D. The hypothesis being tested is that Gla-300 is noninferior to IDeg-100 in adults with T1D based on percentage TIR at week 12. Here, we describe the methodology that will be used.
Methods
Study Design
InRange (study ID no.: LPS14947) is a multicentre, randomised, active-controlled, parallel-group, 12-week, open-label, phase 4 study to compare the glucose values and variability between Gla-300 and IDeg-100 using CGM. This study has been registered on the US National Library of Medicine ClinicalTrials.gov database (NCT04075513).
Study Population
Approximately 338 males and females will be recruited from multiple centres following provision of signed informed consent. Eligibility criteria are: 18–70 years of age with T1D; HbA1c ≥ 7% (48 mmol/mol) to ≤ 10% (86 mmol/mol) at screening; previous treatment with multiple daily injections using a basal insulin analogue once daily and rapid-acting insulin analogues for at least 1 year; and treatment with a stable dose of basal/bolus insulin regimen for 30 days prior to screening. Exclusion criteria include: presence of any clinically significant abnormality or adverse event (AE) that may preclude safe completion of the study or constrain efficacy assessment; end-stage renal disease; recent (within 3 months) or planned intravitreal injections, laser treatment or vitrectomy surgery for retinopathy or maculopathy; pregnancy; and bilirubin levels > 1.5-fold the upper limit of normal or fasting C-peptide levels > 0.2 nmol/L. Further, participants must not have experienced a change in body weight of ≥ 5 kg within 3 months prior to screening, used an insulin pump within 6 months prior to screening or have received Gla-300 or IDeg-100 as basal insulin within 30 days prior to screening, glucose-lowering drugs other than rapid-acting insulin analogues within 3 months prior to screening or glucocorticoids for > 10 days within 3 months prior to screening.
Prior to enrolment, the protocol will be reviewed and approved to be in accordance with the ethical standards of the Institutional Review Boards and Independent Ethics Committees of each participating institution in compliance with the Helsinki Declaration. The full list of Institutional Review Boards and Independent Ethics Committees can be found in the Supplementary Material. This study will be conducted in accordance with the Declaration of Helsinki with all relevant amendments, the Council for International Organizations of Medical Sciences (CIOMS) International Ethical Guidelines, the applicable ICH Good Clinical Practice Guidelines and all applicable laws and regulations.
Randomisation and Study Intervention
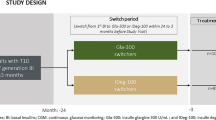
Participants are to be followed for approximately 18 weeks, which includes 1–2 weeks of screening followed by a 4-week run-in period, a 12-week treatment period and a 2- to 4-day follow-up period (Fig. 1). During the 4-week run-in period (weeks – 4 to – 1), participants undergo stabilisation of their current basal and mealtime insulin treatment and are trained on study-related procedures (weeks – 4 to – 3), and baseline CGM values are determined using the blinded CGM device (weeks – 3 to – 1). To be eligible for randomisation, a minimum of 10 days (not necessarily consecutive) of useable CGM data must be generated during the run-in period. During the run-in period, participants titrate their basal and mealtime insulin to a fasting self-monitored plasma glucose (SMPG) target of ≥ 70 to < 100 mg/dL (≥ 3.9 to < 5.6 mmol/L), and 2-h postprandial SMPG to a target of ≥ 130 to ≤ 180 mg/dL (≥ 7.2 to ≤ 10 mmol/L) [Electronic Supplementary Material (ESM) Table 1]. At week 0, participants satisfying the inclusion/exclusion criteria, including CGM handling and compliance with manufacturer’s instructions, are randomised 1:1 (stratified by screening HbA1c values of < 8.0 vs. ≥ 8.0%) to receive once-daily Gla-300 or IDeg-100 by subcutaneous injection in the morning. The randomised treatment period (weeks 0–12) includes an insulin titration period (weeks 0–8) and a CGM data collection period (over 20 consecutive days during weeks 9–12). During the titration period, doses of Gla-300 or IDeg-100 are titrated at least weekly (but no more often than every 3 days) until participants achieve the target fasting SMPG of ≥ 70 to < 100 mg/dL (≥ 3.9 to < 5.6 mmol/L) while avoiding hypoglycaemia episodes (ESM Table 1). Dose adjustments are based on median fasting SMPG values from the last 3 days, including values recorded on the day of titration, as measured by the participant using glucometers and other provided accessories. CGM data is blinded to both the investigators and participants. Participants are followed-up 2–4 days after their last dose to collect post-treatment safety information.

