
Abstract
Metabolic rewiring and epigenetic remodeling, which are closely linked and reciprocally regulate each other, are among the well-known cancer hallmarks. Recent evidence suggests that many metabolites serve as substrates or cofactors of chromatin-modifying enzymes as a consequence of the translocation or spatial regionalization of enzymes or metabolites. Various metabolic alterations and epigenetic modifications also reportedly drive immune escape or impede immunosurveillance within certain contexts, playing important roles in tumor progression. In this review, we focus on how metabolic reprogramming of tumor cells and immune cells reshapes epigenetic alterations, in particular the acetylation and methylation of histone proteins and DNA. We also discuss other eminent metabolic modifications such as, succinylation, hydroxybutyrylation, and lactylation, and update the current advances in metabolism- and epigenetic modification-based therapeutic prospects in cancer.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
PATHWAYS LEADING TO THE INTEGRATION OF METABOLISM AND EPIGENETIC MODIFICATION DURING CANCER DEVELOPMENT
Metabolic reprogramming is one of the major features of cancer, during which characteristics of metabolic enzymes, upstream regulating molecules and downstream metabolic products, known as metabolites, are altered (DeBerardinis et al., 2008b; Heiden et al., 2009; Jones and Thompson, 2009; Hanahan and Weinberg, 2011; DeBerardinis and Thompson, 2012; Hirschey et al., 2015; DeBerardinis and Chandel, 2016; Pavlova and Thompson, 2016; Sun et al., 2018; Thompson, 2019; Dai et al., 2020; Faubert et al., 2020). Recently, metabolism has been regarded as a major player and context-dependent regulator of epigenetic modifications, and increasing evidence suggests that intermediary metabolites drive chromatin dynamics through chemical posttranslational modifications (PTMs) that alter chromatin structures and functions (Kaelin and McKnight, 2013; Janke et al., 2015; Keating and El-Osta, 2015; Parker and Metallo, 2016; Reid et al., 2017; Chisolm and Weinmann, 2018; Wang and Lei, 2018; Zheng et al., 2020). Cellular metabolism and the epigenome interact in a bidirectional manner and interact with the genetic and molecular drivers that regulate cancer (Fig. 1). However, a comprehensive understanding of the interactions between molecular drivers, metabolic reprogramming, and epigenetic modifications in cancer are lacking, and thus, further elucidation of the associations is both necessary and pressing for more effective cancer therapy.

Crosstalks between metabolic reprogramming, epigenetic modifications, and transcriptional regulation. The cell metabolome and epigenome interact in a two-way manner and with genetic and molecular drivers that regulate cancer. A comprehensive understanding of the interactions between molecular drivers, metabolic reprogramming, and epigenetic modifications in cancer will further elucidate their connections and contribute to the development of effective cancer therapies
Cellular chromatin is composed of DNA and histones. Histones can undergo a wide range of PTMs such as phosphorylation, methylation, acetylation, and other acylation modifications. Similar to histones, DNA and RNA can be chemically modified by methylation to regulate gene expression. Epigenetic characteristics are usually abnormal in cancer cells. Human cancers often exhibit characteristic changes in DNA methylation, including genome-wide hypomethylation and site-specific hypermethylation (Jones and Baylin, 2002; Feinberg and Tycko, 2004). Global DNA hypomethylation in cancer was first observed by the Bert Vogelstein group in 1983 (Feinberg and Vogelstein, 1983). In mice, DNA hypomethylation is sufficient to induce aggressive T-cell lymphomas with a high frequency of chromosome 15 trisomy (Eden et al., 2003; Gaudet et al., 2003), whereas tumor suppressor genes are usually silenced by site-specific DNA hypermethylation at their promoters (Esteller et al., 2001). Similarly, the loss of histone 4 lysine 16 acetylation or histone 4 lysine 20 trimethylation is a common hallmark of human cancers (Fraga et al., 2005). Low levels of histone 3 lysine 4 dimethylation are associated with poor prognosis for patients with prostate (Seligson et al., 2005; Bianco-Miotto et al., 2010), lung (Barlesi et al., 2007; Seligson et al., 2009), breast (Elsheikh et al., 2009), pancreas (Manuyakorn et al., 2010), or kidney cancer (Ellinger et al., 2010). In addition, many oncogenes and tumor suppressors such as hypoxia-inducible factors (HIFs) (Watson et al., 2010; Prickaerts et al., 2016; Nanduri et al., 2017), von Hippel-Lindau tumor suppressor (VHL) (Herman et al., 1994; Schmitt et al., 2009; Vanharanta et al., 2013), Myc (Dang, 2012; Stine et al., 2015; Poole and van Riggelen, 2017; Topper et al., 2017; Poli et al., 2018; Li et al., 2020), p53 (Vrba et al., 2008; Su et al., 2009; Saldana-Meyer and Recillas-Targa, 2011), phosphatase and tensinhomolog (PTEN) (Salvesen et al., 2001; Kang et al., 2002; Soria et al., 2002; Garcia et al., 2004; Alvarez-Nunez et al., 2006), liver kinase B1 (LKB1) (Esteller et al., 2000; Trojan et al., 2000), AMP-activated protein kinase (AMPK) (Ruderman et al., 2010; Gongol et al., 2018; Yuan et al., 2020), and mechanistic target of rapamycin kinase (mTOR) (Laribee, 2018; Zeng et al., 2019), drive epigenetic reprogramming and are regulated by epigenetic modifications (Fig. 1).
Epigenetic abnormalities regulate the expression of many metabolic genes, thus playing important roles in metabolic rewiring and redox homeostasis of cancer cells (Wong et al., 2017). In contrast, metabolic flux is involved in epigenetic regulation by affecting the biosynthesis of macromolecules and energy production (Zheng et al., 2020). All these events are synergistically involved in the path to cancer. For example, in addition to regulating glucose, glutamine and serine metabolism at the transcriptional level (Gao et al., 2009; Stine et al., 2015; Sun et al., 2015; Wu et al., 2017), cMyc increases SDHA (succinate dehydrogenase complex, subunit A) acetylation by promoting SKP2 (S-phase kinase-associated protein 2)-mediated sirtuin3 degradation, leading to SDHA deactivation and succinate accumulation. Increased succinate inhibits the activity of histone demethylases, which triggers histone 3 lysine 4 trimethylation and the expression of tumor-specific genes and subsequent tumor progression (Li et al., 2020). During pancreatic ductal adenocarcinoma (PDAC) progression, 6-phosphogluconate dehydrogenase (6PGD) -mediated oxidative pentose phosphate pathway (oxPPP) supports the reprogramming of histone H3K9 and DNA methylation, thereby promoting N-cadherin (epithelial-mesenchymal transition marker) transcription and N-cadherin-mediated distant metastasis (McDonald et al., 2017). SETD2 (SET domain-containing 2, a histone lysine methyltransferase) integrates EZH2 (enhancer of zeste homolog 2) and the AMPK signaling pathway to restrict prostate cancer metastasis by linking metabolism with epigenetic modifications (Yuan et al., 2020). H3.3K27M (histone H3.3 lysine 27-to-methionine) mutation in diffuse intrinsic pontine gliomas (DIPGs) results in global H3K27me3 reduction by multiple mechanisms, such as the aberrant PRC2 interactions or hampered H3K27me3 spreading (Bender et al., 2013; Chan et al., 2013; Lewis et al., 2013; Stafford et al., 2018; Harutyunyan et al., 2019). However, by integrating metabolic and epigenetic pathways, Chung et al. found that H3.3K27M mutations promote glycolysis, glutaminolysis, and TCA cycle-derived α-KG (α-ketoglutarate) accumulation, leading to α-KG-dependent activation of H3K27 demethylases KDM6A/6B, H3K27 hypomethylation, and tumor progression (Chung et al., 2020; Zhao and Miao, 2020). Histone acetylation regulates cell proliferation and tumor progression (Cai et al., 2011; Donohoe et al., 2012; Lee et al., 2014), as well as other cellular biological behaviors not covered in this review article, such as intracellular pH (McBrian et al., 2013), hippocampal memory (Mews et al., 2017), cell fate decisions (Yadav et al., 2018), and cellular differentiation (Chisolm and Weinmann, 2018).
Notably, all these events and reactions require metabolites, including acetyl-CoA, NAD+ (nicotinamide adenine dinucleotide), SAM (S-adenosyl methionine), α-KG, FAD (flavin adenine dinucleotide), ATP, and succinate, as substrates or cofactors (Fig. 2). The dysregulation of histone PTMs and DNA/RNA modifications is associated with the occurrence of many diseases. Although all these metabolites play crucial roles in energy metabolism, cell cycle progression, cell growth and death, neuroregeneration, circadian rhythm, and the pluripotency of stem cells, in this review, we discuss the current understanding of how essential metabolites, as well as their regulating molecules, control the epigenome by dynamically regulating the metabolic states of DNA, histones and other proteins during cancer development.

An overview of metabolic connections to epigenetic remodeling. Nutrients such as glucose, fatty acids, and amino acids are metabolized by cells to produce a variety of metabolites, such as acetyl-CoA, NAD+, SAM, α-KG, ATP, and succinate, which function as substrates or cofactors to modify chromatin and proteins. Specifically, 1) UDP-GlcNAc, as a donor substrate derived from the HBP pathway integrating glucose, glutamine, fatty acid (acetyl-CoA), and nucleotide metabolism (UDP), is catalyzed by OGT for GlcNAcylation modification, and OGA controls the reverse reaction. 2) Lactate generates lactyl-CoA, which contributes a lactyl group to lysine residues of histone proteins through p300, generating a novel modification called lactylation. 3) Glucose-, fatty acid-, amino acid-, and acetate-derived acetyl-CoA are widely involved in acetylation modification. Histone acetylation is catalyzed by HATs, and the reverse reaction is mediated by lysine deacetylases (HDAC and SIRT). 4) Based on the ratio of ATP:AMP, AMPK is required for the phosphorylation of histones under various stress conditions. 5) Histone lysine β-hydroxybutyrylation (Kbhb) depends on the metabolite β-hydroxybutyrate (βOHB), which is produced by the ketone body metabolic pathway. The enzymes involved in acetylation modification mediate this reversible reaction. 6) Citrulline is categorized into two types: free citrulline from the arginine-coupled urea cycle and the guanidine dehydration of arginine residues on proteins to create citrulline residues. Histone citrullination is a PTM that converts arginine residues to citrulline via PAD enzymes, which are Ca2+-dependent. 7) TCA cycle-derived succinyl-CoA is the major substrate for succinylation, and the opposite reaction is mediated by KAT2A, CPT1A, and SIRT5. 8) Reversible chromatin methylation is coupled with SSP, the folate cycle, and the methionine cycle. SAM is the substrate for HMTs and DNMTs, leading to the production of SAH. Succinate, fumarate, and 2-HG inhibit the demethylases HDMs and TETs, which catalyze the demethylation reaction in an α-KG-dependent manner. In addition, NAD+ and NADH transitions are involved in modifications such as acetylation, β-hydroxybutyrylation, and succinylation
METABOLITES PLAY KEY ROLES IN EPIGENETIC REMODELING ON THE PATH TO CANCER
Acetyl-CoA metabolism in acetylation regulation
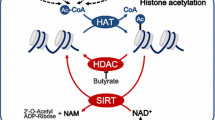
Writers, readers, and erasers of protein acetylation
Protein (histone) acetylation is a chemical reaction catalyzed by lysine (histone) acetyltransferases (KATs/HATs), during which an acetyl group donated by acetyl-CoA is added to a lysine residue of the protein (histone). Three major families of KATs, GNAT (G protein subunit alpha transducin), MYST (Moz, Ybf2/Sas3, Sas2, and Tip60), and p300/CBP (E1A-binding protein p300/CREB-binding protein), have been identified (Sabari et al., 2017). All these KATs require acetyl-CoA, the sole donor of the acetyl group in eukaryotic cells (Choudhary et al., 2014). Bromodomain proteins (e.g., BRD4 and BRDT), YEATS domain proteins (e.g., MLLT3 and Taf14), and double PHD finger (DPF) domain proteins (e.g., MOZ and DPF2) are readers that interact with acetyl-lysine residues and recognize the lysine acetylation (Kacetyl) (Sabari et al., 2017) to recruit transcription factors and/or super elongation complexes to support transcriptional activation (Fujisawa and Filippakopoulos, 2017; Gates et al., 2017; Zhao et al., 2017; Haws et al., 2020). Lysine deacetylases are erasers critical for removing acetyl groups. Zinc-dependent histone deacetylases (zinc-dependent HDACs) and NAD+-dependent sirtuins are two major families of lysine deacetylases (De Ruijter et al., 2003; Jing and Lin, 2015). Class I (HDAC1, 2, 3, and 8), class II (HDAC4, 5, 6, 7, 9, and 10), and class IV (HDAC11) HDACs are zinc-dependent enzymes, and class III HDACs, also called sirtuins, are dependent on the NAD+ concentration (Fig. 2).
In most mammalian cells, acetyl-CoA is a central metabolite that is primarily generated from glucose-derived pyruvate by the pyruvate dehydrogenase complex (PDC) in mitochondria. Fatty acid β-oxidation (Rufer et al., 2009), the catabolism of branched amino acids (BCAAs) (Harris et al., 2005), and free acetate all contribute to the generation of mitochondrial acetyl-CoA (Pietrocola et al., 2015) (Fig. 2); however, there is no acetyl-CoA transporter on the mitochondrial membrane. In rapidly proliferating cells, citrate, upon synthesis due to acetyl-CoA and oxaloacetate (OAA) condensation in mitochondria, is quickly exported to the cytosol by the citrate carrier SLC25A1, where it is converted back to acetyl-CoA and OAA by ATP citrate lyase (ACLY) (Icard et al., 2012; Zaidi et al., 2012). Both ACLY and all the subunits of PDC are present in the nucleus of mammalian cells and promote the generation of acetyl-CoA (Wellen et al., 2009; Sutendra et al., 2014). Acetyl-CoA synthesis from acetate is mediated by acyl-CoA synthetase short-chain family members (ACSSs), including ACSS1 and ACSS3 in mitochondria and ACSS2 in the cytoplasm and nucleus (Luong et al., 2000; Fujino et al., 2001; Perez-Chacon et al., 2009; Ariyannur et al., 2010; Choudhary et al., 2014; Comerford et al., 2014). Acetyl-CoA functions as a carbon source for histone acetylation, cell growth and proliferation (Cai et al., 2011) and regulates autophagy (Eisenberg et al., 2014) and intracellular pH (McBrian et al., 2013). Here, we focus on localized acetyl-CoA production mediated by PDC, ACLY, and ACSSs in different organelles and its regulation of chromatin and other proteins.
The roles of compartmentalized acetyl-CoA metabolism in chromatin regulation and protein acetylation
PDC Glucose-derived cytosolic pyruvate enters mitochondria by the mitochondrial pyruvate carrier (MPC), a heterodimer of MPC1 and MPC2 (Herzig et al., 2012). Mitochondrial pyruvate is decarboxylated to generate acetyl-CoA by PDC, a large multicomponent composed of pyruvate dehydrogenase (PDH), dihydrolipoamide S-acetyltransferase (DLAT), dihydrolipoamide dehydrogenase (DLD), pyruvate dehydrogenase kinase (PDK), pyruvate dehydrogenase phosphatase (PDP), and pyruvate dehydrogenase complex, component X (PDHX). Among these proteins, PDH, DLAT, and DLD are directly involved in CoA- and NAD+-dependent pyruvate decarboxylation; PDK and PDP are two regulatory components; and PDHX is a nonenzymatic subunit (Patel et al., 2014).
Once mitochondrial activity is suppressed by Bcl-xL (B-cell lymphoma-2-like 1, also known as BCL2L1) overexpression, the levels of citrate and acetyl-CoA are decreased, but there is no obvious decrease in histone H3 or H4 acetylation (Yi et al., 2011). By isolating the nuclear components and confocal microscopy, Sutendra et al. found the presence of PDH, DLAT, and DLD in the nucleus in different types of cells. These components are required for acetyl-CoA generation and the acetylation of the core histones H2B, H3, and H4. Increased nuclear PDC proteins are translocated from mitochondria upon serum stimulation, epidermal growth factor stimulation, or mitochondrial stress during S phase. The inhibition of nuclear PDC by implementing novel strategies decreased the acetylation levels of specific histone lysine residues that are vital for cell cycle progression and S phase entry (de Boer and Houten, 2014; Sutendra et al., 2014) (Fig. 3).

Compartmentalized acetyl-CoA metabolism in chromatin regulation. Under stimulation or stress conditions, mitochondrial-localized PDC and cytosol-localized ACLY and ACSS2 may translocate into the nucleus for the generation of the nuclear acetyl-CoA pool, mediating global histone acetylation (left). In certain cases, PDC binds with PKM2 and p300 to generate a large complex in the nucleus. In this large nuclear complex, the pyruvate kinase activity of PKM2 controls the production of pyruvate from PEP, and nuclear PDC further catalyzes the reaction in which pyruvate produces local acetyl-CoA to support the histone acetylation modification at special gene enhancers controlled by p300 (right)
The role of PDC in cancer progression remains inconclusive (Kim et al., 2006; Papandreou et al., 2006; Hitosugi et al., 2011; Kaplon et al., 2013; Sutendra et al., 2014). In mouse and human prostate cancer models, Chen et al. found that mitochondrial PDC provides cytosolic citrate for lipid synthesis, whereas nuclear PDC is critical for the acetylation of H3K9 and the expression of sterol regulatory element-binding transcription factor (SREBF) target genes, such as ACLY and squalene epoxidase (SQLE). Therefore, PDCs located in different organelles promote lipogenesis and prostate cancer progression by providing substrates and upregulating lipid metabolic enzymes at epigenetically modified levels, respectively (Chen et al., 2018a). The E2 subunit of PDC (also known as DLAT) binds with PKM2 (pyruvate kinase isozyme M2) and p300 to generate a large complex in the nucleus that includes aryl hydrocarbon receptor (AhR), a transcription factor involved in xenobiotic metabolism such as CYP1A1 (cytochrome P4501A1). In this large nuclear complex, the pyruvate kinase activity of PKM2 controls the production of pyruvate from PEP, and nuclear PDC catalyzes pyruvate to produce local acetyl-CoA for histone acetylation at the gene enhancer controlled by p300 (Matsuda et al., 2016) (Fig. 3). A novel oncogene with kinase-domain (NOK), a potent oncogene, promotes histone acetylation by inducing the translocation of PDC from mitochondria to the nucleus, thus causing the occurrence and metastasis of tumors (Shi et al., 2017).
ACLY ACLY, which catalyzes the conversion of citrate to acetyl-CoA and OAA, is overexpressed in many cancers and links energy metabolism, biosynthesis, and epigenetic modification (Chypre et al., 2012; Zaidi et al., 2012; Icard et al., 2020). The structural basis for ACLY function was recently revealed (Verschueren et al., 2019). SiRNA knockdown of ACLY or pharmacologic inhibitor SB-204990 inhibiting ACLY activity can significantly increase the mitochondrial membrane potential and inhibit lipid synthesis, cell cycle entry, and cell growth (Hatzivassiliou et al., 2005). By deconvolution microscopy and subcellular fractionation, ACLY was found to exist not only in the cytoplasm but also in the nucleus. Nuclear localized ACLY is the major source of acetyl-CoA accumulation required for histone acetylation and homologous recombination-mediated DNA repair (Wellen et al., 2009; Linder and Mostoslavsky, 2017; Sivanand et al., 2017) (Fig. 3).
During growth factor stimulation or adipocyte differentiation, glucose affects histone acetylation and fatty acid synthesis in an ACLY-dependent manner (Wellen et al., 2009; Lee et al., 2014; Martinez Calejman et al., 2020). The ratio of acetyl-CoA and coenzyme A is glucose-sensitive and determines histone acetylation levels in cancer cells. Activated AKT (AKT serine/threonine kinase) phosphorylates ACLY, resulting in sustained histone acetylation under glucose deprivation conditions, and pAKT (Ser473) was positively correlated with histone acetylation levels in human glioma and prostate cancers (Lee et al., 2014). The AKT-ACLY axis also supports the proliferation of KRAS (Kirsten rat sarcoma 2 viral oncogene homolog)-mutant pancreatic acinar cells, and inhibition of AKT reduces histone acetylation and suppresses acinar-to-ductal metaplasia (ADM). Pancreas-specific deletion of ACLY inhibits ADM and pancreatic tumorigenesis without overt metabolic abnormalities (Carrer et al., 2019). Recently, ACLY was identified as a novel substrate of caspase-10, which is cleaved by caspase-10 at the conserved Asp1026 site. Under metabolic stress conditions, such as glucose starvation, increased caspase-10 downregulates intracellular lipid levels and represses GCN5-mediated histone H3 and H4 acetylation by ACLY cleavage, ultimately inhibiting the expression of tumor-related proliferative genes and metastatic genes as well as tumor progression (Kumari et al., 2019). In patient-derived acute myeloid leukemia (AML) cells, both the substrate and product of phosphoinositide 3-kinase (PI3K), phosphatidylinositol-(4,5)-bisphosphate (PIP2), and phosphatidylinositol-(3,4,5)-trisphosphate (PIP3), respectively, bind to ACLY. The Src-family kinase (SFK) Lyn directly interacts and phosphorylates the tyrosine residues of ACLY. Inhibitors of PI3K, Lyn, and ACLY action suppress the growth of AML cells by decreasing H3K9 acetylation levels (Basappa et al., 2020).
Macrophage activation or polarization can be finely tuned by metabolic shifts. Upon interleukin-4 (IL-4) stimulation, AKT is activated to enhance glucose utilization in murine bone marrow-derived M2 macrophages. Histone acetylation levels at select M2 genes such as Arg1, Retnla and Mgl2, are increased through AKT-phosphorylated ACLY. SB-204990, the inhibitor of ACLY, indeed suppressed the induction of IL-4/AKT-dependent M2 genes (Covarrubias et al., 2016; Williams and O’Neill, 2018). However, in human monocyte-derived macrophages, ACLY is not required for IL-4-induced macrophage polarization, although pharmacological ACLY inhibitors suppress IL-4-induced target gene expression, suggesting off-target effects of ACLY inhibitors (Namgaladze et al., 2018). It’s known that tumor-associated macrophages (TAMs) create an inflammatory environment that facilitates survival and proliferation of tumor cells, but the role of ACLY-mediated metabolic rewiring of macrophages in tumorigenesis remains unclear. Understanding what conditions within tumors affect the IL-4-AKT-ACLY signaling axis may provide new insights into the role of macrophages in tumor progression. Therefore, tumor microenvironment plays an important role in determining macrophage activity.
Toll-like receptor 4 (TLR4) is an important sensor that recognizes lipopolysaccharide (LPS). Upon LPS recognition, TLR4 promotes the secretion of inflammatory factors and interferon by recruiting four signaling adaptor molecules, including MyD88 (myeloid differentiation primary-response protein 88), MAL (MyD88-adaptor-like protein, also called TIR domain-containing adaptor protein (TIRAP)), TRIF (TIR-domain-containing adaptor protein-inducing IFNB), and TRAM (TRIF-related adaptor molecule). LPS stimulation induces the metabolic reprogramming of glycolysis and the TCA cycle, leading to the accumulation of synthetic citrate and an increase in the acetyl-CoA pool in bone marrow-derived macrophages (BMDMs). MyD88 and TRIF signaling drives LPS-induced ACLY phosphorylation and histone acetylation, and ACLY activation is critical for histone acetylation at the IL12b gene locus and for facilitating enhancer chromatin accessibility in response to LPS stimulation (Lauterbach et al., 2019; Williams and O’Neill, 2020). IL-2-induced ACLY phosphorylation and ACLY activation are required for T-cell proliferation, and inhibition of ACLY by SB-204990 induces G1/S cell cycle arrest and suppresses the accumulation of histone acetylation levels in IL-2-treated T cells (Osinalde et al., 2016). This study suggests that activation of ACLY in T cells can inhibit tumor growth by promoting the proliferation of T cells.
ACSS Glucose-derived pyruvate is the major source of acetyl-CoA generation. In rapidly proliferating cells or hypoxic cells, however, pyruvate preferentially converts to lactate and does not enter mitochondria to produce acetyl-CoA. With findings similar to those showing ACLY-deficient budding yeast reliance on acetate for acetyl-CoA synthesis (De Virgilio et al., 1992; van den Berg et al., 1996; Takahashi et al., 2006), Comerford et al. showed that ACSS2 is the major enzyme required for acetate uptake and utilization and further incorporation into lipids and for histone acetylation in mammalian cells. ACSS2-knockout (KO) reduced the tumorigenesis of hepatocellular carcinoma in a mouse model, and ACSS2 expression was significantly elevated in hepatocellular tumors of mice and in a variety of human tumor samples, including breast, ovarian, and lung cancer tissues, as determined by immunohistochemical (IHC) staining (Comerford et al., 2014). Glucose oxidation contributes less than 50% of the carbon to the acetyl-CoA pool in human brain tumors (Maher et al., 2012), and 13C-acetate is oxidized in primary and metastatic mouse glioblastomas (GBMs) in situ even with the simultaneous coinfusion of available 13C-glucose. ACSS2 expression is required for the incorporation of 13C-acetate into glutamate and is positively correlated with the malignancy of GBM (Lyssiotis and Cantley, 2014; Mashimo et al., 2014).
Under metabolic stress, such as hypoxia and lipid-depleted conditions, induced ACSS2 expression promotes the uptake and utilization of acetate to produce acetyl-CoA, which further contributes to fatty acids and supports the biosynthesis of membrane phospholipids. Nuclear-localized ACSS2 maintains the levels of histone acetylation (Schug et al., 2015; Bulusu et al., 2017) (Fig. 3). Exogenous acetate addition rescues the hypoxia-induced decrease in histone acetylation and epigenetically activates lipogenic genes, such as fatty acid synthase (FASN) and acetyl-CoA carboxylase 1 (ACACA). The high expression of ACSS1 and ACSS2 in hepatocellular carcinoma is critical for acetate-mediated histone acetylation and de novo lipogenesis (Gao et al., 2016). AMPK phosphorylates ACSS2 at S659 and promotes its nuclear translocation under glucose deprivation conditions. In the nucleus, ACSS2 binds to transcription factor EB (TFEB) at the promoter regions of lysosomal and autophagy-associated genes and further promotes H3 acetylation and the expression of these genes by locally producing acetyl-CoA from acetate (Li et al., 2017a; Li et al., 2017b). Similar to the yeast system, ACLY-deficient cancer cells primarily use acetate to supply abundant acetyl-CoA by upregulating ACSS2 (Zhao et al., 2016).
Lactate promotes histone acetylation and gene expression in cell culture as an endogenous HDAC inhibitor. Latham et al. found that the effect of lactate, trichostatin A (TSA) and butyrate on gene expression was similar, suggesting that the three of them had a common HDAC inhibition mechanism (Latham et al., 2012). Lactate is known to promote tumorigenesis by providing ATP, acidifying microenvironment, recycling, and immunosuppression. Therefore, the role and contribution of lactate-mediated histone acetylation in tumorigenesis still need further study. Butyrate is a short-chain fatty acid produced by the fermentation of dietary fiber by the gut microbiota in the colon (Scheppach and Weiler, 2004; Hamer et al., 2008). High levels of butyrate in the lumen are the major energy sources that are metabolized to acetyl-CoA by ACLY for the proliferation of normal colonocytes and cancerous colonocytes (Roediger, 1982; Fleming et al., 1991; Donohoe et al., 2012). Butyrate-derived acetyl-CoA induces histone acetylation and regulates gene expression by stimulating HATs and inhibiting HDACs in an ACLY-dependent and ACLY-independent manner, respectively (Donohoe et al., 2012). β-Hydroxybutyrate (β-OHB) is a byproduct of the oxidation of fatty acids. In addition to serving as energetic metabolites, β-OHB has been increasingly shown to promote protein acetylation as a signaling metabolite in two ways. On one hand, the catabolism of β-OHB into acetyl-CoA increases the intracellular acetyl-CoA concentration, which favors the acetylation of histone and nonhistone proteins. On the other hand, under fasting or calorie restriction conditions, endogenous β-OHB binds and inhibits class I histone deacetylase, promotes the acetylation of Lys9 and Lys14 of histone H3 and activates gene transcription controlled by the transcription factor FOXO3a (forkhead box O3A), which is associated with the longevity of a variety of organisms (Shimazu et al., 2013). These findings support the increase in β-OHB concentration observed in mammals during caloric restriction and the resistance of cells to oxidative stress under these conditions. In studies of Drosophila, nematodes, and yeast, class I HDACs have been implicated in the life-extending effects of caloric restriction, suggesting that an environment that increases the β-OHB concentration (e.g., caloric restriction) may extend life by inhibiting class I HDACs.
NAD+ metabolism and acetylation regulation
NAD+ serves as a cofactor of sirtuins during the deacetylation of lysine residues, and it plays important roles in enhancing mitochondrial function and protecting liver and kidney tissues from injury (Katsyuba et al., 2018). NAD+ is mainly synthesized from the tryptophan, Preiss-Handler, or nicotinamide (NAM) salvage pathways, with the latter pathway contributing the majority of NAD+ (Verdin, 2015; Yang and Sauve, 2016). The NAD+/NADH ratio is closely related to the acetylation state and energy state. High glycolytic cells often generate a low NAD+/NADH ratio, thereby resulting in the repressed activity of sirtuins, especially SIRT6 which binds NAD+ with relatively high affinity (K(d) = 27 ± 1 μmol/L) in the absence of an acetylated substrate (Pan et al., 2011; Madsen et al., 2016). Under stress and nutrient restriction conditions, NAM phosphoribosyltransferase (NAMPT) is induced and protects cells against death induced by genotoxic stress in a SIRT3- and SIRT4-dependent manner (Yang et al., 2007). In Ndufs4 (NADH dehydrogenase [ubiquinone] iron-sulfur protein 4)-KO mice, mitochondrial complex I loss leads to reduced NAD+ levels. The addition of nicotinamide mononucleotide (NMN), the precursor of NAD+, or cell-permeable α-KG increases the lifespan of Ndufs4-KO mice by promoting protein hyperacetylation (Lee et al., 2019). In addition to affecting acetylation, NAD levels also regulate methylation status. Lozoya et al. found that depletion of mitochondrial DNA (mtDNA) leads to DNA hypermethylation by reprogramming the methionine cycle and increasing SAM levels, almost all of which can be rescued by maintaining mitochondrial NADH oxidation (Lozoya et al., 2018).
Acetylation regulates the location, activity and function of transcription factors and metabolic enzymes
Using 13C-labeled glucose and gas chromatography mass spectrometry (GC/MS) analysis, the oncogene c-Myc was demonstrated to promote fatty acid biosynthesis and H4K16 acetylation by inducing mitochondrial acetyl-CoA generation (Morrish et al., 2010; Edmunds et al., 2014). C-Myc interacts with p300 through its TAD (transcription activation domain), and the Myc-Max complex can be acetylated by p300 and GCN5 (general control of amino acid synthesis 5-like 2 in Yeast). In addition, p300 is recruited by c-Myc to the promoter as a coactivator of the human telomerase reverse transcriptase (hTERT) gene to promote transcription (Faiola et al., 2005). Hypoxia-inducible factor 1α (HIF-1α), the master regulator of the hypoxic microenvironment, is acetylated by p300/CBP-associated factor (PCAF) and deacetylated by SIRT1. SIRT1 inhibits HIF-1α activity by blocking p300 recruitment, leading to downregulated glycolysis and retarded tumor growth (Lim et al., 2010).
In addition, the activities of many metabolic enzymes are regulated by acetylation modulation (Choudhary et al., 2009; Zhao et al., 2010). For example, the enzyme activity of glyceraldehyde-3-phosphate dehydrogenase (GAPDH) is increased when it is acetylated at K254 by PCAF acetyltransferase (Li et al., 2014). PCAF-mediated K117 and K251 acetylation of GAPDH is necessary for its nuclear localization after apoptotic stimulation (Ventura et al., 2010). Mitogenic and oncogenic signaling induces p300-mediated acetylation of PKM2 at the K433 residue, promoting PKM2 protein kinase activity and nuclear translocation by preventing its dimer-to-tetramer transition (Lv et al., 2013). Acetylation regulates glucogenesis and PPP by modulating the activity of phosphoenolpyruvate carboxykinase (PEPCK) and 6PGD, respectively (Jiang et al., 2011; Shan et al., 2014). Glucose at high levels stabilize ACLY by inducing PCAF-mediated acetylation at lysine 540, 546, and 554. Acetylated ACLY promotes de novo lipid synthesis, cell proliferation, and tumor progression in lung cancer (Lin et al., 2013). Long-chain acyl-CoA dehydrogenase (LCAD), a key mitochondrial fatty acid oxidation enzyme, is a direct target of SIRT3. Hyperacetylation of LCAD at lysine 42 in SIRT3-knockout mice reduced LCAD enzyme activity (Hirschey et al., 2010), and acetylation of lysine residue 318 and 322 of LCAD are two other SIRT3-targeted sites (Bharathi et al., 2013). Branched-chain amino acid transaminase 2 (BCAT2), the rate-limiting enzyme of BCAA metabolism, is acetylated at K44 by CREB-binding protein (CBP). This PTM modulation of BCAT2 promotes its degradation and suppresses BCAA catabolism and pancreatic cancer progression (Lei et al., 2020). Please see Table 1 for more details.
Metabolic control of (histones) proteins and DNA methylation
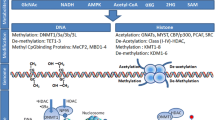
Writers, readers, and erasers of proteins and DNA methylation
Methylation extensively regulates cellular physiology by modulating the status and activity of proteins (histone) but also of DNA and RNA. Histone methylation ranging from mono- to trimethylation occurs at lysine or arginine residues in H3 and H4 (Di Lorenzo and Bedford, 2011; Kinnaird et al., 2016; Guccione and Richard, 2019). There are only eight residues (H3K4/9/18/23/27/36/79 and H4K20) that undergo significant methylation modulation, but each lysine can support mono-, di-, or trimethylation (Haws et al., 2020). These histone methylation marks can activate or repress gene expression, depending on the types of residues, the number of methyl group(s) added, and the location within the N-terminal regions of H3 or H4 involved (Greer and Shi, 2012). For instance, the methylation of H3K4 and H3K79 is generally associated with transcriptional activation, while H3K9 and H3K27 methylation suppresses gene transcription (Etchegaray and Mostoslavsky, 2016).
Histone methyltransferases (HMTs), as writers of histone methylation, catalyze the methylation reaction in a site-specific manner, mainly on the ε-amino group of lysine residues. Histone demethylases (HDMs) serve as erasers that remove methyl groups from histones. The first identified HDM was lysine-specific histone demethylase 1 (LSD1, also known as KDM1A) in 2004. LSD1 utilizes FAD (also known as vitamin B2) as a cofactor to oxidize the methylated lysine ε-amino to remove methyl groups and produce FADH2 (Shi et al., 2004; Anand and Marmorstein, 2007; Greer and Shi, 2012), but LSD1 catalyzes only the removal of methyl groups on mono- or dimethylated lysine residues. Jumonji C (JmjC) domain-containing demethylases (JHDM or Jmj-KDM) are other HDMs that function through a ferrous (Fe2+)- and α-KG-dependent dioxygenase mechanisms and are critical for the removal of methyl groups in all three forms (Anand and Marmorstein, 2007; Dimitrova et al., 2015). Methylated lysine residues are recognized by “reader” proteins containing methyl-lysine-binding motifs, including PHD, Tudor, PWWP, WD40, BAH (bromo adjacent homology), ADD (ATRX-DNMT3-DNMT3L), chromodomain (CD), double chromodomain (DCD), tandem Tudor domain (TTD), ankyrinrepeat, MBT (malignant brain tumor), and zn-CW (zinc finger CW) domains. These “reader” proteins have the ability to distinguish target methyl-lysines based on their methylation state and surrounding amino acid sequences (Yun et al., 2011; Musselman et al., 2014; Hyun et al., 2017) (Fig. 2).
In human DNA, the DNA base, especially in CpG islands, can be methylated at the fifth carbon of cytosine (5mC/5-methylcytosine) by DNA methyltransferases (DNMTs), usually resulting in transcriptional repression (Bergman and Cedar, 2013). DNA methyltransferase (writer) enzymes, including DNMT1, DNMT3a, and DNMT3b, are major players in the methylation of 5mC at gene promoters (Greenberg and Bourc’his, 2019). Similar to histone methylation, DNA methylation is also a reversible epigenetic modification. Ten-eleven translocation enzymes TET1, TET2, and TET3 (erasers), which rely on Fe2+ and α-KG as co-substrate and cofactor, drive the demethylation of DNA (Pastor et al., 2013). DNA methylation is recognized by methyl-binding proteins (MBPs) (readers), including methyl-CpG-binding protein 2 (MeCP2), MBD1, MBD2, MBD3, MBD4, MBD5, MBD6, SET domain bifurcated 1/2 (SETDB1/2), or bromodomain adjacent to zinc finger domain 2A/B (BAZ2A/B) (Mahmood and Rabbani, 2019) (Fig. 2).
All HMTs and DNMTs require the intermediary metabolite SAM as a methyl donor for both histones and DNA (Takusagawa et al., 1996). Ferrous and α-KG are essential substrates and cofactors for HDMs and TETs. Other TCA cycle intermediary metabolites, such as 2-HG (2-hydroxyglutarate), succinate and fumarate, are also involved in the methylation regulation of histones and DNA by inhibiting HDM and TET activity (Etchegaray and Mostoslavsky, 2016; Haws et al., 2020) (Fig. 2). Although RNA can also be methylated, we focus on discussing how histone and DNA methylation are regulated by SAM derived from one-carbon metabolism (methionine, threonine, and serine metabolism) and other intermediary metabolites derived from the TCA cycle.
One-carbon metabolism is directly linked to chromatin dynamics
S-adenosyl methionine (SAM) and methylation Major components of one-carbon metabolism are the methionine cycle and folate cycle. SAM, generated in the methionine cycle, is the primary methyl group donor for histone or DNA methylation (Fig. 2). In mammalian cells, intracellular SAM biosynthesis depends on the condensation of methionine and ATP, which is catalyzed by the rate-limiting enzyme methionine adenosyltransferase Iα or IIα (MATIα or MATIIα) (Sakata et al., 1993; Markham and Pajares, 2009; Reytor et al., 2009). MATIα is expressed specifically in the liver, whereas MATIα is ubiquitously expressed in various tissues (Markham and Pajares, 2009). SAM is demethylated to form S-adenosylhomocysteine (SAH), which is further converted to homocysteine after deadenylation by S-adenosyl homocysteine hydrolase (SAHH). Homocysteine accepts carbon from the folate cycle through 5-methyltetrahydrofolate (mTHF) to generate methionine, resulting in a full turn of the methionine cycle (Locasale, 2013).
SAM is a universal methyl donor and is utilized by methyltransferases to methylate DNA, RNA, metabolites, and proteins, including histones. Methionine metabolism regulates the genomic architecture, chromatin dynamics and gene expression by dynamically modulating trimethylation at lysine 4 on histone H3 (H3K4me3) in both mice with normal physiology and human cancer cells (Dai et al., 2018). Under methionine-limiting conditions, SAM, SAH and the SAM/SAH ratio are dynamically regulated, which reduces the H3K4me3 level and affects the expression of methylation-related enzymes (Mentch et al., 2015). In immortalized mouse embryonic fibroblasts (iMEFs), THP-1 cells, and mouse hepatoma (Hepa-1) cells, MATIIα represses cyclooxygenase 2 (COX-2) expression at the mRNA level. Specifically, MATIIα interacts with the histone H3K9 methyltransferase SETDB1, leading to the accumulation of H3K9me3 at the COX-2 locus and the repression of the COX-2 gene (Kera et al., 2013). MATIIα also interacts with the transcription factor MafK in the nucleus and acts as a transcriptional corepressor of MafK by affecting the levels of H3K4me2 and H3K9me2. MATIIα is involved in the MafK-mediated suppression of heme oxygenase-1 (HO-1) (Katoh et al., 2011). Moreover, SAM was reported to be involved in innate immunity by regulating H3K4me3 levels in C. elegans (Ding et al., 2015). Recently, Bian et al. found that tumor cells can absorb a large amount of methionine through the methionine transporter SLC43A2, and competition results in methionine deficiency in T cells, thus affecting epigenetic changes, including loss of H3K79me2 in T cells and impairing the effector function of T cells (Bian et al., 2020).
Glycine N-methyltransferase (GNMT), the most abundant liver methyltransferase, is a SAM-buffering enzyme that catalyzes the transfer of a methyl group from SAM to glycine to form sarcosine, leading to SAM depletion and sarcosine accumulation (Obata et al., 2014; Serefidou et al., 2019). Martinez-Chantar et al. showed that deletion of GNMT in mice induces the hypermethylation of DNA and histones, resulting in steatosis, fibrosis, and hepatocellular carcinoma (Martinez-Chantar et al., 2008). However, Liao et al. found global hypomethylation of DNA in GNMT-knockout mice. In their opinion, decreased DNA methylation is associated with decreased DNMT activity and aberrant DNMT1 and DNMT3b expression (Liao et al., 2009). Hughey et al. also found that elevated SAM promotes polyamine synthesis, polyamine catabolism, transsulfuration, and de novo lipogenesis in GNMT-knockout mice (Hughey et al., 2018). Threonine, as the only amino acid critical for the pluripotency of mouse embryonic stem cells (mESCs), regulates stem cell fate by regulating their methylation status. Depletion of threonine from the culture medium or knocking down threonine dehydrogenase (TDH) by shRNAs in mESCs decreased the levels of SAM and H3K4me3, leading to slowed growth and increased differentiation (Shyh-Chang et al., 2013).
Folate is a well-documented metabolite in DNA methylation (Crider et al., 2012; Ly et al., 2012). Diets low in folate cause genomic DNA hypomethylation, which can affect DNA stability and gene expression and increase the risk of neoplasia. Physiological intake of folic acid can reverse this phenomenon in patients with colorectal adenocarcinoma (Pufulete et al., 2005). Folate supplementation effectively decreased the degree of DNA hypomethylation of the rectal mucosa, but only in patients with a single polyp (Cravo et al., 1998). Similar to many other metabolites, folate can be detected in the nucleus (Zamierowski and Wagner, 1977). In the nucleus, folate is bound to LSD1 and protects LSD1 from inhibition by formaldehyde (Luka et al., 2011; Luka et al., 2014). In mice treated with a folate-deficient diet, reduced folate levels in the liver are associated with increased methylated H3K4 levels due to decreased LSD1 activity (Garcia et al., 2016).
Serine and glycine metabolism in methylation regulation Serine and glycine, which are involved in nucleotide synthesis, methylation reactions, and the generation of GSH (glutathione) and NADPH (the reduced form of nicotinamide adenine dinucleotide phosphate), are additional important one-carbon donors that are integrated with the folate cycle. In most cultured cells, serine donates its β-carbon atom to tetrahydrofolate (THF) via serine hydroxymethyltransferases (SHMTs), generating glycine and 5,10-methylene-THF (me-THF), which initiates the folate cycle. Serine and glycine regulate methylation by linking with the folate cycle, which is coupled to the methionine cycle (Fig. 2). me-THF can also be produced by the glycine cleavage system (glycine dehydrogenase (GLDC) is the major component), in which glycine is cleaved into ammonia, carbon dioxide, and a carbon unit, which is involved in the methylation of THF (Locasale, 2013; Yang and Vousden, 2016). Moreover, other nutrient sources, including threonine, choline, betaine, dimethylglycine, and sarcosine (N-methylglycine), regulate methylation reactions via their convertion into glycine (Wang et al., 2009; Locasale, 2013).