Study design. Asterisk indicates telephone calls by investigators to monitor insulin titration weekly between site visits for all participants, unless investigators are attending the study site for sensor replacement. Participant has option to come to site on day −10 and day 74 for sensor replacement. CGM Continuous glucose monitoring, Gla-300 insulin glargine 300 U/mL, HbA1c glycated haemoglobin, IDeg-100 insulin degludec 100 U/mL, MDI multiple daily injection, QD once daily, R randomisation, SMPG self-monitored plasma glucose, T1D type 1 diabetes
Planned Outcomes
Study Objectives
The primary objective is to demonstrate that Gla-300 is noninferior to IDeg-100 for TIR [glucose ≥ 70 to ≤ 180 mg/dL (≥ 3.9 to ≤ 10 mmol/L)] and variability in adults with T1D. The secondary aims are to evaluate glycaemic control and other variability parameters in each treatment group at week 12 using CGM in accordance with the International Hypoglycaemia Study Group (IHSG) [21] and Advanced Technologies and Treatments for Diabetes consensus (ATTD) [22] criteria for classification of hypoglycaemia, and to compare the treatment safety profiles.
Endpoints
The primary endpoint is percentage time spent in the glucose range of ≥ 70 to ≤ 180 mg/dL (≥ 3.9 to ≤ 10 mmol/L) at week 12. Secondary endpoints include: glucose total CV and glucose within-day and between-day CV at week 12; change from baseline to week 12 in HbA1c and central laboratory fasting plasma glucose (FPG); percentage time and mean hours per day with glucose < 70 mg/dL [anytime and during the night (0000–0559 hours)] and > 180 mg/dL; number of participants with AEs; and number of participants with at least one hypoglycaemic event and number of hypoglycaemic events per participant-year from baseline to week 12. Exploratory endpoints include further analysis of glycaemic control and variability parameters at week 12 using CGM; daily insulin doses; patient-reported outcomes in terms of treatment satisfaction, perception of blood glucose control, sleep, and productivity; and 7-point SMPG profiles (further details provided in ESM Information file).
Data Collection
Continuous Glucose Monitoring
Baseline and investigational CGM values are assessed during weeks 0–3 and 9–12, respectively, using the Dexcom G6® Pro CGM System (DexCom, Inc., San Diego, CA, USA). This CGM device records interstitial glucose levels at 5-min intervals for up to 10 continuous days via a subcutaneous sensor inserted into the abdomen at least 8 cm (3 inches) from the insulin injection sites. During each CGM period, participants wear the CGM device for 20 consecutive days to ensure collection of at least 10 days (not necessarily consecutive) of useable data. Useable CGM data for any given day are defined as > 80% time of records per 24 h and no missing data for ≥ 2 h per 24 h excluding the warm-up time for the sensor after insertion. Participants, investigators and study personnel are blinded to the study CGM data. The data will be uploaded to a separate server that uses vendor software (QuintilesIMS Study Management Suite; IQVIA, Durham, NC, USA) to maintain data anonymity; these data are used to generate a data acceptability report with daily CGM performance. The investigator will use the CGM summary to review each participant’s daily performance and to determine the quality of CGM records.
Self-Monitored Plasma Glucose
Participants self-monitor plasma glucose levels from week – 4 using provided Roche Accu-check® glucometers (Roche Diabetes Care, Inc., Mannheim, Germany) and corresponding accessories (lancet, control solutions and test strips). Insulin dose and injection time for basal insulin (prior to randomisation) and during the study treatment period (after randomisation) and all doses of mealtime insulin are documented daily in a provided diary. Pre-injection SMPG is measured 30 min prior to Gla-300 or IDeg-100 before breakfast from day 1 until up-titration has been completed and fasting pre-breakfast SMPG is stable in the target range, and then ≥ 3 times per week. Seven-point SMPG is carried out before (pre-injection after randomisation) and 2 h after all meals, and at bedtime at least once during weeks –1 and 11.
SMPG diary entries are reviewed by the investigators at each visit for episodes of hypoglycaemia and data from glucometers. Weekly telephone calls will be uploaded to site computers at weeks 0, 8 and 12. SMPG data will be used to guide adjustments in basal and mealtime insulin titrations to reach glucose targets.
Glycated Haemoglobin
Glycated haemoglobin levels are assessed at week – 6/– 5 (screening), week 0 (randomisation) and week 12. HbA1c is analysed according to a central certified level I National Glycohemoglobin Standardization Program.