Serine can provide one-carbon units to generate methionine from homocysteine; in addition, ATP (purine) generated by serine-mediated de novo synthesis is also involved in the production of SAM from methionine (Fig. 2). In colorectal cancer cells, methionine is the major methyl donor, and serine does not directly provide one-carbon units for methylation under conditions of methionine supplementation. However, serine availability controls the methyl transfer from methionine to DNA and RNA because this process is impeded during serine starvation. In brief, serine-contributed ATP synthesized de novo (based on serine availability) is critical for the SAM cycle regardless of whether methionine is present, and the role of serine is highlighted in supporting DNA/RNA methylation through the maintenance of nucleotide levels (Maddocks et al., 2016; Parker and Metallo, 2016). The serine-responsive SAM-containing metabolic enzyme complex (SESAME) is a supercomplex consisting of pyruvate kinase, serine metabolic enzyme, and SAM synthetases in yeast. The interaction of SESAME with the Set1 H3K4 methyltransferase complex regulates H3K4 methylation and H3T11 phosphorylation (H3pT11) by sensing glycolysis and glucose-derived serine metabolism (Li et al., 2015b). LKB1 (also known as STK11) loss and KRAS activation (KRASG12D) synergistically potentiate glycolysis, serine metabolism, and tumorigenesis. In LKB1-deficient cells, the activated de novo serine biosynthesis pathway promotes DNA methylation. LKB1 loss decreases phosphoserine aminotransferase 1 (PSAT1)-mediated DNA methylation and retrotransposon expression, important modulators of host gene expression. Tumor-bearing mice with LKB1 loss and human LKB1-mutant pancreatic tumor cells are more sensitized to DNMT knockdown or DNMT inhibitor decitabine treatment, which inhibits serine biosynthesis and DNA methylation (Kottakis et al., 2016). More recently, another serine biosynthesis enzyme, SHMT2, was reported to initiate lymphoma development by epigenetically silencing tumor suppressors. The SHMT2 gene is amplified in human B cells. Elevated SHMT2 expression in human and mouse follicular lymphoma (FL), the most common form of B-cell lymphoma, is controlled by MYC, and a similar mechanism has been reported in hepatoma carcinoma cells (Sun et al., 2015). SHMT2 activation induces SAM synthesis to promote DNA and histone methylation, leading to promoter silencing of previously unappreciated tumor suppressor genes, such as SAM and SH3 domain-containing protein 1 (SASH1) and protein tyrosine phosphatase receptor type M (PTPRM), and the initiation of lymphomagenesis (Parsa et al., 2020).
TCA cycle-derived intermediary metabolites regulate methylation status
α-KG regulates histone and DNA methylation Although the TCA cycle is known to play central roles in ATP production, it is also now appreciated as a source of biosynthetic precursors and chemical intermediates (DeBerardinis et al., 2008a). α-KG, also known as 2-oxoglutarate (2-OG), is generated from isocitrate in a reaction catalyzed by cytoplasmic IDH1 (isocitrate dehydrogenase 1) or mitochondrial IDH2 and IDH3, accompanied by the production of NADPH from NADP. α-KG is a cosubstrate required for the histone demethylase JHDM and DNA demethylase TETs, as described above (Fig. 2). In addition to isocitrate, other amino acids, such as arginine, histidine, proline, and glutamate from glutamine-derived glutaminolysis also mediate α-KG synthesis (Wise et al., 2011; Metallo et al., 2012; Mullen et al., 2012; Kaelin and McKnight, 2013). The core region of solid tumors, such as melanoma and breast cancer, displayed low glutamine levels compared with the tumor periphery, as determined by liquid chromatography-mass spectrometry (LC-MS) analysis. In patient-derived V600EBRAF melanoma cells, treatment to ensure low glutamine levels significantly decreased α-KG levels, which led to the hypermethylation of histone H3, H3K27-mediated tumor dedifferentiation, and resistance to BRAF inhibitor treatment. Knocking down the H3K27-specific demethylase KDM6B mimics the low-glutamine condition and mediates resistance to PLX4032 (BRAF inhibitor) treatment, and the opposite results are obtained when H3K27 methyltransferase EZH2 is knocked down (Pan et al., 2016). Epigenetic and metabolic reprogramming coordinates the polarization of macrophages and contributes to their functional plasticity (Ivashkiv, 2013; O’Neill and Pearce, 2016). Glutamine-derived α-KG is also important for the alternative (M2) activation of macrophages. A high α-KG/succinate ratio is found in IL-4-induced M2 macrophages compared to LPS-induced M1 macrophages. M2 polarization depends on the α-KG–JMJD3-mediated demethylation of H3K27 (Liu et al., 2017). Moreover, intracellular α-KG derived from glucose or glutamine promotes H3K27 demethylation and TET-dependent DNA demethylation, contributing to the maintenance of embryonic stem cell (ESC) pluripotency (Carey et al., 2015). PSAT1, a serine biosynthesis transaminase, mediates the production of α-KG. PSAT1 knockdown is sufficient to reduce intracellular α-KG and accelerate the differentiation of mouse ESCs by modulating DNA 5’-hydroxymethylcytosine (5’-hmC) and histone methylation levels (Hwang et al., 2016).
IDH mutation-induced 2HG regulates DNA and histone methylation Two independent groups undertaking cancer genome sequencing projects identified IDH1 mutations in both glioblastoma multiforme and acute myeloid leukemia in 2008 and 2009, respectively (Parsons et al., 2008; Mardis et al., 2009). A missense mutation in a single arginine residue, R132, in the enzyme active site is sufficient to cause IDH1-related disease alteration. Mutations in IDH2 are also apparent in GBM and other cancers (Yan et al., 2009). The R132H substitution of IDH1 (Parsons et al., 2008; Mardis et al., 2009) and the R172K and R140Q substitutions of IDH2 (Ward et al., 2010) constitute the majority of mutational events and lead to the occurrence of GBM, AML, chondrosarcoma, cholangiocarcinoma, and angioimmunoblastic T-cell lymphoma (Cairns and Mak, 2013; Lu et al., 2013). Mutant IDH1 and IDH2 are oncogenes that catalyze the conversion of α-KG to 2HG in an NADPH-dependent manner (Dang et al., 2009; Losman and Kaelin, 2013). There are two enantiomeric forms of 2HG, D-(or R-) and L-(or S-) type 2-HG, all of which are α-KG inhibitors that inhibit α-KG-dependent histone lysine demethylases, such as FIH (factor inhibiting HIF), PHD2 (prolyl hydroxylase domain-containing protein, also known as HIF prolyl-hydroxylase 2), and JMJDs (Chowdhury et al., 2011).
D2HG is the major form in diseases with IDH1 or IDH2 mutants (Dang et al., 2009; Gross et al., 2010). FAD-dependent D-2-hydroxyglutarate dehydrogenase (D2HGDH) regulates the generation of D2HG in E. coli, yeast, and human cancer cells (Zhao and Winkler, 1996; Fan et al., 2015; Lin et al., 2015; Becker-Kettern et al., 2016; Ye et al., 2018). Leukemic IDH1 and IDH2 mutants induce global DNA hypermethylation, destroy TET2 function, impair hematopoietic differentiation, increase the expression of stem/progenitor cell markers, and ultimately promote malignant transformation (Figueroa et al., 2010). In nontransformed cells, adipocytes, and immortalized astrocytes, the introduction of either mutant IDH or cell-permeable 2HG blocks cell differentiation by inducing global and promoter-specific H3K9 and H3K27 methylation (Lu et al., 2012).
D2HG regulates the HIF-1 signaling axis HIF-1 protein levels are precisely controlled by PHDs, also known as Eglnine homologs (EGLNs), which are α-KG-dependent dioxygenases that function as cellular oxygen sensors. The R132H mutant of tumor-derived IDH1 showed decreased catalytic activity due to impaired isocitrate binding and reduced α-KG levels, leading to elevated HIF-1α protein levels in human glioblastoma cells (Zhao et al., 2009). An increase in HIF in IDH-mutant tumors is usually present in necrotic areas and is presumed to be due to severe hypoxia (Williams et al., 2011). Losman et al. found that D2HG, but not L2HG, promotes leukemic transformation in a dose- and passage-dependent manner. In TF-1 human erythroleukemia cells overexpressing the IDH1 R132H mutant, HIF-1α is diminished due to the agonistic effect of D2HG on PHDs (Losman et al., 2013; McCarthy, 2013; Ye et al., 2013). In immortalized human astrocytes and HCT116 colorectal cancer cells, D2HG stimulates PHD activity by acting as its cosubstrate, resulting in reduced HIF levels and ultimately enhancing cell proliferation and transformation (Koivunen et al., 2012). This regulatory complexity indicates that D2HG-regulated HIF stability is cell type- and context-dependent (Losman and Kaelin, 2013).
The roles of L2HG in tumor cells and immune cells In renal cell carcinoma (RCC), accumulated L2HG mediates epigenetic modifications by serving as an oncometabolite and an epigenetic modifier. Lower expression of L-2-hydroxyglutarate dehydrogenase (L2HGDH) in RCC results in the accumulation of L2HG and reduces 5hmC levels on DNA. This outcome is consistent with the 2HG-mediated suppression of TET enzymes, which convert 5mC to 5hmC. The re-expression of L2HGDH promotes 5hmC accumulation, reduces H3K27me3 and H3K9me3 levels, and inhibits the proliferation of RCC cells (Shim et al., 2014). Moreover, enhanced L2HG production is also found under hypoxic conditions (Intlekofer et al., 2015; Oldham et al., 2015), and in turn, L2HG stabilizes HIF-1 protein levels by inhibiting PHD activity (Koivunen et al., 2012). 13C-labeled glucose or glutamine assays demonstrated that glutamine-derived α-KG is critical for hypoxia-induced L2HG generation. Although IDH controls the generation of D2HG, knocking down IDH1 or IDH2 did not affect L2HG levels in response to hypoxia. L2HG levels are modestly decreased by knocking down MDH1 or MDH2 (malate dehydrogenase), which are known to convert α-KG to L2HG (Rzem et al., 2007), but knocking down LDHA (lactate dehydrogenase A) strikingly decreased L2HG accumulation in hypoxic cells (Intlekofer et al., 2015). L2HG accumulation is necessary and sufficient for the activation of H3K9me3 and repressive histone methylation (Intlekofer et al., 2015) and inhibits electron transport and glycolysis to alleviate reductive stress (Oldham et al., 2015). In response to T-cell receptor triggering, the accumulation of L2HG in mouse CD8+ T cells depends on the VHL-HIF-LDHA axis and PDK-PDH signaling. In turn, L2HG stabilizes HIF-1α and modulates the global histone H3K27me3. L2HG induction or supplementation enhances the proliferation, long-term persistence and antitumor capacity of adoptively transferred CD8+ T cells (Tyrakis et al., 2016; Cairns and Mak, 2017).
Fumarate and succinate antagonize the roles of α-KG In addition to IDH1 and IDH2, germinal and somatic mutations of fumarate hydratase (FH) and succinate dehydrogenases (SDHA, SDHB, SDHC, SDHD, and SDHAF2), encoding FH and SDH enzymes, are common in a number of human cancers (Baysal et al., 2000; Astuti et al., 2001; Hao et al., 2009; Kaelin, 2009; Bayley et al., 2010; Oermann et al., 2012). Accumulated fumarate and succinate resulting from FH and SDH mutations share structural similarity with α-KG. Both fumarate and succinate increase global histone methylation and HIF-1α protein levels and reduce endostatin in cultured cells by inhibiting the activity of α-KG-dependent KDMs. In addition, TET-mediated 5hmC production is decreased by knocking down FH or SDH or supplementation with fumarate or succinate. These epigenetic alterations induced by FH or SDH loss contribute to tumorigenesis (Xiao et al., 2012). The epithelial-to-mesenchymal transition (EMT) has been implicated in tumor progression and metastasis. In human FH-deficient UOK262 cells, mesenchymal markers, including Snai2, Zeb1 and Zeb2, are induced, and the re-expression of FH reverses the expression of these markers. Fumarate inhibits the TET-mediated demethylation of anti-metastatic miR-200, leading to the induction of EMT-related transcription factors and enhanced migratory potential of renal cancer (Sciacovelli et al., 2016).
Oncometabolites hinder DNA repair Recently, two studies by Sulkowsk et al. suggested that IDH, FH, or SDH mutation-induced accumulation of 2-HG, fumarate, or succinate suppresses the homologous recombination (HR) DNA repair pathway in gliomas and AML with mutant IDH, hereditary leiomyomatosis and renal cell cancer (HLRCC), and succinate dehydrogenase-related hereditary paraganglioma and pheochromocytoma (SDH PGL/PCC) (Sulkowski et al., 2017; Sulkowski et al., 2018). In 2020, Sulkowsk et al. further revealed the pathways in which metabolites (2HG, succinate, and fumarate) interfere with DNA repair. By inhibiting the activity of the histone demethylase KDM4B, tumor cell metabolites cause the hypermethylation of H3K9me3 at DNA break sites, thus affecting DNA homology-dependent repair (HDR). Subsequently, the enrichment of key HDR molecules TIP60 (tat-interacting protein, also known as histone acetyltransferase KAT5) and ATM (ataxia telangiectasia mutated) and downstream repair factors at DNA fracture sites was reduced. This oncometabolite-induced HDR defect confers intensive sensitivity to poly (ADP-ribose) polymerase (PARP) inhibitors being tested in clinical trials (Chen and Xiong, 2020; Sulkowski et al., 2020). Therefore, this study explains a molecular mechanism of tumor metabolite-induced HDR inhibition and suggests a potential therapeutic strategy for tumor therapy.
Succinyl-CoA and (histone) succinylation
Research on succinylation stemmed from its role in inhibiting antibody formation and testing allergic skin responses in animals that were sensitive to dinitrophenyl-polyline in 1962 (Parker et al., 1962). In the following years, the succinylation of pepsinogen (Gounaris and Perlmann, 1967), ovalbumin (Kidwai et al., 1976), and histone amino groups (Pineiro et al., 1992) was studied in succession. However, it was not until 2011 that succinylation was identified as a natural PTM of lysine residues in bacteria by affinity purification with anti-succinyl lysine antibody (Zhang et al., 2011; Alleyn et al., 2018; Sreedhar et al., 2020). Succinylation of lysine (Ksucc) residues converts the cationic lysine side chain into an anionic chain with large potential impacts on protein structures, charges, and functions, and this modification is reversible, dynamic, and evolutionarily conserved in both prokaryotes and eukaryotes (Xie et al., 2012; Weinert et al., 2013; Wang et al., 2017b; Kurmi et al., 2018; Wang et al., 2019a).
TCA cycle-derived succinyl-CoA is the major substrate for succinylation. Succinyl-CoA can be generated from the TCA cycle, lipids, and amino acid metabolism (histidine, proline, glutamine, glutamate, methionine, and the BCAAs isoleucine, leucine, and valine) (Hirschey and Zhao, 2015) and then synthesized by succinyl-CoA synthetase. As early as 1992, Pineiro et al. noticed that the transcriptional properties of succinylated nucleosomal cores are similar to those of acetylated particles, which had been observed in 1991 (Pineiro et al., 1991; Pineiro et al., 1992). Defects in the TCA cycle by the depletion of SDH increase succinyl-CoA, and subsequent histone hypersuccinylation correlates with active gene expression (Smestad et al., 2018). The α-ketoglutarate dehydrogenase complex (α-KGDH) in the nucleus can bind to lysine acetyl transferase 2A (KAT2A) in gene promoter regions, and KAT2A binds to succinyl CoA and acts as a succinyltransferase to succinylate histone H3 on lysine 79. If the α-KGDH complex is blocked from entering the nucleus or KAT2A protein expression is inhibited, the expression of downstream target genes can be reduced, thus inhibiting tumor growth (Wang et al., 2017b; Wang et al., 2018b; Xu et al., 2021). Moreover, carnitine palmitoyltransferase 1A (CPT1A) is found to have lysine succinyltransferase activity upon the succinylation of S100A10 in gastric cancer (Kurmi et al., 2018; Wang et al., 2019a). These studies showed that the nonmetabolic functions of α-KGDH and CPT1A play important roles in tumor progression. Although succinyl-CoA is mainly synthesized in mitochondria, cytosolic succinate is converted back to succinyl-CoA (Alarcon et al., 2002), a result that reasonably explains how proteins undergo lysine succinylation in the cytoplasm and nucleus.
KAT2A and CPT1A are writers of protein succinylation, and SIRT5 has been identified as an eraser of Ksucc by catalyzing the hydrolysis of succinyl lysine in vitro and desuccinylating several mammalian proteins, such as glutamate dehydrogenase (GDH), malate dehydrogenase, and citrate synthase (CS), that were identified by mass spectrometry to have been modified by succinylation (Du et al., 2011) (Fig. 2). SIRT7 was identified as another histone desuccinylase, especially in response to DNA damage (Li et al., 2016). The YEATS domain of GAS41 recognizes succinylation as a pH-dependent reader of Ksucc (Wang et al., 2018a). Park et al. identified 2,565 succinylation sites on 779 proteins regulated by SIRT5 in mammalian fibroblasts and liver tissues, and found that diverse mitochondrial and nonmitochondrial metabolic enzymes can be succinylated (Park et al., 2013). Most of the 2,565 succinylation sites do not overlap with acetylation sites; however, thousands of succinylation sites mapped by Weinert et al. in diverse organisms including bacteria, yeast, and human cells, extensively overlap with acetylation sites (Weinert et al., 2013). SIRT5 is a mitochondrial protein, and in SIRT5-KO mice, the mitochondrial lysine succinylome in liver tissues is significantly changed. The metabolic pathways of fatty acid β-oxidation and ketogenesis are highly targeted by SIRT5. Lack of SIRT5 impairs β-oxidation and promotes the accumulation of acylcarnitines. 3-Hydroxy-3-methylglutaryl-CoA synthase 2 (HMGCS2), the rate-limiting enzyme of ketogenesis, is hypersuccinylated in the absence of SIRT5, but this modification of HMGCS2 inhibits its activity and reduces ketone body production (Rardin et al., 2013). IDH1 mutant-induced production of 2-HG inhibits SDH activity and promotes hypersuccinylation in mitochondria, which induces cancerous metabolism and apoptosis resistance (Li et al., 2015a). These results demonstrate that SIRT5 is a global regulator of lysine succinylation in the cytosol, mitochondria, and nucleus and indicate mechanisms for preventing tumorigenesis by modulating protein succinylation (Park et al., 2013; Rardin et al., 2013; Stram and Payne, 2016; Carrico et al., 2018).
PKM2, a notable metabolic enzyme, is succinylated at lysine 498 (K498), which increases PKM2 activity and sensitizes cells to oxidative damage by decreasing cellular NADPH production. These processes are reversed by SIRT5, which is critical for PKM2 desuccinylation by binding to PKM2 at K498 (Xiangyun et al., 2017). However, under nutrient stress conditions, such as glucose starvation, K433 succinylation of PKM2 promotes its translocation to mitochondria, whereas succinylation of K498 has no effect on this process. Mitochondrial PKM2 prevents the degradation of voltage-dependent anion channel 3 (VDAC3) and increases mitochondrial permeability to generate more ATP for cell survival under glucose starvation conditions (Qi et al., 2019). In addition, UCP1 (mitochondrial uncoupling protein 1) is a novel target of SIRT5-mediated desuccinylation recently identified in brown fat tissues in SIRT5-KO mice (Wang et al., 2019b). ACOX1 (acyl-CoA oxidase 1) and IDH2 are desuccinylated by SIRT5 in response to oxidative damage (Zhou et al., 2016; Chen et al., 2018b). SIRT5 also inhibits ubiquitin-mediated glutaminase (GLS) degradation by desuccinylating glutaminase, thereby regulating mitophagy and tumorigenesis (Polletta et al., 2015; Greene et al., 2019).
SDHA, which mediates succinate dehydrogenation, is a direct target of SIRT5, and desuccinylation of SDHA by SIRT5 suppresses SDH activity and cellular respiration (Park et al., 2013). Succinate has been shown to act as a proinflammatory metabolite that accumulates in LPS- or interferon-γ (IFN-γ)-treated macrophages (Tannahill et al., 2013; Jha et al., 2015). LPS-induced glutamine-derived succinate stabilizes HIF-1α protein expression, which is critical for LPS-induced interleukin-1β (IL-1β) transcription. A twofold increase in protein succinylation, such as MDH, was discovered in LPS-treated macrophages, a result that can be explained by an increase in succinate and a decrease in the expression of desuccinylase SIRT5 in macrophages (Tannahill et al., 2013). Analogous to its status in tumors, PKM2 is succinylated in macrophages. The succinylation of PKM2 at K311 inhibits PKM2 enzyme activity and promotes its nuclear translocation in dimer form. In the nucleus, by performing nonmetabolic functions, the dimeric form of PKM2 binds to HIF-1α to promote the transcription of IL-1β. Desuccinylation of PKM2 by SIRT5 blocks LPS-induced IL-1β expression to prevent DSS (dextran sodium sulfate)-induced colitis in mice (Wang et al., 2017a).
Ketone body-derived β-hydroxybutyrate modulates protein hydroxybutyrylation
Ketone bodies contain three different molecules, acetone, acetoacetate (AcAc), and β-OHB, which are byproducts of the oxidation of fatty acids in the liver to provide energy for the heart and brain under fasting conditions (Newman and Verdin, 2014a, b, 2017). In addition to serving as energy metabolites and promoting protein acetylation, as mentioned above, Xie et al. discovered a novel epigenetic modification, histone lysine β-hydroxybutyrylation (Kbhb), which is closely related to ketone body metabolism (Fig. 2). The researchers found significant increases in histone lysine β-hydroxybutyrylation modification in mouse liver cells but no change in acetylation modification that is mainly derived from glucose metabolism under fasting conditions. Further ChIP-qPCR (chromatin immunoprecipitation (ChIP) coupled with quantitative PCR) assays and ChIP-seq data showed that with the increase in histone Kbhb modification, the expression of some genes related to physiological responses to fasting was upregulated, such as amino acid catabolism, redox balance, circadian rhythm, and PPAR (peroxisome proliferator-activated receptor) signaling (Xie et al., 2016). Histone acyltransferase p300 acts as a writer to mediate histone β-hydroxybutyrylation (Kaczmarska et al., 2017), and human SIRT3 acts as an eraser to selectively remove histone β-hydroxybutyrylation with a preference for H3K4, K9, K18, K23, K27, and H4K16 but has no activity with H4K5, K8, and K12, which distinguishes it from Zn-dependent class I HDACs (Abmayr and Workman, 2019; Zhang et al., 2019b). The Kbhb levels of Lys9 in histone H3 (H3K9bhb) were reduced in the brains of depressed mice, and exogenous β-OHB rescued this phenomenon and ameliorated depressive behaviors of these mice (Chen et al., 2017).
Naïve CD8+ T (Tn) cells can differentiate into CD8+ effector T (Teff) cells after receiving antigen stimulation. Some Teff cells persist and develop into long-lived CD8+ memory T (Tmem) cells. Fatty acid-mediated oxidative phosphorylation and phosphoenolpyruvate carboxykinase 1 (PCK1)-mediated glycogen metabolism control the formation and maintenance of CD8+ Tmem cells (Pearce et al., 2009; Ma et al., 2018). Recently, Zhang et al. found that acetyl-CoA in mitochondria indirectly regulates PCK1 expression through the ketogenesis pathway, and epigenetic modification of histones through β-OHB is also critical for the formation of CD8+ Tmem cells (Zhang et al., 2020). Accumulated β-OHB and AcAc, but not acetone, were found in CD8+ Tmem cells compared to Tn or Teff cells, as determined by LC-MS analysis. After β-OHB and AcAc treatment of induced T cells, it was found that only β-OHB was specifically involved in the memory formation of CD8+ Tmem cells, and similar results were observed in mice treated with a carbohydrate-free ketogenic diet. Furthermore, β-OHB facilitates CD8+ Tmem cell formation by upregulating H3K9bhb of Foxo1 and Ppargc1a (which encodes PGC-1α), two transcription factors critical for Pck1 expression. These results provide new insights into how CD8+ Tmem cells balance energy supply and redox homeostasis and into their potential clinical applications for regulating T cell memory.
In addition to the hydroxybutyrylation modification of histones, p53 can undergo hydroxybutyrylation modification in the presence of β-OHB, and this modification is catalyzed by histone acetyltransferase CBP/p300 (Kaczmarska et al., 2017).This modification of p53 was apparent in cells treated with β-OHB and in thymic tissues of mice whose fasting resulted in increased serum β-OHB concentration. Hydroxybutyrylation of p53 inhibits its acetylation and reduces the expression of its target genes, including p21 and PUMA, ultimately promoting cell proliferation (Liu et al., 2019).
Lactylation is a novel PTM mediated by lactate and plays roles in addition to lactate metabolism
Lactic acid has long been considered a waste product of glycolytic metabolism; however, in 2017, two independent groups recently found that it can be reused as the primary carbon source for the mitochondrial TCA cycle in normal tissues and tumors (Faubert et al., 2017; Hui et al., 2017; Sun et al., 2018). Recently, it was found that lactic acid is integrated into cell metabolism by inducing dynamic endoplasmic reticulum-mitochondrial Mg2+ changes (Daw et al., 2020). Lactate was also found to promote histone acetylation and regulate gene expression as an HDAC inhibitor (Latham et al., 2012). Furthermore, inspired by the wide acylation of histones by intracellular metabolites, Zhang et al. recently found that lactic acid can also modify the lysine residues of histones in a new epigenetic modification known as lactylation (Zhang et al., 2019a) (Fig. 2). Twenty-eight lysine lactylation (Kla) sites on core histones, including H3, H4, H2A, and H2B, were identified in human HeLa cells and mouse BMDMs. The lactylation of H3 and H4 is p53-dependent and p300-mediated (Fig. 2). Hypoxia and macrophage polarization, which are associated with increased lactate derived from activated glycolysis, can enhance intracellular histone lactylation. In the late phase of M1 macrophage polarization, increased histone Kla directly promotes gene transcription and induces homeostatic genes, including Arg1, a marker of M2 macrophages. Interestingly, the researchers also detected histone lactylation in macrophages isolated from mouse melanoma and lung tumors and observed a positive correlation between histone lactylation and oncogenic production by reparative M2 macrophages. These findings suggest that high lactate and histone lactylation levels in macrophages may contribute to tumor formation and progression.
It is surprising that a single metabolite can have such a powerful effect on immune cell function. The discovery of histone lactylation and its impact on macrophage biology is a blueprint for understanding how lactic acid changes other cell types, unlocking the mysteries of the Warburg effect and understanding its impact on human disease. Whether cancer cells and other immune cells, such as T cells, can be regulated through this mechanism is unclear. In addition to cancer, the Warburg effect has been observed in other diseases, including sepsis, autoimmune diseases, atherosclerosis, diabetes, and aging. Therefore, more studies are needed on the role and regulation of this newly discovered histone modification.
ATP-, O-GlcNAc-, citrulline-, and itaconate-mediated phosphorylation, O-GlcNAcylation, citrullination, and itaconation
ATP and phosphorylation
In addition to lysine and arginine, serine and threonine can be extensively modified, such as by phosphorylation and O-GlcNAcylation. Histone phosphorylation is a dynamic modification in which phosphate groups are added to residues of serine or threonine. Phosphorylation was first discovered in the late 1960s (Kleinsmith et al., 1966; Gutierrez and Hnilica, 1967), and the first histone kinase was discovered in 1968 (Langan, 1968). Only these serine and threonine residues have established as residues for histone phosphorylation, but subsequent data suggest that histone tyrosine residues can also be phosphorylated (Cook et al., 2009; Dawson et al., 2009; Singh et al., 2009b; Xiao et al., 2009). AMPK is a sensor of cellular energy status consistent with the ratios of AMP:ATP and ADP:ATP, and this kinase is activated by an increase in AMP or ADP but inactivated by ATP (Hardie, 2011; Hardie et al., 2016). Yeast AMPK homolog Snf1 kinase is required for the phosphorylation of histone H3 at serine 10 (H3pS10) in the promoter of the INO1 gene (Lo et al., 2001). Mammalian AMPK was reported to phosphorylate histone H2B at serine 36 (H2BpS36) (Bungard et al., 2010) (Fig. 2). As a nutrient sensor, AMPK can be activated under a variety of stress conditions. During glucose deprivation, AMPK is localized to the promoter of p53 and activates p53-responsive genes, which are essential for cell survival under metabolic stress. Currently, an increasing number of studies have recognized the importance of AMPK in tumorigenesis; therefore, methods to activate AMPK activity, such as exercise, calorie restriction and metformin treatment, have been identified, and some have been tested in preclinical models (Steinberg and Carling, 2019).
Histone phosphorylation is associated with many different cellular processes, such as transcriptional activation, mitosis, meiosis, DNA repair, and apoptosis (Cohen et al., 2011). Although all histones H1, H2A, H2B, H3, and H4 can be phosphorylated at multiple sites, H3 phosphorylation has been most widely and intensively studied (Prigent and Dimitrov, 2003; Nowak and Corces, 2004). H3S10 can be phosphorylated by a variety of kinases, such as mitogen- and stress-activated protein kinase 1 and 2 (MSK1 and MSK2) (Soloaga et al., 2003), ribosomal S6 kinase 2 (RSK2) (Sassone-Corsi et al., 1999), I κB kinase-α (IKK-α) (Anest et al., 2003), and PIM1 kinase (Zippo et al., 2007). PIM1 kinase-mediated H3S10 phosphorylation at the E-box of Myc target genes is involved in gene activation and contributes to cell transformation and tumor growth (Zippo et al., 2007). The overexpression of histone H3 enhanced the neoplastic cell transformation induced by epidermal growth factor and cancer development, and histone H3 phosphorylation-mediated c-jun and c-fos induction is critical for these processes (Choi et al., 2005). The phosphorylation of H3S10 is a hallmark of mitosis, starting in prophase, reaching its highest level in metaphase, and then decreasing toward the end of the cell cycle. Mitotic H3S10 phosphorylation is mainly controlled by Aurora B (Adams et al., 2001; Giet and Glover, 2001; Richie and Golden, 2005), Aurora A (Kim et al., 2016), and polo-like kinase 1 (PLK1). Moreover, histone phosphorylation regulates tumorigenesis by participating in DNA damage repair and apoptosis.
O-GlcNAc and O-GlcNAcylation
O-linked β-N-acetylglucosamine glycosylation (O-GlcNAcylation) is a PTM that regulates basic cellular processes. UDP-GlcNAc (uridine diphosphate GlcNAc), the final product of the hexosamine biosynthetic pathway (HBP) that integrates glucose, glutamine, amino acid, fatty acid, and nucleotide metabolism, serves as the donor substrate for O-GlcNAcylation (Hart et al., 2007; Hart et al., 2011; Ferrer et al., 2016; Yang and Qian, 2017) (Fig. 2). In contrast to other PTMs, which are regulated by many writers and erasers, glycosylation is regulated by a single pair of enzymes, O-GlcNAc transferase (OGT) and O-GlcNAcase (OGA). OGT catalyzes the transfer of GlcNAc from UDP-GlcNAc to the Ser and Thr residues of target proteins, whereas OGA catalyzes the hydrolysis of this PTM (Hart et al., 2007; Hart et al., 2011; Slawson and Hart, 2011) (Fig. 2). In addition to relying on nutrient metabolism to provide a substrate, O-GlcNAcylation is sensitive to various types of cellular stresses, such as heat shock, hypoxia, and nutrient deprivation (Hart et al., 2011; Ferrer et al., 2014; Yang and Qian, 2017). However, the detailed mechanism by which cells sense these stresses to induce O-GlcNAcylation and how various stimuli trigger dynamic changes in protein O-GlcNAcylation are far from clear. After heat shock, the O-GlcNAcylation of histones increases rapidly, and this increase is correlated with an increase in DNA compaction (Sakabe et al., 2010). By modifying proteins such as nuclear factor-κB (NF-κB), nuclear factor of activated T cells cytoplasmic 1 (NFATC1) (Golks et al., 2007), CREB-regulated transcription coactivator 2 (CRTC2) (Dentin et al., 2008), forkhead box O1 (FOXO1) (Housley et al., 2008), PPARG coactivator 1α (PGC1α) (Ruan et al., 2012), and RNA polymerase II (Pol II) (Kelly et al., 1993) or by proteins interacting with HDAC (Yang et al., 2002), EZH2 (Chu et al., 2014), PRC2 (Gambetta et al., 2009), or TET2 (Dehennaut et al., 2014; Lewis and Hanover, 2014; Singh et al., 2015), O-GlcNAcylation regulates fundamental cellular processes such as transcription, epigenetic programming, and cell signaling pathway activation (Hardiville and Hart, 2014; Hart, 2019). Therefore, in view of its importance in cellular processes, the disruption of O-GlcNAcylation is related to the pathological processes of many kinds of tumors. Several studies showed decreased O-GlcNAc levels in some tumor samples compared with matched adjacent tissues (Slawson et al., 2001), whereas other studies found accumulated O-GlcNAc, enhanced O-GlcNAcylation and increased OGT and OGA expression in breast, lung, and colon tumor tissues compared with the respective corresponding control tissues (Gu et al., 2010; Mi et al., 2011). A similar phenomenon was evident in patients with chronic lymphocytic leukemia (Shi et al., 2010).
There is extensive crosstalk between O-GlcNAcylation components and those of other common PTMs, such as phosphorylation, ubiquitylation, acetylation, and methylation. The interaction of O-GlcNAcylation and phosphorylation components has been well studied due to the similarly modified Ser and Thr residues (Hart et al., 2011; Song et al., 2019). O-GlcNAcylation is found on the Ser and Thr residues on core histones H2A Thr101, H2B Ser36, and H4 Ser47 which have been identified as phosphorylation sites (Maile et al., 2004; Olson et al., 2006; Mayya et al., 2009; Sakabe et al., 2010). Several studies have shown that O-GlcNAcylation can prevent the degradation of target proteins by inhibiting their ubiquitination through different mechanisms. For instance, overexpression of OGA in HepG2 cells resulted in the decreased O-GlcNAcylation of AKT and increased phosphorylation and activity of AKT. O-GlcNAcylation sites in AKT have been identified as Thr308 and Ser473, key phosphorylation sites for AKT activation. These observations suggest that the O-GlcNAcylation of AKT competes directly with its phosphorylation (Soesanto et al., 2008; Ruan et al., 2012; Li et al., 2013; Ruan et al., 2013; Shi et al., 2015). Because OGA has both a C-terminal histone acetyltransferase-like (HAT-like) domain and an N-terminal O-GlcNAc hydrolase domain, O-GlcNAcylation and acetylation can be regulated reciprocally (Allison et al., 2012; Hayakawa et al., 2013). Other studies have shown that OGT regulates transcription, especially at transcriptional start sites, in a TET2-dependent manner, suggesting that O-GlcNAcylation and DNA methylation synergistically regulate gene transcription (Chen et al., 2013b; Deplus et al., 2013; Shi et al., 2013; Vella et al., 2013; Dehennaut et al., 2014; Zhang et al., 2014).
Citrulline and citrullination
Citrulline is a noncoding amino acid and its metabolism is categorized into two types: free citrulline from the arginine-coupled urea cycle and citrullinated proteins. Here, we focus on the second type of citrulline and discuss the mechanism and significance of citrullination (also known as arginine deimination) (Fig. 2). Histone citrulllination affects approximately 10% of all histone molecules. This less-noticed PTM converts arginine residues to citrulline by peptidyl-arginine deiminase (PAD or PADI) enzymes, resulting in reduced hydrogen bonding and a looser chromatin structure. PADs contain five Ca2+-dependent enzymes (PAD 1-4 and PAD6) (Witalison et al., 2015; Audia and Campbell, 2016; Yuzhalin, 2019) (Fig. 2). Citrullination modification is known to play essential roles in autoimmune and inflammatory diseases such as rheumatoid arthritis (Darrah and Andrade, 2018), periodontitis (Konig et al., 2016), autoimmune encephalomyelitis (Carrillo-Vico et al., 2010), systemic lupus erythematosus (Knight et al., 2015), and other diseases such as multiple sclerosis, atherosclerosis, thrombosis, and inflammatory bowel disease (Pritzker et al., 2000; Moscarello et al., 2007; Caprariello et al., 2018). PAD4 has been found to be a prognostic biomarker due to its increased expression in various solid tumors compared to that on the respective normal tissues (Chang et al., 2009; Chang et al., 2011) and in the peripheral blood of patients with lung cancer (Ulivi et al., 2013). In malignant lymphomas, PAD4 is also expressed in approximately 40% of cells, suggesting that its expression is associated with the development of this blood disease (Chang et al., 2009). Benign and nontumorous inflammatory tissues do not express PAD4, while metastatic tumors show higher PAD4 levels than corresponding primary tumors, suggesting that citrullination plays a role in the progression of benign tumors to aggressive malignancies (Yuzhalin et al., 2018). In colon cancer cells, knocking down PAD4 increases the expression of p53 and its target genes, leading to cell cycle arrest and cell apoptosis (Li et al., 2008). Inhibition of PADs by their inhibitor CI-amidine leads to the upregulation of OKL38 (oxidative stress-induced growth inhibitor 1), which promotes cell apoptosis and mitochondrial dysfunction in breast and osteosarcoma cells (Yao et al., 2008). Another PAD inhibitor, YW3-56, inhibits tumor growth by interfering with autophagy and regulating the Sestrin 2-mTORC1 signaling axis (Wang et al., 2012).
Among the PAD family members, PAD4 is the only protein with a nuclear localization signal (NLS) and has been reported to citrullinate histone H3 (Arg 2, 8, 17, 26), H2A, H4 (Arg 3), and H1 (Arg 54) (Wang et al., 2004; Tanikawa et al., 2012; Christophorou et al., 2014). The citrullination of H3 has been reported to open chromatin and promote gene transcription (Fuhrmann and Thompson, 2016), and citrullinated histone H3, which is mediated by PAD4, plays a vital role in the release of neutrophil extracellular traps (NETs) (Li et al., 2010). NETs have been detected in several human cancer types (Berger-Achituv et al., 2013; Merza et al., 2015; Yang et al., 2015; Tohme et al., 2016) and have been found to promote the proliferation and metastasis of cancer cells (Demers et al., 2016; Albrengues et al., 2018; Monti et al., 2018). Thålin et al. found that a 3-fold increase in citrullinated histone H3 is associated with neutrophil activation markers neutrophil elastase (NE) and myeloperoxidase (MPO) and the inflammatory cytokines IL-6 and IL-8 in 60 patients with different advanced cancers (Thalin et al., 2018). Mauracher et al. found that NET formation is associated with H3 citrullination and the occurrence of venous thromboembolism (VTE) in cancer patients (Mauracher et al., 2018). These studies suggest that citrullinated histone H3 is a novel prognostic marker for cancer detection. Upon chemotherapy treatment, PAD4 regulates cell apoptosis by interacting with p53 to citrullinate histone H4 (Arg 3) (Tanikawa et al., 2012). In addition to p53, calcium homeostasis also regulates PAD activity, and PAD-mediated citrullination is involved in calcium-mediated apoptosis (Asaga et al., 1998; Mattson and Chan, 2003; Hsu et al., 2014).
Itaconate and itaconation
Nearly 200 years after Swiss chemist Samuel Baup first described itaconate (itaconic acid or methylenesuccinic acid) as a product of citric acid distillation in 1836 (Luan and Medzhitov, 2016), itaconate was recently rediscovered by three groups as a microbial metabolite in the mouse lungs infected by Mycobacterium tuberculosis (MTB) (Shin et al., 2011), in the supernatant and cell lysates of LPS-activated RAW264.7 macrophages (Sugimoto et al., 2011) and in the intracellular compartment of glia-like VM-M3 cells (Strelko et al., 2011). Itaconate is synthesized from cis-aconitate in the TCA cycle of macrophages and is activated by a variety of factors, including LPS and other Toll-like receptor ligands and cytokines, such as type I and type II interferons (Shin et al., 2011; Strelko et al., 2011; Sugimoto et al., 2011; Michelucci et al., 2013). These stimuli increase the expression of aconitate decarboxylase 1 (ACOD1; also known as CAD), previously known as immune-responsive gene 1 (IRG1), which is critical for itaconate production (Lee et al., 1995; Michelucci et al., 2013). IRG1 was recently found to be induced by steroid hormone progesterone (Cheon et al., 2003), heme oxygenase-1/carbon monoxide (Uddin et al., 2016), interferon regulatory factor 1 (IRF1), the ZBP1/RIPK1/RIPK3/IRF1 axis (Tallam et al., 2016; Daniels et al., 2019) and BCAT1 (Papathanassiu et al., 2017), and suppressed by nuclear receptor Nur77 (Nr4a1) (Koenis et al., 2018) or A20 (a negative regulator of NF-κB signaling) (Van Quickelberghe et al., 2018). Itaconate, as a signal transducer, exerts anti-inflammatory effects (Murphy and O’Neill, 2018). The itaconate signal transduction mechanism is as follows: 1) it inhibits SDH and counteracts the proinflammatory signals of succinate (Cordes et al., 2016; Lampropoulou et al., 2016); 2), modifies KEAP1 to activate Nrf2 (Mills et al., 2018); and 3), and induces the ATF3-IκBζ axis, another anti-inflammatory signaling pathway (Bambouskova et al., 2018). The IRG1-itaconate-SDH axis also links innate immune tolerance with trained immunity (Dominguez-Andres et al., 2019), and IRG1 expression is important to prevent immunopathology during MTB infection (Nair et al., 2018). In glioma pathogenesis, IRG1 was identified as a novel oncogene that promotes the growth and tumorigenesis of glioma (Pan et al., 2014); IRG1-mediated itaconate production potentiates peritoneal tumor growth, and IRG1 in peritoneal tissue-resident macrophages (pResMϕ) represents a potential therapeutic target for peritoneal tumors (Weiss et al., 2018).
Recently, itaconate was found to modify KEAP1 and glutathione (Bambouskova et al., 2018; Mills et al., 2018), NLRP3 (Hooftman et al., 2020; Qin et al., 2020), ALDOA, GAPDH and LDHA (Qin et al., 2019), and other proteins involved in inflammasome, toll-like signaling, cell death, and DNA damage (Qin et al., 2020). Itaconate contains an electrophilic α,β-unsaturated carboxylic acid group, which can alkylate the cysteine residues of proteins to form a 2,3-dicarboxypropyl adduct via Michael addition. Qin et al. used a specific thiol-reactive probe, 1-OH-Az, for quantitative chemoproteomic profiling of cysteine modifications by itaconate and identified a total of 260 itaconate-modified cysteines in macrophage proteomes and showed that itaconate can modify key cysteines in glycolytic enzymes to inhibit glycolysis, which indicates its anti-inflammatory function (Qin et al., 2019; Yang, 2019). By developing a specific and cell-permeable bioorthogonal probe, itaconate-alkyne (ITalk), Qin et al. further characterized the extensive landscape of itaconate substrates (1,926 protein targets) in living cells and described this Michael addition reaction as “itaconation” (Qin et al., 2020). Moreover, itaconyl-CoA, the coenzyme A (CoA) derivative of itaconate, inhibits B12-dependent methylmalonyl-CoA mutase (MCM) by forming a stable biradical in MCM and derails its activity and repair ability (Shen et al., 2017; Ruetz et al., 2019). Although itaconate plays a decisive inflammatory role in activated macrophages and modifies a variety of proteins on cysteine residues (Luan and Medzhitov, 2016; Murphy and O’Neill, 2018; Nonnenmacher and Hiller, 2018; O’Neill and Artyomov, 2019; Yu et al., 2019), it is not clear whether itaconate affects the epigenetic remodeling of immune cells or tumor cells through itaconation, itaconyl-CoA, or other itaconate-related metabolites; hence, future studies are warranted in this regard.
TARGETING CHROMATIN MODIFICATIONS RELATED TO METABOLISM FOR CANCER THERAPY
Targeting acetylation for cancer therapy
Targeting the epigenetic modulation of histones or DNA to realize cancer therapy has attracted increased interest for decades. The most widely studied epigenetic inhibitors are probably HDAC inhibitors. HDAC inhibitors represent various compounds that inhibit the activity of HDACs, leading to the increased acetylation of lysine residues on histones and nonhistone proteins. The key effects of HDAC inhibitors on tumor cells are to induce cell death, cell cycle arrest, senescence, differentiation, autophagy, and tumor immunogenicity (Falkenberg and Johnstone, 2014). The short fatty acid n-butyrate was found in the mid-1970s by Riggs and colleagues to induce the differentiation of Friend erythroleukemia cells and strong histone hyperacetylation in HeLa cells (Riggs et al., 1977), and then, the groups of Allfrey and Davie reported that n-butyrate was an HDAC inhibitor (Candido et al., 1978; Vidali et al., 1978). The first US Food and Drug Administration (FDA)-approved acetylating modifier, vorinostat (also known as suberanilohydroxamic acid (SAHA)), was initially identified as a drug that induces tumor cell differentiation in vitro and was subsequently identified as an HDAC inhibitor (Richon et al., 1998; Mann et al., 2007). Vorinostat and another HDAC inhibitor, romidepsin, have been approved for use in cutaneous T-cell lymphoma (CTCL). Romidepsin was also approved for the treatment of peripheral T-cell lymphoma (PTCL). Their application in solid tumors has progressed slowly and remains an active research area. In addition, TSA is a commonly used HDAC inhibitor but is used only in laboratory experiments due to its high toxicity.