Fasting Plasma Glucose
Fasting plasma glucose levels are measured in blood samples taken at week – 6/– 5 (screening), week 0 (randomisation) and week 12 at a central laboratory.
Safety Evaluation
All AEs are collected from the time of consent form signing to the end of follow-up to ensure patient safety. An AE is defined as any untoward medical occurrence in a participant that is temporally associated with the use of the study intervention, whether or not it is considered to be related to the study intervention. A serious AE (SAE) is defined as any untoward medical occurrence that, at any dose, results in death, is life-threatening, requires hospitalisation or results in persistent disability/incapacity. The investigator is obligated to assess the relationship between study intervention and each occurrence of every AE/SAE and record all AEs leading to discontinuation. All hypoglycaemic episodes are documented in a hypoglycaemia-specific form in the electronic case report form (eCRF), and SMPG diary entries are reviewed by investigators for episodes of hypoglycaemia. The incidence and distribution of hypoglycaemia, proportion of participants experiencing hypoglycaemia and diurnal distribution of hypoglycaemia are analysed according to American Diabetes Association (ADA) criteria for classification of hypoglycaemia (severe, documented symptomatic, asymptomatic, probable and relative) [23]. Hypoglycaemia fulfilling the seriousness criteria are additionally documented on the SAE form in the eCRF.
Statistical Analysis
Sample Size
The sample size is based on the percentage of time spent in the glucose range of ≥ 70 to ≤ 180 mg/dL (≥ 3.9 to ≤ 10 mmol/L) at week 12, as assessed using the CGM measurements obtained during weeks 10–12. Noninferiority will be tested with a one-sided type I error, and a relative noninferiority margin of 10% is considered. The sample size per treatment group is calculated to provide at least 90% power, assuming a percentage TIR of 56% in the control arm, a common standard deviation (SD) of 14.7% and a non-evaluability rate of 22%.
Analysis of Primary Endpoint
The percentage of time in the glucose range of ≥ 70 to ≤ 180 mg/dL (≥ 3.9 to ≤ 10 mmol/L) at week 12 will be assessed in both arms using 20 days of blinded CGM data during weeks 0–3 and 9–12. An analysis of covariance (ANCOVA) model including the fixed categorical effects of each treatment group and randomisation stratum of screening HbA1c (< 8.0 vs. ≥ 8.0%), and percentage TIR value as continuous fixed covariate is fitted to provide baseline adjusted least-squares means estimates at week 12 for both treatment groups and the differences between these estimates with corresponding standard errors and 95% confidence intervals (CI). Noninferiority is assessed in the intent-to-treat (ITT) population from the adjusted difference estimate of m1 – 0.9 m0 at week 12 and will be demonstrated if the lower bound of the two-sided 95% CI for this estimate is superior to 0.
Analysis of Secondary Endpoints
The CGM endpoints are analysed using the same model described for the primary endpoint using the ITT population. Changes in HbA1c are analysed using an ANCOVA model that includes the fixed categorical effects of Gla-300 and IDeg-100 treatments and the continuous fixed covariate of baseline HbA1c. Changes in FPG will be analysed using an ANCOVA model that includes the randomisation stratum of HbA1c at screening (< 8.0%, ≥ 8.0%), Gla-300 and IDeg-100 treatments and the continuous fixed covariate of baseline FPG. Changes from baseline in the 7-point SMPG profiles (preprandial and 2-h postprandial plasma glucose at all mealtimes, and at bedtime) to week 12 are described by treatment group using the mean and SD. A hierarchical step-down testing procedure will be applied to the primary efficacy endpoint (noninferiority for percentage TIR) and main secondary endpoints [noninferiority (10% margin) for glucose total CV, and superiority for percentage TIR] to control for type I error [24]. For other secondary endpoints and other efficacy variables, no multiplicity adjustments will be made, and any 95% CI and p values will be presented for descriptive purposes only.