The pan-inhibitor nicotinamide inhibits all class III HDAC sirtuins. Sirtinol, cambinol, and EX-527 are specific SIRT1 and SIRT2 inhibitors (Ceccacci and Minucci, 2016; Eckschlager et al., 2017). They can play roles in different types of neurodegeneration and cancer (Lavu et al., 2008). In 2003, Sinclair and colleagues first found that resveratrol, a polyphenol abundant in red wine, is an activator of SIRT1 and was later named STAC (sirtuin-activating compound). STACs are chemical compounds that use NAD+ to remove acetyl groups from proteins. Additional results showed that resveratrol mimics caloric restriction and delays aging by extending the lifespan of metazoans and yeast (Howitz et al., 2003; Wood et al., 2004). Hubbard et al. showed that a single amino acid, Glu230 located in a structured N-terminal domain of SIRT1, is critical for activation of SIRT1 induced by STACs through a common allosteric mechanism (Hubbard et al., 2013). SIRT6, activated by free fatty acids such as oleic and linoleic acids, is a tumor suppressor that suppresses glycolysis (Feldman et al., 2013). The development of small-molecule activators of SIRT6 may be able to target specific tumors with low SIRT6 expression. Therefore, the field of drug development regarding SIRTs may gradually shift from inhibitors to activators, such as activating SIRT1 or SIRT6 to target age-related degenerative diseases and tumors with specific phenotypes, respectively.
In contrast to targeting writers and erasers by inhibiting their catalytic domains, effective targeting of readers needs to disrupt the protein-protein interaction. JQ-1 and I-BET are the first two compounds that can inhibit bromodomain-containing BET (bromodomain and extraterminal) proteins by binding to the bromodomain of the BET proteins (Filippakopoulos et al., 2010; Nicodeme et al., 2010; Zuber et al., 2011). JQ-1 binds the first bromodomain of the BET protein BRD4, which is a key tethering factor that interacts with cyclin T1 and cdk9 to form a core positive transcription elongation factor b (P-TEFb) (Jang et al., 2005). A more exciting effect of BET inhibition is its ability to downregulate previously undruggable MYC oncogene (Delmore et al., 2011; Mertz et al., 2011; Sun and Gao, 2017). JQ-1 or I-BET treatment leads to the transcriptional suppression of MYC target genes, resulting in antitumor effects in MYC-driven models of AML (Dawson et al., 2011; Zuber et al., 2011), Burkitt’s lymphoma (Mertz et al., 2011) and multiple myeloma (Delmore et al., 2011). BET inhibition also promotes immunotherapy by inhibiting the interaction of BRD2 and/or BRD4 with CDK9 and suppressing the expression of inflammatory cytokines (Nicodeme et al., 2010; Bandukwala et al., 2012).
ACLY, ACSS2 and PDC have been previously mentioned as direct effectors of acetyl-coenzyme producers in the nucleus for histone acetylation. Therefore, ACLY, ACSS2 and PDC inhibitors may now be considered new drugs for targeting the metabolic-epigenomic axis in addition to their current uses as metabolic regulators. As described above, SB-204990 is a preclinical inhibitor that specifically targets the ACLY enzyme (Pearce et al., 1998; Hatzivassiliou et al., 2005). BMS-303141 (preclinical) (Li et al., 2007), ETC-1002 (phase II clinical trial) (Ballantyne et al., 2013; Filippov et al., 2014; Gutierrez et al., 2014), and hydroxycitrate (phase IV clinical trial) (Onakpoya et al., 2011; Madeo et al., 2014) are other ACLY inhibitors in clinical trials. 1-(2,3-Di(thiophen-2-yl)quinoxalin-6-yl)-3-(2-methoxyethyl)urea is the most potent and specific inhibitor of ACSS2, which was first reported by Comerford and his colleagues (Comerford et al., 2014). Dichloroacetate (DCA), a classic PDK inhibitor, is considered a PDC activator that inhibits PDK activity. The reactivation of PDC by DCA therapy rectifies the balance between the demand and supply of oxygen, leading to cancer cell death (Michelakis et al., 2010; Dunbar et al., 2014; Chu et al., 2015; Zhang et al., 2015). Please see Table S1 for more details about the clinical trials of targeting acetylation for cancer therapy, including the inhibitors not mentioned in this review article due to space limitations.
Targeting the methylation of DNA and histones for cancer therapy
As indicated above, the frequent hypermethylation of tumor suppressor genes further promotes cancer development; therefore, the demethylation of DNA by blockading DNMTs constitutes an interesting treatment strategy due to its reversibility. Nucleoside analogs and nonnucleoside inhibitors are the two main types of DNMT inhibitors (DNMTi), and nucleoside analogs have been known and studied for many years. Azacitidine (also known as 5-azacytidine) and decitabine (also known as 5-aza-2’-deoxycytidine) are the oldest DNMTis and are nucleoside analogs that incorporate DNA during the S-phase of the cell cycle to form an irreversible complex with DNMTs, leading to the degradation of the DNMTs. They have been approved by FDA as DNA-demethylating drugs used to treat myelodysplastic syndrome (MDS), chronic myelomonocytic leukemia, and a range of other malignancies (Kaminskas et al., 2005a; Kaminskas et al., 2005b) and represent two of the most successful and long-standing inhibitors to target epigenetic processes. Azacitidine and decitabine were first synthesized almost 60 years ago (Evans and Mengel, 1964; Sorm et al., 1964; Christman, 2002). Azacitidine has been shown to have extensive antimetabolic activity in cultured cancer cells and is an effective chemotherapeutic agent in the treatment of acute myelogenous leukemia. However, one limitation of these drugs is that they have a short half-life of approximately 30 min, which limits their exposure to diseased cells, which may thus impair their effectiveness (Marcucci et al., 2005; Karahoca and Momparler, 2013), resulting in no major responses observed in their treatment of solid tumors. Zebularine, a novel nucleoside, is a cytidine analog that is less toxic and can therefore be taken in high doses consistently (Holleran et al., 2005). Guadecitabine (SGI-110), a second-generation DNMT inhibitor, acts as a prodrug of decitabine, conferring better stability and reduced toxicity in tumor-infected nude mice compared to decitabine (Yoo et al., 2007; Chuang et al., 2010), and it has been tested in phase II clinical trials for the treatment of MDS and AML (ClinicalTrials.gov Identifier: NCT01261312).
The identification of small nonnucleoside DNMT inhibitors such as flavonoids (or bioflavonoids, such as epigallocatechin-3-gallate (EGCG)) (Galeotti et al., 2008; Yang et al., 2009; Singh et al., 2011), hydralazine (Chuang et al., 2005; Singh et al., 2009a), procainamide and procaine (Yoo and Medina-Franco, 2011), curcumin (Liu et al., 2009; Shu et al., 2011), RG108 (phthalimido-L-tryptophan) (Siedlecki et al., 2003; Brueckner et al., 2005), SGI-1027 (Denny et al., 1979; Datta et al., 2009), and MG98 (Goffin and Eisenhauer, 2002; Amato, 2007), which bind directly to DNMT catalytic regions without binding to DNA, has also attracted considerable attention. However, in vitro studies have shown that nonnucleoside compounds induce limited epigenetic changes in living cells (Ren et al., 2011), and none of the afore mentioned nonnucleoside DNMT inhibitors has entered clinical development; thus, there is still a long way to go before novel, selective, nonnucleoside DNMT inhibitors will be available for clinical research.
There are fewer effective inhibitors of HMTs and KDMs than inhibitors of acetylation and DNA methylation. Over the past few years, the number of histone methylation small-molecule modulators has increased rapidly through significantly increased efforts in academia and the pharmaceutical industry (Liu et al., 2014; Liu et al., 2015; McGrath and Trojer, 2015). Inhibitors of the histone methyltransferases DOT1L (Yu et al., 2012; Daigle et al., 2013) and EZH2 (Knutson et al., 2012; McCabe et al., 2012) have exciting potential for cancer treatment.
DOT1L (disruptor of telomeric silencing 1-like) was identified as a human homolog of Dot1, a gene found in the yeast Saccharomyces cerevisiae (Singer et al., 1998). DOT1L catalyzes histone H3 lysine 79 (H3K79) methylation by transferring a methyl group from its substrate SAM to the amino group of lysine residues. It is the only enzyme critical for the monomethylation, dimethylation, and trimethylation of the ε-amino group on H3K79 (Feng et al., 2002; Lacoste et al., 2002). EPZ004777, the first SAM-competitive inhibitor of DOT1L, was able to kill biphenotypic (mixed-lineage) leukemia (MLL)-rearranged leukemia cells and prolong the survival time of mice with MLL-rearranged leukemia, but it had little killing effect on non-MLL translocated cells (Daigle et al., 2011; Chen et al., 2013a). By occupying the SAM-binding pocket and inducing conformational changes in DOT1L, EPZ-5676, a derivative of EPZ004777, is another effective DOT1L inhibitor that led to tumor regression in rat xenograft models of MLL-rearranged leukemia (Daigle et al., 2013).
H3K27 methyltransferase EZH2 is the catalytic component of PRC2, an established transcriptional repressor (Cao et al., 2002; Levine et al., 2004). Several studies have shown that EZH2 disorders are often associated with the progression, metastasis, and poor clinical outcomes of patients with lymphoma and solid tumors, including prostate, breast, kidney, and lung cancers (Varambally et al., 2002; Moss and Wallrath, 2007; Yu et al., 2007; Varambally et al., 2008; Wagener et al., 2010; Takawa et al., 2011; McCabe et al., 2012; Volkel et al., 2015). GSK126 effectively inhibits wild-type and mutant EZH2 methyltransferase activity by competing with SAM. GSK126 can inhibit the proliferation of EZH2-mutant diffuse large B-cell lymphoma (DLBCL) cells and markedly inhibit the growth of EZH2-mutant DLBCL xenografts in mice (McCabe et al., 2012). Subsequently, other EZH2 inhibitors, including EPZ005687 (Knutson et al., 2012), EPZ-6438 (tazemetostat) (Knutson et al., 2014), and CPI-1205 (Taplin et al., 2018), have been developed and are currently in phase I or II clinical trials.
IDH mutations indirectly inhibit extensive histone demethylases and lead to histone hypermethylation throughout the genome. Targeting IDH mutations for glioma and AML appears to be a promising therapeutic approach. The IDH1R132H inhibitor (AGI-5198) impairs the growth and promotes the differentiation of glioma cells with the IDH1R132H mutation by inducing the demethylation of histone H3K9me3 and the expression of genes critical for gliogenic differentiation (Rohle et al., 2013). AGI-6780, a potent and selective inhibitor of the tumor-associated mutant IDH2R140Q, binds IDH2 at the dimer interface. Treatment of TF-1 human erythroleukemia cells and primary human AML cells with AGI-6780 induces cellular differentiation (Wang et al., 2013). AG-221, another selective inhibitor of the mutant IDH2R140Q enzyme, suppresses 2HG production and induces the differentiation of both human AML cells and mouse model cells (Yen et al., 2017). These inhibitors provide potential applications as differentiation therapies for cancer, at least for AML. In addition, the frequent deletion of methylthioadenosine phosphorylase (MTAP) as a consequence of 9p21 loss in cancer cells leads to the dysregulation of methionine metabolism and makes tumor cells more sensitive to protein arginine N-methyltransferase 5 (PRMT5) inhibitors, creating a new therapeutic opportunity based on methionine metabolism and epigenomic interactions (Kryukov et al., 2016; Marjon et al., 2016; Mavrakis et al., 2016).
DNA methylation and histone acetylation are the earliest epigenetic targets for drug development. As described above, epigenetic drugs, including DNMT and HDAC inhibitors, have been approved by FDA for clinical use with hematologic malignancies and other cancers. In contrast, there is still considerable room for further development of targeted histone methylation in terms of mechanistic discovery and drug intervention. Moreover, the success of FDA-approved drugs for use with solid tumors has been limited because of the specific tumor microenvironment of these cancers, such as hypoxia and immune cells. Nevertheless, to date, there are many epigenetic targets of proteins and DNA that have not been properly assessed with inhibitors; therefore, epigenetic therapies, along with other therapies, may offer many opportunities for tumor therapy. For more information about the usage of HDAC inhibitors, IDH1/2 inhibitors, SAM cycle inhibitors and other inhibitors or activators of epigenetic modulation in cancer, please see the excellent reviews by Falkenberg et al., Eckschlager et al., Wong et al. and Cheng et al. (Falkenberg and Johnstone, 2014; Eckschlager et al., 2017; Wong et al., 2017; Cheng et al., 2019). Please see Table S2 for more details about the clinical trials of targeting methylation for cancer therapy, including the inhibitors not mentioned in this review article due to space limitations.
Dietary interventions that target epigenetically modified proteins and metabolic molecules as cancer therapy
Calorie restriction and fasting have been shown to extend lifespan and confer health-promoting effects. Recently, an increasing number of studies have discovered that the disruption of the dietary components of methionine (Xu et al., 2020), serine and glycine (Maddocks et al., 2017; Muthusamy et al., 2020), ketone bodies (Xia et al., 2017; Hopkins et al., 2018), choline (Romano et al., 2017), arginine (Poillet-Perez et al., 2018), glutamine (Ishak Gabra et al., 2020), fructose (Goncalves et al., 2019; Zhao et al., 2020), or cysteine (Badgley et al., 2020) mediates the progression of multiple types of cancer. Here, we review the latest progress in dietary controls in cancer therapy at the epigenetic level (Fig. 4).

Dietary-based approaches for cancer therapy. Genetic and environmental factors, including gene mutation, radiation, smoking, and excessive drinking, can cause a variety of human diseases, such as glioma, liver cancer, lung cancer, pancreatic cancer, kidney cancer, and colorectal cancer, which are associated with metabolic dysregulation and epigenetic remodeling. Dietary intake regulates nutrient availability, metabolite generation, and epigenetic modifications. Dietary changes in the composition of ketones (low carb, high fat diet, such as yogurt, eggs), glutamine (dietary fish, soybean), choline (dietary eggs, meat, fish), methionine (methionine restriction: dietary less proteins, it is only tested on animals), or serine (serine restriction: serine- and glycine-free diet, it is only tested on animals), may extend lifespan and have health-promoting effects by reshaping the homeostasis of metabolism and epigenetics such as methylation, acetylation, succinylation, and β-hydroxybutyrylation
Ketogenic diets are high-fat low-carbohydrate diets that inhibit cancer progression, in contrast to high-fat, high-carbohydrate diets that induce obesity and promote cancer progression (Branco et al., 2016). A ketogenic diet promotes the β-oxidation of fatty acids and the generation of ketone bodies, such as AcAc, β-OHB, and acetone, in the livers. Ketogenic diets function as “insulin-suppressing diets” by reducing circulating insulin and insulin-like growth factor-1 (IGF-1) levels, thereby limiting the aberrant activation of oncogenes (Nencioni et al., 2018; Klement, 2019). Insulin feedback induced by PI3K inhibitor treatment reactivates the PI3K-mTOR signaling axis in tumors, which may significantly negate the beneficial therapeutic effects of inhibitors. Feeding ketogenic diets together with a PI3K inhibitor to decrease hyperglycemia and lower insulin release resulted in the improved survival of the mice bearing PI3K-driven tumors (Hopkins et al., 2018). However, ketogenic diets alone had varying effects on different tumor models. For example, accelerated disease progression was shown for AML mouse models fed ketogenic diets alone (Hopkins et al., 2018), and these diets also accelerated tumor growth in other rodent tumor models (Liskiewicz et al., 2016; Xia et al., 2017). Whether ketone body-derived acetyl-CoA and β-hydroxybutyrate and the subsequent acetylation and β-hydroxybutyrylation are involved in the confusing effect of ketogenic diets on different tumor growth remains unclear.
Although glutamine is the most abundant amino acid in the culture medium and is essential for immune cell function and tumor development (Gao et al., 2009; Altman et al., 2016; Kelly and Pearce, 2020), Ishak Gabra et al. found that glutamine supplementation in the diet suppresses melanoma tumor growth independent of BRAF status. Dietary glutamine-derived α-KG levels in vivo led to the hypomethylation of H3K4me3, thereby inhibiting epigenetically activated oncogenic pathways in melanoma (Ishak Gabra et al., 2020). This study showed the potential of dietary intervention with glutamine to block melanoma tumor growth by epigenetic reprogramming. Choline is an essential methyl donor in one-carbon metabolism for the methylation of histones and DNA. Romano et al. found that choline-utilizing bacteria compete with the host to consume choline, affecting the plasma and hepatic levels of methyl donor metabolites in the host. Mice with choline-consuming bacteria exhibit increased susceptibility to metabolic disease upon depletion of methyl donor metabolites when fed a high-fat diet (Romano et al., 2017). Excessive consumption of fructose increases the incidence of obesity and nonalcoholic fatty liver disease (Hannou et al., 2018; Jensen et al., 2018). In vivo isotope tracing revealed that the gut microbiota decomposes dietary fructose into acetate, providing acetyl-CoA for lipogenesis and H3K27 acetylation within the ACSS2 genomic locus. Depletion of the microbiota or hepatic ACSS2 suppresses acetyl-CoA generation and the synthesis of fatty acids (Zhao et al., 2020).
Dietary restriction of methionine as a therapeutic approach was proposed over 60 years ago (Sugimura et al., 1959). A methionine-limited diet suppresses tumor invasion and metastasis (Breillout et al., 1987; Guo et al., 1996; Jeon et al., 2016). The combination of a methionine-limited diet and one-carbon metabolism inhibitors, such as 5-fluorouracil (5-FU), has a synergistic effect on tumor inhibition (Hoshiya et al., 1997; Xiao et al., 2001). Methionine supplementation in tumors restores T-cell function in B16F10 tumor-bearing mice and retards tumor growth (Bian et al., 2020). However, notably, methionine restriction together with choline depletion promotes hepatic injury in some rodent models (Caballero et al., 2010). Dietary restriction of serine and glycine decelerates tumor growth and increases mouse survival (Maddocks et al., 2013; Gravel et al., 2014; Maddocks et al., 2017; Muthusamy et al., 2020). However, it is not clear whether the antitumor effect of serine and glycine starvation depends on the alteration of methylation status. Please see Table S3 for more details about the clinical trials of targeting metabolic enzymes for cancer therapy, including the inhibitors not mentioned in this review article due to space limitations. Moreover, Table S4 summarizes the information of the clinical trials in combination with epigenetic drugs and metabolism-targeting drugs for cancer therapy in recent years.
CONCLUSIONS AND FUTURE PERSPECTIVES
Substantial progress has been made in the understanding and research on the intersection of tumor metabolism and epigenetics in recent decades. However, there are still many important scientific questions to be answered. For example, what are the specific action and mechanism of nonacetyl histone acylation modulations, including propionylation, butyrylation, crotonylation, malonylation, 2-hydroxyisobutyrylation and glutarylation, in the development of different cancers (Sabari et al., 2017; Zhao et al., 2018). When multiple modification pathways target the same amino acid residues, there may be competitive antagonism between different modifications. This is especially true for the ε group of lysine, which can be acetylated, methylated, or ubiquitinated (Bannister and Kouzarides, 2011; Sadakierska-Chudy and Filip, 2015; Zheng et al., 2020), such as the balance between H3K9 acetylation and methylation (Nicolas et al., 2003), dynamic competition of H4 K5K8 acetylation and butyrylation (Goudarzi et al., 2016), and the overlap of succinylation sites and acetylation sites in diverse organisms including bacteria, yeast, and human cells (Weinert et al., 2013). Itaconate, produced mainly by stimulated macrophages, can modify protein alkylation and protein itaconation (Lampropoulou et al., 2016; Bambouskova et al., 2018; Mills et al., 2018; Qin et al., 2019; Qin et al., 2020), but it is unclear whether it directly modifies histones or indirectly modify histones through succinate to regulate gene expression and cancer progression. Another largely unanswered question is, are certain regions of chromatin more susceptible to metabolic rewiring than other regions.
Localized metabolites, especially in the nucleus and mitochondria, reveal the importance of enzyme translocation in the regulation of epigenetics. Thus, discovering the roles of metabolites in other organelles, such as lysosomes, the endoplasmic reticulum, Golgi apparatus, and storage granules, will be crucial to understanding how metabolism and epigenetics interact with each other. Of particular importance, α-KGDH localized in the nucleus serves as a succinyltransferase to succinylate histones (Wang et al., 2017b; Xu et al., 2021), and CPT1A can also serve as a lysine succinyltransferase upon the succinylation of S100A10 (Kurmi et al., 2018; Wang et al., 2019a). Other aforementioned nuclear localized metabolism enzymes, such as PKM2, ACLY, and PDC, not only provide the corresponding substrates or metabolites for chromatin modulation but also interact with other proteins to form complexes. Detailed analyses of the interacting partners and elucidation of their nonmetabolic or moonlighting roles are underappreciated opportunities but important for us to understand and battle cancers.
Many clinical trials are testing epigenetic molecular inhibitors, such as HDAC inhibitors and DNMT inhibitors. The IDH1-mutant inhibitor ivosidenib and IDH2-mutant inhibitor enasidenib are granted by FDA for the treatment of AML with IDH1 or IDH2 mutations, respectively (Table S3). Dietary therapy is another promising antitumor approach that seems to be more convenient and economical and, despite desirable outcomes in animal studies, requires significant research before reaching the clinical stage.
The metabolic rewiring of tumor and immune cells regulates tumor progression by shaping the epigenome in the tumor microenvironment. While several typical PTMs modulated by metabolic enzymes and metabolites are reviewed here, the metabolome-epigenome crosstalk paradigm is continuously expanding, and the elucidation of the molecular basis of these PTMs will provide us with exciting opportunities to efficiently combat cancer.
References
Abmayr SM, Workman JL (2019) Histone lysine de-beta-hydroxybutyrylation by SIRT3. Cell Res 29:694–695
Adams RR, Maiato H, Earnshaw WC, Carmena M (2001) Essential roles of Drosophila inner centromere protein (INCENP) and aurora B in histone H3 phosphorylation, metaphase chromosome alignment, kinetochore disjunction, and chromosome segregation. J Cell Biol 153:865–879
Alarcon C, Wicksteed B, Prentki M, Corkey BE, Rhodes CJ (2002) Succinate is a preferential metabolic stimulus-coupling signal for glucose-induced proinsulin biosynthesis translation. Diabetes 51:2496–2504
Albrengues J, Shields MA, Ng D, Park CG, Ambrico A, Poindexter ME, Upadhyay P, Uyeminami DL, Pommier A, Kuttner V et al (2018) Neutrophil extracellular traps produced during inflammation awaken dormant cancer cells in mice. Science 361:eaao4227
Alleyn M, Breitzig M, Lockey R, Kolliputi N (2018) The dawn of succinylation: a posttranslational modification. Am J Physiol Cell Physiol 314:C228–C232
Allison DF, Wamsley J, Kumar M, Li D, Gray LG, Hart GW, Jones DR, Mayo MW (2012) Modification of RelA by O-linked N-acetylglucosamine links glucose metabolism to NF-kappa B acetylation and transcription. Proc Natl Acad Sci USA 109:16888–16893
Altman BJ, Stine ZE, Dang CV (2016) From Krebs to clinic: glutamine metabolism to cancer therapy. Nat Rev Cancer 16:619–634
Alvarez-Nunez F, Bussaglia E, Mauricio D, Ybarra J, Vilar M, Lerma E, de Leiva A, Matias-Guju X, Grp TNS (2006) PTEN promoter methylation in sporadic thyroid carcinomas. Thyroid 16:17–23
Amato RJ (2007) Inhibition of DNA methylation by antisense oligonucleotide MG98 as cancer therapy. Clin Genitourinary Cancer 5:422–426
Anand R, Marmorstein R (2007) Structure and mechanism of lysine-specific demethylase enzymes. J Biol Chem 282:35425–35429
Anest V, Hanson JL, Cogswell PC, Steinbrecher KA, Strahl BD, Baldwin AS (2003) A nucleosomal function for IkappaB kinase-alpha in NF-kappaB-dependent gene expression. Nature 423:659–663
Ariyannur PS, Moffett JR, Madhavarao CN, Arun P, Vishnu N, Jacobowitz DM, Hallows WC, Denu JM, Namboodiri AMA (2010) Nuclear-cytoplasmic localization of acetyl coenzyme A synthetase-1 in the rat brain. J Comp Neurol 518:2952–2977
Asaga H, Yamada M, Senshu T (1998) Selective deimination of vimentin in calcium ionophore-induced apoptosis of mouse peritoneal macrophages. Biochem Biophys Res Commun 243:641–646
Astuti D, Latif F, Dallol A, Dahia PLM, Douglas F, George E, Skoldberg F, Husebye ES, Eng C, Maher ER (2001) Gene mutations in the succinate dehydrogenase subunit SDHB cause susceptibility to familial pheochromocytoma and to familial paraganglioma. Am J Hum Genet 69:49–54
Audia JE, Campbell RM (2016) Histone Modifications and Cancer. Cold Spring Harb Perspect Biol 8:
Badgley MA, Kremer DM, Maurer HC, DelGiorno KE, Lee HJ, Purohit V, Sagalovskiy IR, Ma A, Kapilian J, Firl CEM et al (2020) Cysteine depletion induces pancreatic tumor ferroptosis in mice. Science 368:85–89
Ballantyne CM, Davidson MH, Macdougall DE, Bays HE, Dicarlo LA, Rosenberg NL, Margulies J, Newton RS (2013) Efficacy and safety of a novel dual modulator of adenosine triphosphate-citrate lyase and adenosine monophosphate-activated protein kinase in patients with hypercholesterolemia: results of a multicenter, randomized, double-blind, placebo-controlled, parallel-group trial. J Am Coll Cardiol 62:1154–1162
Bambouskova M, Gorvel L, Lampropoulou V, Sergushichev A, Loginicheva E, Johnson K, Korenfeld D, Mathyer ME, Kim H, Huang LH et al (2018) Electrophilic properties of itaconate and derivatives regulate the IkappaBzeta-ATF3 inflammatory axis. Nature 556:501–504
Bandukwala HS, Gagnon J, Togher S, Greenbaum JA, Lamperti ED, Parr NJ, Molesworth AM, Smithers N, Lee K, Witherington J et al (2012) Selective inhibition of CD4+ T-cell cytokine production and autoimmunity by BET protein and c-Myc inhibitors. Proc Natl Acad Sci USA 109:14532–14537
Bannister AJ, Kouzarides T (2011) Regulation of chromatin by histone modifications. Cell Res 21:381–395
Barlesi F, Giaccone G, Gallegos-Ruiz MI, Loundou A, Span SW, Lefesvre P, Kruyt FA, Rodriguez JA (2007) Global histone modifications predict prognosis of resected non small-cell lung cancer. J Clin Oncol 25:4358–4364
Yu WY, Chory EJ, Wernimont AK, Tempel W, Scopton A, Federation A, Marineau JJ, Qi J, Barsyte-Lovejoy, D, Yi JN et al (2012) Catalytic site remodelling of the DOT1L methyltransferase by selective inhibitors. Nat Commun 3
Basappa J, Citir M, Zhang Q, Wang HY, Liu X, Melnikov O, Yahya H, Stein F, Muller R, Traynor-Kaplan A et al (2020) ACLY is the novel signaling target of PIP2/PIP3 and Lyn in acute myeloid leukemia. Heliyon 6:
Bayley JP, Kunst HPM, Cascon A, Sampietro ML, Gaal J, Korpershoek E, Hinojar-Gutierrez A, Timmers HJLM, Hoefsloot LH, Hermsen MA et al (2010) SDHAF2 mutations in familial and sporadic paraganglioma and phaeochromocytoma. Lancet Oncol 11:366–372
Baysal BE, Ferrell RE, Willett-Brozick JE, Lawrence EC, Myssiorek D, Bosch A, van der Mey A, Taschner PEM, Rubinstein WS, Myers EN et al (2000) Mutations in SDHD, a mitochondrial complex II gene, in hereditary paraganglioma. Science 287:848–851
Becker-Kettern J, Paczia N, Conrotte JF, Kay DP, Guignard C, Jung PP, Linster CL (2016) Saccharomyces cerevisiae forms d-2-hydroxyglutarate and couples its degradation to d-lactate formation via a cytosolic transhydrogenase. J Biol Chem 291:6036–6058
Bender S, Tang Y, Lindroth AM, Hovestadt V, Jones DT, Kool M, Zapatka M, Northcott PA, Sturm D, Wang W et al (2013) Reduced H3K27me3 and DNA hypomethylation are major drivers of gene expression in K27M mutant pediatric high-grade gliomas. Cancer Cell 24:660–672
Berger-Achituv S, Brinkmann V, Abu Abed U, Kuhn LI, Ben-Ezra J, Elhasid R, Zychlinsky A (2013) A proposed role for neutrophil extracellular traps in cancer immunoediting. Front Immunol 4
Bergman Y, Cedar H (2013) DNA methylation dynamics in health and disease. Nat Struct Mol Biol 20:274–281
Bharathi SS, Zhang YX, Mohsen AW, Uppala R, Balasubramani M, Schreiber E, Uechi G, Beck ME, Rardin MJ, Vockley J et al (2013) Sirtuin 3 (SIRT3) protein regulates long-chain Acyl-CoA dehydrogenase by deacetylating conserved lysines near the active site. J Biol Chem 288:33837–33847
Bian Y, Li W, Kremer DM, Sajjakulnukit P, Li S, Crespo J, Nwosu ZC, Zhang L, Czerwonka A, Pawlowska A et al (2020) Cancer SLC43A2 alters T cell methionine metabolism and histone methylation. Nature
Bianco-Miotto T, Chiam K, Buchanan G, Jindal S, Day TK, Thomas M, Pickering MA, O’Loughlin MA, Ryan NK, Raymond WA et al (2010) Global levels of specific histone modifications and an epigenetic gene signature predict prostate cancer progression and development. Cancer Epidemiol Biomarkers Prev 19:2611–2622
Branco AF, Ferreira A, Simoes RF, Magalhaes-Novais S, Zehowski C, Cope E, Silva AM, Pereira D, Sardao VA, Cunha-Oliveira T (2016) Ketogenic diets: from cancer to mitochondrial diseases and beyond. Eur J Clin Invest 46:285–298
Breillout F, Hadida F, Echinard-Garin P, Lascaux V, Poupon MF (1987) Decreased rat rhabdomyosarcoma pulmonary metastases in response to a low methionine diet. Anticancer Res 7:861–867
Brueckner B, Boy RG, Siedlecki P, Musch T, Kliem HC, Zielenkiewicz P, Suhai S, Wiessler M, Lyko F (2005) Epigenetic reactivation of tumor suppressor genes by a novel small-molecule inhibitor of human DNA methyltransferases. Cancer Res 65:6305–6311
Bulusu V, Tumanov S, Michalopoulou E, van den Broek NJ, MacKay G, Nixon C, Dhayade S, Schug ZT, Voorde JV, Blyth K et al (2017) Acetate recapturing by nuclear acetyl-CoA synthetase 2 prevents loss of histone acetylation during oxygen and serum limitation. Cell Rep 18:647–658
Bungard D, Fuerth BJ, Zeng PY, Faubert B, Maas NL, Viollet B, Carling D, Thompson CB, Jones RG, Berger SL (2010) Signaling kinase AMPK activates stress-promoted transcription via histone H2B phosphorylation. Science 329:1201–1205
Caballero F, Fernandez A, Matias N, Martinez L, Fucho R, Elena M, Caballeria J, Morales A, Fernandez-Checa JC, Garcia-Ruiz C (2010) Specific contribution of methionine and choline in nutritional nonalcoholic steatohepatitis: impact on mitochondrial S-adenosyl-L-methionine and glutathione. J Biol Chem 285:18528–18536
Cai L, Sutter BM, Li B, Tu BP (2011) Acetyl-CoA induces cell growth and proliferation by promoting the acetylation of histones at growth genes. Mol Cell 42:426–437
Cairns RA, Mak TW (2013) Oncogenic isocitrate dehydrogenase mutations: mechanisms, models, and clinical opportunities. Cancer Discov 3:730–741
Cairns RA, Mak TW (2017) S-2HG is an immunometabolite that shapes the T-cell response. Cell Death Differ 24:195–196
Candido EP, Reeves R, Davie JR (1978) Sodium butyrate inhibits histone deacetylation in cultured cells. Cell 14:105–113
Cao R, Wang L, Wang H, Xia L, Erdjument-Bromage H, Tempst P, Jones RS, Zhang Y (2002) Role of histone H3 lysine 27 methylation in Polycomb-group silencing. Science 298:1039–1043
Caprariello AV, Rogers JA, Morgan ML, Hoghooghi V, Plemel JR, Koebel A, Tsutsui S, Dunn JF, Kotra LP, Ousman SS et al (2018) Biochemically altered myelin triggers autoimmune demyelination. Proc Natl Acad Sci USA 115:5528–5533
Carey BW, Finley LWS, Cross JR, Allis CD, Thompson CB (2015) Intracellular alpha-ketoglutarate maintains the pluripotency of embryonic stem cells. Nature 518:413–416
Carrer A, Trefely S, Zhao S, Campbell SL, Norgard RJ, Schultz KC, Sidoli S, Parris JLD, Affronti HC, Sivanand S et al (2019) Acetyl-CoA metabolism supports multistep pancreatic tumorigenesis. Cancer Discov 9:416–435
Carrico C, Meyer JG, He W, Gibson BW, Verdin E (2018) The mitochondrial acylome emerges: proteomics, regulation by sirtuins, and metabolic and disease implications. Cell Metab 27:497–512
Carrillo-Vico A, Leech MD, Anderton SM (2010) Contribution of myelin autoantigen citrullination to T cell autoaggression in the central nervous system. J Immunol 184:2839–2846
Ceccacci E, Minucci S (2016) Inhibition of histone deacetylases in cancer therapy: lessons from leukaemia. Br J Cancer 114:605–611
Chan KM, Fang D, Gan H, Hashizume R, Yu C, Schroeder M, Gupta N, Mueller S, James CD, Jenkins R et al (2013) The histone H3.3K27M mutation in pediatric glioma reprograms H3K27 methylation and gene expression. Genes Dev 27:985–990
Chang XT, Han JX, Pang L, Zhao Y, Yang Y, Shen ZL (2009) Increased PADI4 expression in blood and tissues of patients with malignant tumors. BMC Cancer 9
Chang XT, Hou XL, Pan JH, Fang KH, Wang L, Han JX (2011) Investigating the pathogenic role of PADI4 in oesophageal cancer. Int J Biol Sci 7:769–781
Chen LL, Xiong Y (2020) Tumour metabolites hinder DNA repair. Nature 582:492–494
Chen L, Deshpande AJ, Banka D, Bernt KM, Dias S, Buske C, Olhava EJ, Daigle SR, Richon VM, Pollock RM et al (2013a) Abrogation of MLL-AF10 and CALM-AF10-mediated transformation through genetic inactivation or pharmacological inhibition of the H3K79 methyltransferase Dot1l. Leukemia 27:813–822
Chen Q, Chen Y, Bian C, Fujiki R, Yu X (2013b) TET2 promotes histone O-GlcNAcylation during gene transcription. Nature 493:561–564
Chen L, Miao ZG, Xu XS (2017) beta-hydroxybutyrate alleviates depressive behaviors in mice possibly by increasing the histone3-lysine9-beta-hydroxybutyrylation. Biochem Biophys Res Commun 490:117–122
Chen J, Guccini I, Di Mitri D, Brina D, Revandkar A, Sarti M, Pasquini E, Alajati A, Pinton S, Losa M et al (2018a) Compartmentalized activities of the pyruvate dehydrogenase complex sustain lipogenesis in prostate cancer. Nat Genet 50:219–228
Chen XF, Tian MX, Sun RQ, Zhang ML, Zhou LS, Jin L, Chen LL, Zhou WJ, Duan KL, Chen YJ et al (2018b) SIRT5 inhibits peroxisomal ACOX1 to prevent oxidative damage and is downregulated in liver cancer. Embo Rep 19.
Cheng Y, He C, Wang MN, Ma XL, Mo F, Yang SY, Han JH, Wei XW (2019) Targeting epigenetic regulators for cancer therapy: mechanisms and advances in clinical trials. Signal Transduction Targeted Ther 4
Cheon YP, Xu XP, Bagchi MK, Bagchi IC (2003) Immune-responsive gene 1 is a novel target of progesterone receptor and plays a critical role during implantation in the mouse. Endocrinology 144:5623–5630
Chisolm DA, Weinmann AS (2018) Connections between metabolism and epigenetics in programming cellular differentiation. Annu Rev Immunol 36(36):221–246
Choi HS, Choi BY, Cho YY, Mizuno H, Kang BS, Bode AM, Dong ZG (2005) Phosphorylation of histone H3 at serine 10 is indispensable for neoplastic cell transformation. Cancer Res 65:5818–5827
Choudhary C, Kumar C, Gnad F, Nielsen ML, Rehman M, Walther TC, Olsen JV, Mann M (2009) Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science 325:834–840
Choudhary C, Weinert BT, Nishida Y, Verdin E, Mann M (2014) The growing landscape of lysine acetylation links metabolism and cell signalling. Nat Rev Mol Cell Biol 15:536–550
Chowdhury R, Yeoh KK, Tian YM, Hillringhaus L, Bagg EA, Rose NR, Leung IKH, Li XS, Woon ECY, Yang M et al (2011) The oncometabolite 2-hydroxyglutarate inhibits histone lysine demethylases. EMBO Rep 12:463–469
Christman JK (2002) 5-Azacytidine and 5-aza-2’-deoxycytidine as inhibitors of DNA methylation: mechanistic studies and their implications for cancer therapy. Oncogene 21:5483–5495
Christophorou MA, Castelo-Branco G, Halley-Stott RP, Oliveira CS, Loos R, Radzisheuskaya A, Mowen KA, Bertone P, Silva JCR, Zernicka-Goetz M et al (2014) Citrullination regulates pluripotency and histone H1 binding to chromatin. Nature 507:104–108
Chu CS, Lo PW, Yeh YH, Hsu PH, Peng SH, Teng YC, Kang ML, Wong CH, Juan LJ (2014) O-GlcNAcylation regulates EZH2 protein stability and function. Proc Natl Acad Sci USA 111:1355–1360
Chu QS, Sangha R, Spratlin J, Vos LJ, Mackey JR, McEwan AJ, Venner P, Michelakis ED (2015) A phase I open-labeled, single-arm, dose-escalation, study of dichloroacetate (DCA) in patients with advanced solid tumors. Invest New Drugs 33:603–610
Chuang JC, Yoo CB, Kwan JM, Li TWH, Liang GN, Yang AS, Jones PA (2005) Comparison of biological effects of non-nucleoside DNA methylation inhibitors versus 5-aza-2 ‘-deoxycytidine. Mol Cancer Ther 4:1515–1520
Chuang JC, Warner SL, Vollmer D, Vankayalapati H, Redkar S, Bearss DJ, Qiu XN, Yoo CB, Jones PA (2010) S110, a 5-Aza-2’-deoxycytidine-containing dinucleotide, is an effective DNA methylation inhibitor in vivo and can reduce tumor growth. Mol Cancer Ther 9:1443–1450
Chung C, Sweha SR, Pratt D, Tamrazi B, Panwalkar P, Banda A, Bayliss J, Hawes D, Yang F, Lee HJ et al (2020) Integrated metabolic and epigenomic reprograming by H3K27M mutations in diffuse intrinsic pontine gliomas. Cancer Cell 38(334–349):
Chypre M, Zaidi N, Smans K (2012) ATP-citrate lyase: a mini-review. Biochem Biophys Res Commun 422:1–4
Cohen I, Poreba E, Kamieniarz K, Schneider R (2011) Histone modifiers in cancer: friends or foes? Genes Cancer 2:631–647
Comerford SA, Huang Z, Du X, Wang Y, Cai L, Witkiewicz AK, Walters H, Tantawy MN, Fu A, Manning HC et al (2014) Acetate dependence of tumors. Cell 159:1591–1602
Cook PJ, Ju BG, Telese F, Wang X, Glass CK, Rosenfeld MG (2009) Tyrosine dephosphorylation of H2AX modulates apoptosis and survival decisions. Nature 458:591–596
Cordes T, Wallace M, Michelucci A, Divakaruni AS, Sapcariu SC, Sousa C, Koseki H, Cabrales P, Murphy AN, Hiller K et al (2016) Immunoresponsive gene 1 and itaconate inhibit succinate dehydrogenase to modulate intracellular succinate levels. J Biol Chem 291:14274–14284
Covarrubias AJ, Aksoylar HI, Yu JJ, Snyder NW, Worth AJ, Iyer SS, Wang JW, Ben-Sahra I, Byles V, Polynne-Stapornkul T et al (2016) Akt-mTORC1 signaling regulates Acly to integrate metabolic input to control of macrophage activation. Elife 5
Cravo ML, Pinto AG, Chaves P, Cruz JA, Lage P, Nobre Leitao C, Costa Mira F (1998) Effect of folate supplementation on DNA methylation of rectal mucosa in patients with colonic adenomas: correlation with nutrient intake. Clin Nutr 17:45–49
Crider KS, Yang TP, Berry RJ, Bailey LB (2012) Folate and DNA methylation: a review of molecular mechanisms and the evidence for folate’s role. Adv Nutr 3:21–38
Dai ZW, Mentch SJ, Gao X, Nichenametla SN, Locasale JW (2018) Methionine metabolism influences genomic architecture and gene expression through H3K4me3 peak width. Nat Commun 9
Dai ZW, Ramesh V, Locasale JW (2020) The evolving metabolic landscape of chromatin biology and epigenetics. Nat Rev Genet 21:737–753
Daigle SR, Olhava EJ, Therkelsen CA, Majer CR, Sneeringer CJ, Song J, Johnston LD, Scott MP, Smith JJ, Xiao YH et al (2011) Selective killing of mixed lineage leukemia cells by a potent small-molecule DOT1L inhibitor. Cancer Cell 20:53–65
Daigle SR, Olhava EJ, Therkelsen CA, Basavapathruni A, Jin L, Boriack-Sjodin PA, Allain CJ, Klaus CR, Raimondi A, Scott MP et al (2013) Potent inhibition of DOT1L as treatment of MLL-fusion leukemia. Blood 122:1017–1025
Dang CV (2012) MYC on the path to cancer. Cell 149:22–35
Dang L, White DW, Gross S, Bennett BD, Bittinger MA, Driggers EM, Fantin VR, Jang HG, Jin S, Keenan MC et al (2009) Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature 462:739–U752
Daniels BP, Kofman SB, Smith JR, Norris GT, Snyder AG, Kolb JP, Gao X, Locasale JW, Martinez J, Gale M et al (2019) The nucleotide sensor ZBP1 and kinase RIPK3 induce the enzyme IRG1 to promote an antiviral metabolic state in neurons. Immunity 50:64–76
Darrah E, Andrade F (2018) Rheumatoid arthritis and citrullination. Curr Opin Rheumatol 30:72–78
Datta J, Ghoshal K, Denny WA, Gamage SA, Brooke DG, Phiasivongsa P, Redkar S, Jacob ST (2009) A new class of quinoline-based DNA hypomethylating agents reactivates tumor suppressor genes by blocking DNA methyltransferase 1 activity and inducing its degradation. Cancer Res 69:4277–4285
Daw CC, Ramachandran K, Enslow BT, Maity S, Bursic B, Novello MJ, Rubannelsonkumar CS, Mashal AH, Ravichandran J, Bakewell TM et al (2020) Lactate elicits ER-mitochondrial Mg(2+) dynamics to integrate cellular metabolism. Cell.
Dawson MA, Bannister AJ, Gottgens B, Foster SD, Bartke T, Green AR, Kouzarides T (2009) JAK2 phosphorylates histone H3Y41 and excludes HP1alpha from chromatin. Nature 461:819–822
Dawson MA, Prinjha RK, Dittmann A, Giotopoulos G, Bantscheff M, Chan WI, Robson SC, Chung CW, Hopf C, Savitski MM et al (2011) Inhibition of BET recruitment to chromatin as an effective treatment for MLL-fusion leukaemia. Nature 478:529–533
de Boer VC, Houten SM (2014) A mitochondrial expatriate: nuclear pyruvate dehydrogenase. Cell 158:9–10
De Ruijter AJM, Van Gennip AH, Caron HN, Kemp S, Van Kuilenburg ABP (2003) Histone deacetylases (HDACs): characterization of the classical HDAC family. Biochem J 370:737–749
De Virgilio C, Burckert N, Barth G, Neuhaus JM, Boller T, Wiemken A (1992) Cloning and disruption of a gene required for growth on acetate but not on ethanol: the acetyl-coenzyme A synthetase gene of Saccharomyces cerevisiae. Yeast 8:1043–1051
DeBerardinis RJ, Chandel NS (2016) Fundamentals of cancer metabolism. Sci Adv 2
DeBerardinis RJ, Thompson CB (2012) Cellular Metabolism and Disease: What Do Metabolic Outliers Teach Us? Cell 148:1132–1144
DeBerardinis RJ, Lum JJ, Hatzivassiliou G, Thompson CB (2008a) The biology of cancer: Metabolic reprogramming fuels cell growth and proliferation. Cell Metab 7:11–20
DeBerardinis RJ, Sayed N, Ditsworth D, Thompson CB (2008b) Brick by brick: metabolism and tumor cell growth. Curr Opin Genet Dev 18:54–61
Dehennaut V, Leprince D, Lefebvre T (2014) O-GlcNAcylation, an epigenetic mark. Focus on the histone code, TET family proteins, and polycomb group proteins. Front Endocrinol (Lausanne) 5:155
Delmore JE, Issa GC, Lemieux ME, Rahl PB, Shi JW, Jacobs HM, Kastritis E, Gilpatrick T, Paranal RM, Qi J et al (2011) BET bromodomain inhibition as a therapeutic strategy to target c-Myc. Cell 146:903–916
Demers, M., Wong, S.L., Martinod, K., Gallant, M., Cabral, J.E., Wang, Y.M., and Wagner, D.D. (2016). Priming of neutrophils toward NETosis promotes tumor growth. Oncoimmunology 5.