Strengths and Limitations
The strengths of this study are that it is a prospective, multicentre, randomised, active-controlled, parallel-group, open-label, comparative study. The study protocol benefits from requiring minimal blood sampling and the use of CGM devices, both of which have been associated with low risk [8, 25, 26]. Moreover, the accuracy of CGM has been previously validated in adults with T1D [8, 27]. Although HbA1c is still considered the key method for evaluating glycaemic control [9, 28], CGM is likely to become the new gold standard for several reasons [29]. HbA1c provides a surrogate marker for average blood glucose over the previous few months and, as such, is unable to provide information on acute glycaemic deviations, be it hyper- or hypoglycaemia [9]. CGM, however, assesses blood glucose either in real-time or at intervals of 5–15 min, thereby facilitating almost immediate surveillance of glycaemic excursions as well as review of daily profiles [7, 9, 28]. The TIR metric is particularly advantageous because it allows for prompt adjustment of targets specific to each individual [9]. Nevertheless, there are limitations to CGM technology, including the 5- to 15-min lag between the sensor measuring glucose in interstitial fluid and blood glucose [7], and accuracy, as assessed by the mean or median absolute relative difference versus reference values obtained in a laboratory, which can be affected by aerobic exercise [30]. However, within the scope of this study, these limitations will be similarly reflected in both treatment arms.
The open-label nature of the study is an inherent limitation; however, as Gla-300 and IDeg-100 are distinguishable, no attempt is made to blind the randomly assigned study treatment. However, the assessment of outcomes is based on objectively collected data, blinded for CGM (primary and secondary endpoints) and open for SMPG data (secondary endpoint); individual CGM data are inaccessible to the participant, investigator and sponsor to avoid treatment decisions being made based on data for the primary endpoint.
Conclusions
The InRange trial is the first to use CGM data to assess primary endpoints exploring the similarities and differences of second-generation basal insulin analogues (Gla-300 and IDeg-100) according to the approved labels in adults with T1D. By providing head-to-head comparisons between the two most recently available long-acting basal insulin formulations, InRange should provide comprehensive glucose profiles, as measured by one of the most accurate methods, for participants with T1D receiving Gla-300 and IDeg-100. Additionally, the results should provide further data on the application of TIR in clinical practice.
Change history
18 November 2021
A peer-reviewed video abstract was retrospectively added to this publication
02 May 2020
A Correction to this paper has been published: https://doi.org/10.1007/s13300-020-00820-2
23 June 2020
A Correction to this paper has been published: https://doi.org/10.1007/s13300-020-00853-7
References
Nathan DM, DCCT/EDIC Research Group. The diabetes control and complications trial/epidemiology of diabetes interventions and complications study at 30 years: overview. Diabetes Care. 2014;37(1):9–16.
Leiter LA, Yale J-F, Chiasson J-L, Harris S, Kleinstiver P, Sauriol L. Assessment of the impact of fear of hypoglycemic episodes on glycemic and hypoglycemia management. Can J Diabetes. 2005;29(3):186–92.
Martyn-Nemeth P, Schwarz Farabi S, Mihailescu D, Nemeth J, Quinn L. Fear of hypoglycemia in adults with type 1 diabetes: impact of therapeutic advances and strategies for prevention—a review. J Diabetes Complic. 2016;30(1):167–77.
Foster NC, Beck RW, Miller KM, et al. State of type 1 diabetes management and outcomes from the T1D exchange in 2016–2018. Diabetes Technol Ther. 2019;21(2):66–72.
Pettus JH, Zhou FL, Shepherd L, et al. Incidences of severe hypoglycemia and diabetic ketoacidosis and prevalence of microvascular complications stratified by age and glycemic control in U.S. adult patients with type 1 diabetes: a real-world study. Diabetes Care. 2019;42(12):2220–7.
Ferrari MTM, Neto AM. Efficacy, safety and clinical use of newer basal insulins analogs. Endocrinol Metab Int J. 2018;6(3):215–21.
Klonoff DC, Ahn D, Drincic A. Continuous glucose monitoring: a review of the technology and clinical use. Diabetes Res Clin Pract. 2017;133:178–92.
Bolinder J, Antuna R, Geelhoed-Duijvestijn P, Kroger J, Weitgasser R. Novel glucose-sensing technology and hypoglycaemia in type 1 diabetes: a multicentre, non-masked, randomised controlled trial. Lancet. 2016;388(10057):2254–63.
Battelino T, Danne T, Bergenstal RM, et al. Clinical targets for continuous glucose monitoring data interpretation: recommendations from the international consensus on time in range. Diabetes Care. 2019;42(8):1593–603.
Sanofi S.A. TOUJEO: prescribing information. https://www.accessdata.fda.gov/drugsatfda_docs/label/2015/206538lbl.pdf. Accessed 23 Sep 2019.
Novo Nordisk A/S. TRESIBA: prescribing information. https://www.accessdata.fda.gov/drugsatfda_docs/label/2015/203314lbl.pdf2015. Accessed 23 Sep 2019.