Denny WA, Atwell GJ, Baguley BC, Cain BF (1979) Potential antitumor agents. 29. Quantitative structure-activity relationships for the antileukemic bisquaternary ammonium heterocycles. J Med Chem 22:134–150
Dentin R, Hedrick S, Xie JX, Yates J, Montminy M (2008) Hepatic glucose sensing via the CREB coactivator CRTC2. Science 319:1402–1405
Deplus R, Delatte B, Schwinn MK, Defrance M, Mendez J, Murphy N, Dawson MA, Volkmar M, Putmans P, Calonne E et al (2013) TET2 and TET3 regulate GlcNAcylation and H3K4 methylation through OGT and SET1/COMPASS. EMBO J 32:645–655
Di Lorenzo A, Bedford MT (2011) Histone arginine methylation. FEBS Lett 585:2024–2031
Dimitrova E, Turberfield AH, Klose RJ (2015) Histone demethylases in chromatin biology and beyond. EMBO Rep 16:1620–1639
Ding W, Smulan LJ, Hou NS, Taubert S, Watts JL, Walker AK (2015) s-Adenosylmethionine levels govern innate immunity through distinct methylation-dependent pathways. Cell Metab 22:633–645
Dominguez-Andres J, Novakovic B, Li Y, Scicluna BP, Gresnigt MS, Arts RJW, Oosting M, Moorlag SJCFM, Groh LA, Zwaag J et al (2019) The itaconate pathway is a central regulatory node linking innate immune tolerance and trained immunity. Cell Metab 29:211–220
Donohoe DR, Collins LB, Wali A, Bigler R, Sun W, Bultman SJ (2012) The Warburg effect dictates the mechanism of butyrate-mediated histone acetylation and cell proliferation. Mol Cell 48:612–626
Du JT, Zhou YY, Su XY, Yu JJ, Khan S, Jiang H, Kim J, Woo J, Kim JH, Choi BH et al (2011) Sirt5 is a NAD-dependent protein lysine demalonylase and desuccinylase. Science 334:806–809
Dunbar EM, Coats BS, Shroads AL, Langaee T, Lew A, Forder JR, Shuster JJ, Wagner DA, Stacpoole PW (2014) Phase 1 trial of dichloroacetate (DCA) in adults with recurrent malignant brain tumors. Invest New Drugs 32:452–464
Eckschlager, T., Plch, J., Stiborova, M., and Hrabeta, J. (2017). Histone Deacetylase Inhibitors as Anticancer Drugs. International Journal of Molecular Sciences 18.
Eden A, Gaudet F, Waghmare A, Jaenisch R (2003) Chromosomal instability and tumors promoted by DNA hypomethylation. Science 300:455
Edmunds LR, Sharma L, Kang A, Lu J, Vockley J, Basu S, Uppala R, Goetzman ES, Beck ME, Scott D et al (2014) c-Myc programs fatty acid metabolism and dictates acetyl-CoA abundance and fate. J Biol Chem 289:25382–25392
Eisenberg T, Schroeder S, Andryushkova A, Pendl T, Kuttner V, Bhukel A, Marino G, Pietrocola F, Harger A, Zimmermann A et al (2014) Nucleocytosolic depletion of the energy metabolite acetyl-coenzyme a stimulates autophagy and prolongs lifespan. Cell Metab 19:431–444
Ellinger J, Kahl P, Mertens C, Rogenhofer S, Hauser S, Hartmann W, Bastian PJ, Buttner R, Muller SC, von Ruecker A (2010) Prognostic relevance of global histone H3 lysine 4 (H3K4) methylation in renal cell carcinoma. Int J Cancer 127:2360–2366
Elsheikh SE, Green AR, Rakha EA, Powe DG, Ahmed RA, Collins HM, Soria D, Garibaldi JM, Paish CE, Ammar AA et al (2009) Global histone modifications in breast cancer correlate with tumor phenotypes, prognostic factors, and patient outcome. Cancer Res 69:3802–3809
Esteller M, Avizienyte E, Corn PG, Lothe RA, Baylin SB, Aaltonen LA, Herman JG (2000) Epigenetic inactivation of LKB1 in primary tumors associated with the Peutz-Jeghers syndrome. Oncogene 19:164–168
Esteller M, Corn PG, Baylin SB, Herman JG (2001) A gene hypermethylation profile of human cancer. Cancer Res 61:3225–3229
Etchegaray JP, Mostoslavsky R (2016) Interplay between metabolism and epigenetics: a nuclear adaptation to environmental changes. Mol Cell 62:695–711
Evans JS, Mengel GD (1964) The reversal of cytosine arabinoside activity in vivo by deoxycytidine. Biochem Pharmacol 13:989–994
Faiola F, Liu X, Lo S, Pan S, Zhang K, Lymar E, Farina A, Martinez E (2005) Dual regulation of c-Myc by p300 via acetylation-dependent control of Myc protein turnover and coactivation of Myc-induced transcription. Mol Cell Biol 25:10220–10234
Falkenberg KJ, Johnstone RW (2014) Histone deacetylases and their inhibitors in cancer, neurological diseases and immune disorders. Nat Rev Drug Discovery 13:673–691
Fan J, Teng X, Liu L, Mattaini KR, Looper RE, Vander Heiden MG, Rabinowitz JD (2015) Human phosphoglycerate dehydrogenase produces the oncometabolite D-2-hydroxyglutarate. ACS Chem Biol 10:510–516
Faubert B, Li KY, Cai L, Hensley CT, Kim J, Zacharias LG, Yang C, Do QN, Doucette S, Burguete D et al (2017) Lactate metabolism in human lung tumors. Cell 171(358–371):
Faubert, B., Solmonson, A., and DeBerardinis, R.J. (2020). Metabolic reprogramming and cancer progression. Science 368.
Feinberg AP, Tycko B (2004) Timeline - The history of cancer epigenetics. Nat Rev Cancer 4:143–153
Feinberg AP, Vogelstein B (1983) Hypomethylation distinguishes genes of some human cancers from their normal counterparts. Nature 301:89–92
Feldman JL, Baeza J, Denu JM (2013) Activation of the protein deacetylase SIRT6 by long-chain fatty acids and widespread deacylation by mammalian sirtuins. J Biol Chem 288:31350–31356
Feng Q, Wang HB, Ng HH, Erdjument-Bromage H, Tempst P, Struhl K, Zhang Y (2002) Methylation of H3-lysine 79 is mediated by a new family of HMTases without a SET domain. Curr Biol 12:1052–1058
Ferrer CM, Lynch TP, Sodi VL, Falcone JN, Schwab LP, Peacock DL, Vocadlo DJ, Seagroves TN, Reginato MJ (2014) O-GlcNAcylation regulates cancer metabolism and survival stress signaling via regulation of the HIF-1 pathway. Mol Cell 54:820–831
Ferrer CM, Sodi VL, Reginato MJ (2016) O-GlcNAcylation in cancer biology: linking metabolism and signaling. J Mol Biol 428:3282–3294
Figueroa ME, Abdel-Wahab O, Lu C, Ward PS, Patel J, Shih A, Li YS, Bhagwat N, Vasanthakumar A, Fernandez HF et al (2010) Leukemic IDH1 and IDH2 mutations result in a hypermethylation phenotype, disrupt TET2 function, and impair hematopoietic differentiation. Cancer Cell 18:553–567
Filippakopoulos P, Qi J, Picaud S, Shen Y, Smith WB, Fedorov O, Morse EM, Keates T, Hickman TT, Felletar I et al (2010) Selective inhibition of BET bromodomains. Nature 468:1067–1073
Filippov S, Pinkosky SL, Newton RS (2014) LDL-cholesterol reduction in patients with hypercholesterolemia by modulation of adenosine triphosphate-citrate lyase and adenosine monophosphate-activated protein kinase. Curr Opin Lipidol 25:309–315
Fleming SE, Fitch MD, DeVries S, Liu ML, Kight C (1991) Nutrient utilization by cells isolated from rat jejunum, cecum and colon. J Nutr 121:869–878
Fraga MF, Ballestar E, Villar-Garea A, Boix-Chornet M, Espada J, Schotta G, Bonaldi T, Haydon C, Ropero S, Petrie K et al (2005) Loss of acetylation at Lys16 and trimethylation at Lys20 of histone H4 is a common hallmark of human cancer. Nat Genet 37:391–400
Fuhrmann J, Thompson PR (2016) Protein arginine methylation and citrullination in epigenetic regulation. ACS Chem Biol 11:654–668
Fujino T, Kondo J, Ishikawa M, Morikawa K, Yamamoto TT (2001) Acetyl-CoA synthetase 2, a mitochondrial matrix enzyme involved in the oxidation of acetate. J Biol Chem 276:11420–11426
Fujisawa T, Filippakopoulos P (2017) Functions of bromodomain-containing proteins and their roles in homeostasis and cancer. Nat Rev Mol Cell Biol 18:246–262
Galeotti F, Barile E, Curir P, Dolci M, Lanzotti V (2008) Flavonoids from carnation (Dianthus caryophyllus) and their antifungal activity. Phytochem Lett 1:44–48
Gambetta MC, Oktaba K, Muller J (2009) Essential role of the glycosyltransferase sxc/Ogt in polycomb repression. Science 325:93–96
Gao, P., Tchernyshyov, I., Chang, T.C., Lee, Y.S., Kita, K., Ochi, T., Zeller, K., De Marzo, A., Van Eyk, J., Mendell, J., et al. (2009). c-Myc suppression of miR-23 enhances mitochondrial glutaminase and glutamine metabolism. Cancer Research 69.
Gao, X., Lin, S.H., Ren, F., Li, J.T., Chen, J.J., Yao, C.B., Yang, H.B., Jiang, S.X., Yan, G.Q., Wang, D., et al. (2016). Acetate functions as an epigenetic metabolite to promote lipid synthesis under hypoxia. Nature Communications 7.
Garcia JM, Silva J, Pena C, Garcia V, Rodriguez R, Cruz MA, Cantos B, Provencio M, Espana P, Bonilla F (2004) Promoter methylation of the PTEN gene is a common molecular change in breast cancer. Genes Chromosom Cancer 41:117–124
Garcia BA, Luka Z, Loukachevitch LV, Bhanu NV, Wagner C (2016) Folate deficiency affects histone methylation. Med Hypotheses 88:63–67
Gates LA, Shi JJ, Rohira AD, Feng Q, Zhu BK, Bedford MT, Sagum CA, Jung SY, Qin J, Tsai MJ et al (2017) Acetylation on histone H3 lysine 9 mediates a switch from transcription initiation to elongation. J Biol Chem 292:14456–14472
Gaudet F, Hodgson JG, Eden A, Jackson-Grusby L, Dausman J, Gray JW, Leonhardt H, Jaenisch R (2003) Induction of tumors in mice by genomic hypomethylation. Science 300:489–492
Giet R, Glover DM (2001) Drosophila Aurora B kinase is required for histone H3 phosphorylation and condensin recruitment during chromosome condensation and to organize the central spindle during cytokinesis. J Cell Biol 152:669–681
Goffin J, Eisenhauer E (2002) DNA methyltransferase inhibitors—state of the art. Ann Oncol 13:1699–1716
Golks A, Tran TT, Goetschy JF, Guerini D (2007) Requirement for O-linked N-acetylglucosaminyltransferase in lymphocytes activation. EMBO J 26:4368–4379
Goncalves MD, Lu CY, Tutnauer J, Hartman TE, Hwang SK, Murphy CJ, Pauli C, Morris R, Taylor S, Bosch K et al (2019) High-fructose corn syrup enhances intestinal tumor growth in mice. Science 363:1345–1349
Gongol, B., Sari, I., Bryant, T., Rosete, G., and Marin, T. (2018). AMPK: An Epigenetic Landscape Modulator. International Journal of Molecular Sciences 19.
Goudarzi A, Zhang D, Huang H, Barral S, Kwon OK, Qi S, Tang Z, Buchou T, Vitte AL, He T et al (2016) Dynamic competing histone H4 K5K8 acetylation and butyrylation are hallmarks of highly active gene promoters. Mol Cell 62:169–180
Gounaris AD, Perlmann GE (1967) Succinylation of pepsinogen. J Biol Chem 242:2739–2745
Gravel SP, Hulea L, Toban N, Birman E, Blouin MJ, Zakikhani M, Zhao YH, Topisirovic I, St-Pierre J, Pollak M (2014) Serine deprivation enhances antineoplastic activity of biguanides. Cancer Res 74:7521–7533
Greenberg MVC, Bourc’his D (2019) The diverse roles of DNA methylation in mammalian development and disease. Nat Rev Mol Cell Biol 20:590–607
Greene KS, Lukey MJ, Wang XY, Blank B, Druso JE, Lin MCJ, Stalnecker CA, Zhang CL, Abril YN, Erickson JW et al (2019) SIRT5 stabilizes mitochondrial glutaminase and supports breast cancer tumorigenesis. Proc Natl Acad Sci USA 116:26625–26632
Greer EL, Shi Y (2012) Histone methylation: a dynamic mark in health, disease and inheritance. Nat Rev Genet 13:343–357
Gross S, Cairns RA, Minden MD, Driggers EM, Bittinger MA, Jang HG, Sasaki M, Jin SF, Schenkein DP, Su SSM et al (2010) Cancer-associated metabolite 2-hydroxyglutarate accumulates in acute myelogenous leukemia with isocitrate dehydrogenase 1 and 2 mutations. J Exp Med 207:339–344
Gu YC, Mi WY, Ge YQ, Liu HY, Fan QO, Han CF, Yang J, Han F, Lu XZ, Yu WG (2010) GlcNAcylation plays an essential role in breast cancer metastasis. Cancer Res 70:6344–6351
Guccione E, Richard S (2019) The regulation, functions and clinical relevance of arginine methylation. Nat Rev Mol Cell Biol 20:642–657
Guo H, Tan Y, Kubota T, Moossa AR, Hoffman RM (1996) Methionine depletion modulates the antitumor and antimetastatic efficacy of ethionine. Anticancer Res 16:2719–2723
Gutierrez RM, Hnilica LS (1967) Tissue specificity of histone phosphorylation. Science 157:1324–1325
Gutierrez MJ, Rosenberg NL, MacDougall DE, Hanselman JC, Margulies JR, Strange P, Milad MA, McBride SJ, Newton RS (2014) Efficacy and Safety of ETC-1002, a novel investigational low-density lipoprotein-cholesterol-lowering therapy for the treatment of patients with hypercholesterolemia and Type 2 diabetes mellitus. Arterioscler Thromb Vasc Biol 34:676–683
Hamer HM, Jonkers D, Venema K, Vanhoutvin S, Troost FJ, Brummer RJ (2008) Review article: the role of butyrate on colonic function. Aliment Pharmacol Ther 27:104–119
Hanahan D, Weinberg RA (2011) Hallmarks of cancer: the next generation. Cell 144:646–674
Hannou SA, Haslam DE, McKeown NM, Herman MA (2018) Fructose metabolism and metabolic disease. J Clin Invest 128:545–555
Hao HX, Khalimonchuk O, Schraders M, Dephoure N, Bayley JP, Kunst H, Devilee P, Cremers CWRJ, Schiffman JD, Bentz BG et al (2009) SDH5, a gene required for flavination of succinate dehydrogenase, is mutated in paraganglioma. Science 325:1139–1142
Hardie DG (2011) AMP-activated protein kinase-an energy sensor that regulates all aspects of cell function. Genes Dev 25:1895–1908
Hardie DG, Schaffer BE, Brunet A (2016) AMPK: an energy-sensing pathway with multiple inputs and outputs. Trends Cell Biol 26:190–201
Hardiville S, Hart GW (2014) Nutrient regulation of signaling, transcription, and cell physiology by O-GlcNAcylation. Cell Metab 20:208–213
Harris RA, Joshi M, Jeoung NH, Obayashi M (2005) Overview of the molecular and biochemical basis of branched-chain amino acid catabolism. J Nutr 135:1527s–1530s
Hart GW (2019) Nutrient regulation of signaling and transcription. J Biol Chem 294:2211–2231
Hart GW, Housley MP, Slawson C (2007) Cycling of O-linked beta-N-acetylglucosamine on nucleocytoplasmic proteins. Nature 446:1017–1022
Hart GW, Slawson C, Ramirez-Correa G, Lagerlof O (2011) Cross talk between O-GlcNAcylation and phosphorylation: roles in signaling, transcription, and chronic disease. Annu Rev Biochem 80(80):825–858
Harutyunyan AS, Krug B, Chen HF, Papillon-Cavanagh S, Zeinieh M, De Jay N, Deshmukh S, Chen CCL, Belle J, Mikael LG et al (2019) H3K27M induces defective chromatin spread of PRC2-mediated repressive H3K27me2/me3 and is essential for glioma tumorigenesis. Nat Commun 10
Hatzivassiliou G, Zhao FP, Bauer DE, Andreadis C, Shaw AN, Dhanak D, Hingorani SR, Tuveson DA, Thompson CB (2005) ATP citrate lyase inhibition can suppress tumor cell growth. Cancer Cell 8:311–321
Haws SA, Leech CM, Denu JM (2020) Metabolism and the epigenome: a dynamic relationship. Trends Biochem Sci
Hayakawa K, Hirosawa M, Tabei Y, Arai D, Tanaka S, Murakami N, Yagi S, Shiota K (2013) Epigenetic switching by the metabolism-sensing factors in the generation of orexin neurons from mouse embryonic stem cells. J Biol Chem 288:17099–17110
Heiden MGV, Cantley LC, Thompson CB (2009) Understanding the warburg effect: the metabolic requirements of cell proliferation. Science 324:1029–1033
Herman JG, Latif F, Weng Y, Lerman MI, Zbar B, Liu S, Samid D, Duan DS, Gnarra JR, Linehan WM et al (1994) Silencing of the VHL tumor-suppressor gene by DNA methylation in renal carcinoma. Proc Natl Acad Sci USA 91:9700–9704
Herzig S, Raemy E, Montessuit S, Veuthey JL, Zamboni N, Westermann B, Kunji ER, Martinou JC (2012) Identification and functional expression of the mitochondrial pyruvate carrier. Science 337:93–96
Hirschey MD, Zhao YM (2015) Metabolic regulation by lysine malonylation, succinylation, and glutarylation. Mol Cell Proteomics 14:2308–2315
Hirschey MD, Shimazu T, Goetzman E, Jing E, Schwer B, Lombard DB, Grueter CA, Harris C, Biddinger S, Ilkayeva OR et al (2010) SIRT3 regulates mitochondrial fatty-acid oxidation by reversible enzyme deacetylation. Nature 464:121–U137
Hirschey MD, DeBerardinis RJ, Diehl AME, Drew JE, Frezza C, Green MF, Jones LW, Ko YH, Le A, Lea MA et al (2015) Dysregulated metabolism contributes to oncogenesis. Semin Cancer Biol 35:S129–S150
Hitosugi T, Fan J, Chung TW, Lythgoe K, Wang X, Xie JX, Ge QY, Gu TL, Polakiewicz RD, Roesel JL et al (2011) Tyrosine phosphorylation of mitochondrial pyruvate dehydrogenase kinase 1 is important for cancer metabolism. Mol Cell 44:864–877
Holleran JL, Parise RA, Joseph E, Eiseman JL, Covey JM, Glaze ER, Lyubimov AV, Chen YF, D’Argenio DZ, Egorin MJ (2005) Plasma pharmacokinetics, oral bioavailability, and interspecies scaling of the DNA methyltransferase inhibitor, zebularine. Clin Cancer Res 11:3862–3868
Hooftman A, Angiari S, Hester S, Corcoran SE, Runtsch MC, Ling C, Ruzek MC, Slivka PF, McGettrick AF, Banahan K et al (2020) The immunomodulatory metabolite itaconate modifies NLRP3 and inhibits inflammasome activation. Cell Metab 32:468–478
Hopkins BD, Pauli C, Du X, Wang DG, Li X, Wu D, Amadiume SC, Goncalves MD, Hodakoski C, Lundquist MR et al (2018) Suppression of insulin feedback enhances the efficacy of PI3K inhibitors. Nature 560:499–503
Hoshiya Y, Kubota T, Inada T, Kitajima M, Hoffman RM (1997) Methionine-depletion modulates the efficacy of 5-fluorouracil in human gastric cancer in nude mice. Anticancer Res 17:4371–4375
Housley MP, Rodgers JT, Udeshi ND, Kelly TJ, Shabanowitz J, Hunt DF, Puigserver P, Hart GW (2008) O-GlcNAc regulates FoxO activation in response to glucose. J Biol Chem 283:16283–16292
Howitz KT, Bitterman KJ, Cohen HY, Lamming DW, Lavu S, Wood JG, Zipkin RE, Chung P, Kisielewski A, Zhang LL et al (2003) Small molecule activators of sirtuins extend Saccharomyces cerevisiae lifespan. Nature 425:191–196
Hsu PC, Liao YF, Lin CL, Lin WH, Liu GY, Hung HC (2014) Vimentin is involved in peptidylarginine deiminase 2-induced apoptosis of activated jurkat cells. Mol Cells 37:426–434
Hubbard BP, Gomes AP, Dai H, Li J, Case AW, Considine T, Riera TV, Lee JE, Yen ES, Lamming DW et al (2013) Evidence for a common mechanism of SIRT1 regulation by allosteric activators. Science 339:1216–1219
Hughey CC, Trefts E, Bracy DP, James FD, Donahue EP, Wasserman DH (2018) Glycine N-methyltransferase deletion in mice diverts carbon flux from gluconeogenesis to pathways that utilize excess methionine cycle intermediates. J Biol Chem 293:11944–11954
Hui S, Ghergurovich JM, Morscher RJ, Jang C, Teng X, Lu W, Esparza LA, Reya T, Le Z, Yanxiang Guo J et al (2017) Glucose feeds the TCA cycle via circulating lactate. Nature 551:115–118