White JR Jr. Advances in insulin therapy: a review of new insulin glargine 300 units/mL in the management of diabetes. Clin Diabetes. 2016;34(2):86–91.
Bailey TS, Pettus J, Roussel R, et al. Morning administration of 0.4 U/kg/day insulin glargine 300 U/mL provides less fluctuating 24-hour pharmacodynamics and more even pharmacokinetic profiles compared with insulin degludec 100 U/mL in type 1 diabetes. Diabetes Metab. 2018;44(1):15–21.
Haahr H, Heise T. A review of the pharmacological properties of insulin degludec and their clinical relevance. Clin Pharmacokinet. 2014;53(9):787–800.
Rosenstock J, Cheng A, Ritzel R, et al. More similarities than differences testing insulin glargine 300 units/mL versus insulin degludec 100 units/mL in insulin-naive type 2 diabetes: the randomized head-to-head BRIGHT trial. Diabetes Care. 2018;41(10):2147–54.
Philis-Tsimikas A, Klonoff DC, Khunti K, et al. Risk of hypoglycaemia with insulin degludec versus insulin glargine U300 in insulin-treated patients with type 2 diabetes: the randomised, head-to-head CONCLUDE trial. Diabetologia. 2020. https://doi.org/10.1007/s00125-019-05080-9.
Kawaguchi Y, Sawa J, Sakuma N, Kumeda Y. Efficacy and safety of insulin glargine 300 U/mL vs insulin degludec in patients with type 2 diabetes: a randomized, open-label, cross-over study using continuous glucose monitoring profiles. J Diabetes Investig. 2019;10(2):343–51.
Yamabe M, Kuroda M, Hirosawa Y, Kamino H, Ohno H, Yoneda M. Comparison of insulin glargine 300 U/mL and insulin degludec using flash glucose monitoring: a randomized cross-over study. J Diabetes Investig. 2019;10(2):352–7.
Monnier L, Colette C, Wojtusciszyn A, et al. Toward defining the threshold between low and high glucose variability in diabetes. Diabetes Care. 2017;40(7):832–8.
Heise T, Norskov M, Nosek L, Kaplan K, Famulla S, Haahr HL. Insulin degludec: lower day-to-day and within-day variability in pharmacodynamic response compared with insulin glargine 300 U/mL in type 1 diabetes. Diabetes Obes Metab. 2017;19(7):1032–9.
International Hypoglycaemia Study Group. Glucose concentrations of less than 3.0 mmol/L (54 mg/dL) should be reported in clinical trials: a joint position statement of the American Diabetes Association and the European Association for the Study of Diabetes. Diabetes Care. 2017;40(1):155–7.
Danne T, Nimri R, Battelino T, et al. International consensus on use of continuous glucose monitoring. Diabetes Care. 2017;40(12):1631–40.
American Diabetes Association. Standards of medical care in diabetes-2019. Diabetes Care. 2019;42(Supplement):1.
Hochberg Y, Tamhane AC. Multiple comparison procedures. Hoboken: Wiley. 1987.
Wojciechowski P, Rys P, Lipowska A, Gaweska M, Malecki MT. Efficacy and safety comparison of continuous glucose monitoring and self-monitoring of blood glucose in type 1 diabetes: systematic review and meta-analysis. Pol Arch Med Wewn. 2011;121(10):333–43.
Klonoff DC. Improving the safety of blood glucose monitoring. J Diabetes Sci Technol. 2011;5(6):1307–11.
Johnson T, Zhang X, Balo A. Assessing glucose trend accuracy with a novel continuous glucose monitoring system. Diabetes. 2018;67[Suppl 1]:950-P.
Wright LA, Hirsch IB. Metrics beyond hemoglobin A1C in diabetes management: time in range, hypoglycemia, and other parameters. Diabetes Technol Ther. 2017;19(S2):S16–26.
Burd JF, Melero FAA, Noetzel V. Hemoglobin A1c (HbA1c) shows improvement in glycemic control in as little as two weeks following the addition of lysulin™, to the treatment of diabetes. Diabetes Manag. 2018;8(3):82–4.
Biagi L, Bertachi A, Quiros C, et al. Accuracy of continuous glucose monitoring before, during, and after aerobic and anaerobic exercise in patients with type 1 diabetes mellitus. Biosensors (Basel). 2018;8(1). doi: https://doi.org/10.3390/bios8010022.
Acknowledgements
Funding
Sponsorship for this study and Rapid Service Fee were funded by Sanofi S.A., Paris, France. All authors take responsibility for the interpretation of the data in this study protocol.
Editorial Assistance
Editorial assistance was provided by Rebecca Lawson, PhD, of Fishawack Communications, and was funded by Sanofi S.A.
Authorship
All named authors meet the International Committee of Medical Journal Editors (ICMJE) criteria for authorship for this article, take responsibility for the integrity of the work as a whole, and have given their approval for this version to be published.
Authorship Contributions
All authors participated in the interpretation of the data, the writing, reviewing and editing of the manuscript, and had final responsibility for approving the published version.
Disclosures
Tadej Battelino has received honoraria for participation on advisory boards for Novo Nordisk, Sanofi, Eli Lilly, Boehringer, Medtronic and Bayer HealthCare, and as a speaker for AstraZeneca, Eli Lilly, Bayer, Novo Nordisk, Medtronic, Sanofi and Roche; owns stocks of DreaMed Diabetes; T.B.’s institution has received research grant support and travel expenses from Abbott Diabetes Care, Medtronic, Novo Nordisk, GluSense, Sanofi, Sandoz and Diamyd.
Thomas Danne has received speaker honoraria and research support and has consulted for Abbott, Bayer, Bristol-Myers Squibb, AstraZeneca, Boehringer Ingelheim, Dexcom, Eli Lilly, Medtronic, Novo Nordisk, Sanofi and Roche. T.D. is a shareholder of DreaMed.
Steve Edelman is a board member of Senseonics and serves on the speakers’ bureau and advisory panel for AstraZeneca, Dexcom, Eli Lilly, MannKind, Merck, Novo Nordisk and Sanofi.
Pratik Choudhary serves on advisory panels for Abbott, Cellnovo, Eli Lilly, Medtronic, Novo Nordisk, Roche and Sanofi; has received research support from Beta‐O2 and Medtronic; and serves on speakers bureaus for Abbott, Eli Lilly, Johnson & Johnson, Medtronic, Merck (MSD), Novo Nordisk, Roche and Sanofi.
Eric Renard has received consultancy honoraria from A. Menarini Diagnostics, Abbott, Becton–Dickinson, Cellnovo, Dexcom, Eli Lilly, Insulet, Johnson & Johnson, Medtronic, Novo Nordisk, Roche and Sanofi, and research support from Abbott, Dexcom, Insulet, and Roche.
Richard Bergenstal has received research support from, consulted for, or has been on a scientific advisory board for Abbott Diabetes Care, Dexcom, Eli Lilly, Johnson & Johnson, Medtronic, Novo Nordisk, Onduo, Roche, Sanofi, and United HealthCare. His research is partly funded by the National Institute of Diabetes and Digestive and Kidney Diseases (National Institutes of Health grant DK108611). R.M.B.’s employer, the nonprofit HealthPartners Institute, contracts for his services and no personal income goes to R.M.B.
Zsolt Bosnyak, Bhaswati Mukherjee and Valerie Pilorget are employees and shareholders of Sanofi S.A.
Compliance with Ethics Guidelines
Prior to enrolment, the protocol will be reviewed and approved by the Institutional Review Boards and Independent Ethics Committees of each participating institution in compliance with the Declaration of Helsinki. The full list of Institutional Review Boards and Independent Ethics Committees can be found in the Supplementary Material. This study will be conducted in accordance with the Declaration of Helsinki with all relevant amendments, the Council for International Organizations of Medical Sciences (CIOMS) International Ethical Guidelines, the applicable ICH Good Clinical Practice (GCP) Guidelines and all applicable laws and regulations.
Data Availability
Sanofi shares clinical trials results on publicly accessible websites, based on company commitments, international and local legal and regulatory requirements, and other clinical trial disclosure commitments established by pharmaceutical industry associations, including https://clinicaltrials.gov, https://www.clinicaltrialsregister.eu, https://www.sanofi.com, and some national registries. Anonymised individual subject data and supporting clinical documents will be available for request at https://clinicalstudydatarequest.com. Details on data sharing criteria and process for requesting access can be found at https://clinicalstudydatarequest.com.
Open Access
This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

Cite this article
Battelino, T., Bosnyak, Z., Danne, T. et al. InRange: Comparison of the Second-Generation Basal Insulin Analogues Glargine 300 U/mL and Degludec 100 U/mL in Persons with Type 1 Diabetes Using Continuous Glucose Monitoring—Study Design. Diabetes Ther 11, 1017–1027 (2020). https://doi.org/10.1007/s13300-020-00781-6
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13300-020-00781-6