Hwang IY, Kwak S, Lee S, Kim H, Lee SE, Kim JH, Kim YA, Jeon YK, Chung DH, Jin X et al (2016) Psat1-Dependent fluctuations in alpha-ketoglutarate affect the timing of ESC differentiation. Cell Metab 24:494–501
Hyun K, Jeon J, Park K, Kim J (2017) Writing, erasing and reading histone lysine methylations. Exp Mol Med 49
Icard P, Poulain L, Lincet H (2012) Understanding the central role of citrate in the metabolism of cancer cells. Biochim Biophys Acta 1825:111–116
Icard P, Wu ZR, Fournel L, Coquerel A, Lincet H, Alifano M (2020) ATP citrate lyase: A central metabolic enzyme in cancer. Cancer Lett 471:125–134
Intlekofer AM, Dematteo RG, Venneti S, Finley LWS, Lu C, Judkins AR, Rustenburg AS, Grinaway PB, Chodera JD, Cross JR et al (2015) Hypoxia induces production of L-2-hydroxyglutarate. Cell Metab 22:304–311
Ishak Gabra MB, Yang Y, Li H, Senapati P, Hanse EA, Lowman XH, Tran TQ, Zhang L, Doan LT, Xu X et al (2020) Dietary glutamine supplementation suppresses epigenetically-activated oncogenic pathways to inhibit melanoma tumour growth. Nat Commun 11:3326
Ivashkiv LB (2013) Epigenetic regulation of macrophage polarization and function. Trends Immunol 34:216–223
Jang MK, Mochizuki K, Zhou MS, Jeong HS, Brady JN, Ozato K (2005) The bromodomain protein Brd4 is a positive regulatory component of P-TEFb and stimulates RNA polymerase II-dependent transcription. Mol Cell 19:523–534
Janke R, Dodson AE, Rine J (2015) Metabolism and epigenetics. Annu Rev Cell Dev Biol 31:473–496
Jensen T, Abdelmalek MF, Sullivan S, Nadeau KJ, Green M, Roncal C, Nakagawa T, Kuwabara M, Sato Y, Kang DH et al (2018) Fructose and sugar: a major mediator of non-alcoholic fatty liver disease. J Hepatol 68:1063–1075
Jeon H, Kim JH, Lee E, Jang YJ, Son JE, Kwon JY, Lim TG, Kim S, Park JH, Kim JE et al (2016) Methionine deprivation suppresses triple-negative breast cancer metastasis in vitro and in vivo. Oncotarget 7:67223–67234
Jha AK, Huang SCC, Sergushichev A, Lampropoulou V, Ivanova Y, Loginicheva E, Chmielewski K, Stewart KM, Ashall J, Everts B et al (2015) Network integration of parallel metabolic and transcriptional data reveals metabolic modules that regulate macrophage polarization. Immunity 42:419–430
Jiang WQ, Wang SW, Xiao MT, Lin Y, Zhou LS, Lei QY, Xiong Y, Guan KL, Zhao SM (2011) Acetylation regulates gluconeogenesis by promoting PEPCK1 degradation via recruiting the UBR5 ubiquitin ligase. Mol Cell 43:33–44
Jing H, Lin HN (2015) Sirtuins in epigenetic regulation. Chem Rev 115:2350–2375
Jones PA, Baylin SB (2002) The fundamental role of epigenetic events in cancer. Nat Rev Genet 3:415–428
Jones RG, Thompson CB (2009) Tumor suppressors and cell metabolism: a recipe for cancer growth. Genes Dev 23:537–548
Kaczmarska Z, Ortega E, Goudarzi A, Huang H, Kim S, Marquez JA, Zhao YM, Khochbin S, Panne D (2017) Structure of p300 in complex with acyl-CoA variants. Nat Chem Biol 13:21–29
Kaelin WG (2009) SDH5 Mutations and Familial Paraganglioma: Somewhere Warburg is Smiling. Cancer Cell 16:180–182
Kaelin WG, McKnight SL (2013) Influence of Metabolism on Epigenetics and Disease. Cell 153:56–69
Kaminskas E, Farrell A, Abraham S, Baird A, Hsieh LS, Lee SL, Leighton JK, Patel H, Rahman A, Sridhara R et al (2005a) Approval summary: Azacitidine for treatment of myelodysplastic syndrome subtypes. Clin Cancer Res 11:3604–3608
Kaminskas E, Farrell AT, Wang YC, Sridhara R, Pazdur R (2005b) FDA drug approval summary: Azacitidine (5-azacytidine, Vidaza) for injectable suspension. Oncologist 10:176–182
Kang YH, Lee HS, Kim WH (2002) Promoter methylation and silencing of PTEN in gastric carcinoma. Lab Invest 82:285–291
Kaplon J, Zheng L, Meissl K, Chaneton B, Selivanov VA, Mackay G, van der Burg SH, Verdegaal EM, Cascante M, Shlomi T et al (2013) A key role for mitochondrial gatekeeper pyruvate dehydrogenase in oncogene-induced senescence. Nature 498:109–112
Karahoca M, Momparler RL (2013) Pharmacokinetic and pharmacodynamic analysis of 5-aza-2’-deoxycytidine (decitabine) in the design of its dose-schedule for cancer therapy. Clin Epigenetics 5:3
Katoh Y, Ikura T, Hoshikawa Y, Tashiro S, Ito T, Ohta M, Kera Y, Noda T, Igarashi K (2011) Methionine adenosyltransferase II serves as a transcriptional corepressor of maf oncoprotein. Mol Cell 41:554–566
Katsyuba E, Mottis A, Zietak M, De Franco F, van der Velpen V, Gariani K, Ryu D, Cialabrini L, Matilainen O, Liscio P et al (2018) De novo NAD(+) synthesis enhances mitochondrial function and improves health. Nature 563:354–359
Keating ST, El-Osta A (2015) Epigenetics and metabolism. Circ Res 116:715–736
Kelly B, Pearce EL (2020). Amino assets: how amino acids support immunity. Cell Metab
Kelly WG, Dahmus ME, Hart GW (1993) RNA polymerase II is a glycoprotein. Modification of the COOH-terminal domain by O-GlcNAc. J Biol Chem 268:10416–10424
Kera Y, Katoh Y, Ohta M, Matsumoto M, Takano-Yamamoto T, Igarashi K (2013) Methionine adenosyltransferase II-dependent histone H3K9 methylation at the COX-2 gene locus. J Biol Chem 288:13592–13601
Kidwai SA, Ansari AA, Salahuddin A (1976) Effect of succinylation (3-carboxypropionylation) on the conformation and immunological activity of ovalbumin. Biochem J 155:171–180
Kim JW, Tchernyshyov I, Semenza GL, Dang CV (2006) HIF-1-mediated expression of pyruvate dehydrogenase kinase: a metabolic switch required for cellular adaptation to hypoxia. Cell Metab 3:177–185
Kim SR, Kim KB, Chae YC, Park JW, Seo SB (2016) H3S10 phosphorylation-mediated transcriptional regulation by Aurora kinase A. Biochem Biophys Res Commun 469:22–28
Kinnaird A, Zhao S, Wellen KE, Michelakis ED (2016) Metabolic control of epigenetics in cancer. Nat Rev Cancer 16:694–707
Kleinsmith LJ, Allfrey VG, Mirsky AE (1966) Phosphoprotein metabolism in isolated lymphocyte nuclei. Proc Natl Acad Sci USA 55:1182–1189
Klement RJ (2019) The emerging role of ketogenic diets in cancer treatment. Curr Opin Clin Nutr Metab Care 22:129–134
Knight JS, Subramanian V, O’Dell AA, Yalavarthi S, Zhao WP, Smith CK, Hodgin JB, Thompson PR, Kaplan MJ (2015) Peptidylarginine deiminase inhibition disrupts NET formation and protects against kidney, skin and vascular disease in lupus-prone MRL/lpr mice. Ann Rheum Dis 74:2199–2206
Knutson SK, Wigle TJ, Warholic NM, Sneeringer CJ, Allain CJ, Klaus CR, Sacks JD, Raimondi A, Majer CR, Song J et al (2012) A selective inhibitor of EZH2 blocks H3K27 methylation and kills mutant lymphoma cells. Nat Chem Biol 8:890–896
Knutson SK, Kawano S, Minoshima Y, Warholic NM, Huang KC, Xiao YH, Kadowaki T, Uesugi M, Kuznetsov G, Kumar N et al (2014) Selective inhibition of EZH2 by EPZ-6438 leads to potent antitumor activity in EZH2-mutant non-hodgkin lymphoma. Mol Cancer Ther 13:842–854
Koenis DS, Medzikovic L, van Loenen PB, van Weeghel M, Huveneers S, Vos M, Evers-van Gogh IJ, Van den Bossche J, Speijer D, Kim Y et al (2018) Nuclear receptor nur77 limits the macrophage inflammatory response through transcriptional reprogramming of mitochondrial metabolism. Cell Reports 24:2127–2140
Koivunen P, Lee S, Duncan CG, Lopez G, Lu G, Ramkissoon S, Losman JA, Joensuu P, Bergmann U, Gross S et al (2012) Transformation by the (R)-enantiomer of 2-hydroxyglutarate linked to EGLN activation. Nature 483:485
Konig MF, Abusleme L, Reinholdt J, Palmer RJ, Teles RP, Sampson K, Rosen A, Nigrovic PA, Sokolove J, Giles JT et al (2016) Aggregatibacter actinomycetemcomitans-induced hypercitrullination links periodontal infection to autoimmunity in rheumatoid arthritis. Sci Transl Med 8
Kottakis F, Nicolay BN, Roumane A, Karnik R, Gu HC, Nagle JM, Boukhali M, Hayward MC, Li YY, Chen T et al (2016) LKB1 loss links serine metabolism to DNA methylation and tumorigenesis. Nature 539:390–395
Kryukov GV, Wilson FH, Ruth JR, Paulk J, Tsherniak A, Marlow SE, Vazquez F, Weir BA, Fitzgerald ME, Tanaka M et al (2016) MTAP deletion confers enhanced dependency on the PRMT5 arginine methyltransferase in cancer cells. Science 351:1214–1218
Kumari R, Deshmukh RS, Das S (2019) Caspase-10 inhibits ATP-citrate lyase-mediated metabolic and epigenetic reprogramming to suppress tumorigenesis. Nat Commun 10
Kurmi K, Hitosugi S, Wiese EK, Boakye-Agyeman F, Gonsalves WI, Lou ZK, Karnitz LM, Goetz MP, Hitosugi T (2018) Carnitine palmitoyltransferase 1A Has a lysine succinyltransferase activity. Cell Rep 22:1365–1373
Lacoste N, Utley RT, Hunter JM, Poirier GG, Cote J (2002) Disruptor of telomeric silencing-1 is a chromatin-specific histone H3 methyltransferase. J Biol Chem 277:30421–30424
Lampropoulou V, Sergushichev A, Bambouskova M, Nair S, Vincent EE, Loginicheva E, Cervantes-Barragan L, Ma XC, Huang SCC, Griss T et al (2016) Itaconate links inhibition of succinate dehydrogenase with macrophage metabolic remodeling and regulation of inflammation. Cell Metab 24:158–166
Langan TA (1968) Histone phosphorylation: stimulation by adenosine 3’,5’-monophosphate. Science 162:579–580
Laribee RN (2018) Transcriptional and epigenetic regulation by the mechanistic target of rapamycin complex 1 pathway. J Mol Biol 430:4874–4890
Latham T, Mackay L, Sproul D, Karim M, Culley J, Harrison DJ, Hayward L, Langridge-Smith P, Gilbert N, Ramsahoye BH (2012) Lactate, a product of glycolytic metabolism, inhibits histone deacetylase activity and promotes changes in gene expression. Nucleic Acids Res 40:4794–4803
Lauterbach MA, Hanke JE, Serefidou M, Mangan MSJ, Kolbe CC, Hess T, Rothe M, Kaiser R, Hoss F, Gehlen J et al (2019) Toll-like receptor signaling rewires macrophage metabolism and promotes histone acetylation via ATP-citrate lyase. Immunity 51(997–1011):
Lavu S, Boss O, Elliott PJ, Lambert PD (2008) Sirtuins - novel therapeutic targets to treat age-associated diseases. Nat Rev Drug Discovery 7:841–853
Lee CG, Jenkins NA, Gilbert DJ, Copeland NG, O’Brien WE (1995) Cloning and analysis of gene regulation of a novel LPS-inducible cDNA. Immunogenetics 41:263–270
Lee JV, Carrer A, Shah S, Snyder NW, Wei SZ, Venneti S, Worth AJ, Yuan ZF, Lim HW, Liu SC et al (2014) Akt-dependent metabolic reprogramming regulates tumor cell histone acetylation. Cell Metab 20:306–319
Lee CF, Caudal A, Abell L, Gowda GAN, Tian R (2019) Targeting NAD(+) metabolism as interventions for mitochondrial disease. Sci Rep 9
Lei MZ, Li XX, Zhang Y, Li JT, Zhang F, Wang YP, Yin M, Qu J, Lei QY (2020) Acetylation promotes BCAT2 degradation to suppress BCAA catabolism and pancreatic cancer growth. Signal Transduct Target Ther 5:70
Levine SS, King IF, Kingston RE (2004) Division of labor in polycomb group repression. Trends Biochem Sci 29:478–485
Lewis BA, Hanover JA (2014) O-GlcNAc and the epigenetic regulation of gene expression. J Biol Chem 289:34440–34448
Lewis PW, Muller MM, Koletsky MS, Cordero F, Lin S, Banaszynski LA, Garcia BA, Muir TW, Becher OJ, Allis CD (2013) Inhibition of PRC2 activity by a gain-of-function H3 mutation found in pediatric glioblastoma. Science 340:857–861
Li JJ, Wang HX, Tino JA, Robl JA, Herpin TF, Lawrence RM, Biller S, Jamil H, Ponticiello R, Chen LP et al (2007) 2-Hydroxy-N-arylbenzenesulfonamides as ATP-citrate lyase inhibitors. Bioorg Med Chem Lett 17:3208–3211
Li PX, Yao HJ, Zhang ZQ, Li M, Luo Y, Thompson PR, Gilmour DS, Wang YM (2008) Regulation of p53 target gene expression by peptidylarginine deiminase 4. Mol Cell Biol 28:4745–4758
Li PX, Li M, Lindberg MR, Kennett MJ, Xiong N, Wang YM (2010) PAD4 is essential for antibacterial innate immunity mediated by neutrophil extracellular traps. J Exp Med 207:1853–1862
Li MD, Ruan HB, Hughes ME, Lee JS, Singh JP, Jones SP, Nitabach MN, Yang XY (2013) O-GlcNAc signaling entrains the circadian clock by inhibiting BMAL1/CLOCK ubiquitination. Cell Metab 17:303–310
Li TT, Liu MX, Feng X, Wang Z, Das I, Xu YP, Zhou X, Sun YP, Guan KL, Xiong Y et al (2014) Glyceraldehyde-3-phosphate dehydrogenase is activated by lysine 254 acetylation in response to glucose signal. J Biol Chem 289:3775–3785
Li F, He XD, Ye DW, Lin Y, Yu HX, Yao CF, Huang L, Zhang JN, Wang F, Xu S et al (2015a) NADP(+)-IDH mutations promote hypersuccinylation that impairs mitochondria respiration and induces apoptosis resistance. Mol Cell 60:661–675
Li SS, Swanson SK, Gogol M, Florens L, Washburn MP, Workman JL, Suganuma T (2015b) Serine and SAM responsive complex sesame regulates histone modification crosstalk by sensing cellular metabolism. Mol Cell 60:408–421
Li L, Shi L, Yang SD, Yan RR, Zhang D, Yang JG, He L, Li WJ, Yi X, Sun LY et al (2016) SIRT7 is a histone desuccinylase that functionally links to chromatin compaction and genome stability. Nat Commun 7
Li XJ, Qian X, Lu ZM (2017a) Local histone acetylation by ACSS2 promotes gene transcription for lysosomal biogenesis and autophagy. Autophagy 13:1790–1791
Li XJ, Yu WL, Qian X, Xia Y, Zheng YH, Lee JH, Li W, Lyu JX, Rao G, Zhang XC et al (2017b) Nucleus-translocated ACSS2 promotes gene transcription for lysosomal biogenesis and autophagy. Mol Cell 66:684–697
Li ST, Huang, Shen S, Cai Y, Xing S, Wu G, Jiang Z, Hao Y, Yuan M, Wang N et al (2020) Myc-mediated SDHA acetylation triggers epigenetic regulation of gene expression and tumorigenesis. Nat Metab 2:256–269
Liao YJ, Liu SP, Lee CM, Yen CH, Chuang PC, Chen CY, Tsai TF, Huang SF, Lee YH, Chen YM (2009) Characterization of a glycine N-methyltransferase gene knockout mouse model for hepatocellular carcinoma: implications of the gender disparity in liver cancer susceptibility. Int J Cancer 124:816–826
Lim JH, Lee YM, Chun YS, Chen J, Kim JE, Park JW (2010) Sirtuin 1 modulates cellular responses to hypoxia by deacetylating hypoxia-inducible factor 1 alpha. Mol Cell 38:864–878
Lin RT, Tao R, Gao X, Li TT, Zhou X, Guan KL, Xiong Y, Lei QY (2013) Acetylation stabilizes ATP-citrate lyase to promote lipid biosynthesis and tumor growth. Mol Cell 51:506–518
Lin AP, Abbas S, Kim SW, Ortega M, Bouamar H, Escobedo Y, Varadarajan P, Qin Y, Sudderth J, Schulz E et al (2015) D2HGDH regulates alpha-ketoglutarate levels and dioxygenase function by modulating IDH2. Nat Commun 6:7768
Linder SJ, Mostoslavsky R (2017) Put your mark where your damage is: Acetyl-CoA production by ACLY promotes DNA repair. Mol Cell 67:165–167
Liskiewicz AD, Kasprowska D, Wojakowska A, Polanski K, Lewin-Kowalik J, Kotulska K, Jedrzejowska-Szypulka H (2016) Long-term high fat ketogenic diet promotes renal tumor growth in a rat model of tuberous sclerosis. Sci Rep 6:21807
Liu ZF, Xie ZL, Jones W, Pavlovicz RE, Liu SJ, Yu JH, Li PK, Lin JY, Fuchs JR, Marcucci G et al (2009) Curcumin is a potent DNA hypomethylation agent. Bioorg Med Chem Lett 19:706–709
Liu Y, Liu K, Qin S, Xu C, Min J (2014) Epigenetic targets and drug discovery: part 1: histone methylation. Pharmacol Ther 143:275–294
Liu K, Liu Y, Lau JL, Min J (2015) Epigenetic targets and drug discovery Part 2: histone demethylation and DNA methylation. Pharmacol Ther 151:121–140
Liu PS, Wang HP, Li XY, Chao T, Christen TTS, Christen S, Di Conza G, Cheng WC, Chou CH, Vavakova M et al (2017) Alpha-ketoglutarate orchestrates macrophage activation through metabolic and epigenetic reprogramming. Nat Immunol 18:985–994
Liu K, Li FZ, Sun QQ, Lin N, Han HC, You KQ, Tian F, Mao ZB, Li TT, Tong TJ et al (2019) p53 beta-hydroxybutyrylation attenuates p53 activity. Cell Death Dis 10
Lo WS, Duggan L, Emre NCT, Belotserkovskya R, Lane WS, Shiekhattar R, Berger SL (2001) Snf1—a histone kinase that works in concert with the histone acetyltransferase Gcn5 to regulate transcription. Science 293:1142–1146
Locasale JW (2013) Serine, glycine and one-carbon units: cancer metabolism in full circle. Nat Rev Cancer 13:572–583
Losman JA, Kaelin WG (2013) What a difference a hydroxyl makes: mutant IDH, (R)-2-hydroxyglutarate, and cancer. Genes Dev 27:836–852
Losman JA, Looper RE, Koivunen P, Lee S, Schneider RK, McMahon C, Cowley GS, Root DE, Ebert BL, Kaelin WG (2013) (R)-2-hydroxyglutarate is sufficient to promote leukemogenesis and its effects are reversible. Science 339:1621–1625
Lozoya OA, Martinez-Reyes I, Wang T, Grenet D, Bushel P, Li J, Chandel N, Woychik RP, Santos JH (2018) Mitochondrial nicotinamide adenine dinucleotide reduced (NADH) oxidation links the tricarboxylic acid (TCA) cycle with methionine metabolism and nuclear DNA methylation. PLoS Biol 16:
Lu C, Ward PS, Kapoor GS, Rohle D, Turcan S, Abdel-Wahab O, Edwards CR, Khanin R, Figueroa ME, Melnick A et al (2012) IDH mutation impairs histone demethylation and results in a block to cell differentiation. Nature 483:474
Lu C, Venneti S, Akalin A, Fang F, Ward PS, DeMatteo RG, Intlekofer AM, Chen C, Ye JB, Hameed M et al (2013) Induction of sarcomas by mutant IDH2. Genes Dev 27:1986–1998
Luan HH, Medzhitov R (2016) Food fight: role of itaconate and other metabolites in antimicrobial defense. Cell Metab 24:379–387
Luka Z, Moss F, Loukachevitch LV, Bornhop DJ, Wagner C (2011) Histone demethylase LSD1 Is a folate-binding protein. Biochemistry 50:4750–4756
Luka Z, Pakhomova S, Loukachevitch LV, Calcutt MW, Newcomer ME, Wagner C (2014) Crystal structure of the histone lysine specific demethylase LSD1 complexed with tetrahydrofolate. Protein Sci 23:993–998
Luong A, Hannah VC, Brown MS, Goldstein JL (2000) Molecular characterization of human acetyl-CoA synthetase, an enzyme regulated by sterol regulatory element-binding proteins. J Biol Chem 275:26458–26466
Lv L, Xu YP, Zhao D, Li FL, Wang W, Sasaki N, Jiang Y, Zhou X, Li TT, Guan KL et al (2013) Mitogenic and oncogenic stimulation of K433 acetylation promotes PKM2 protein kinase activity and nuclear localization. Mol Cell 52:340–352
Ly A, Hoyt L, Crowell J, Kim YI (2012) Folate and DNA methylation. Antioxid Redox Signal 17:302–326
Lyssiotis CA, Cantley LC (2014) Acetate fuels the cancer engine. Cell 159:1492–1494
Ma RH, Ji TT, Zhang HF, Dong WQ, Chen XF, Xu PW, Chen DG, Liang XY, Yin XN, Liu YY et al (2018) A Pck1-directed glycogen metabolic program regulates formation and maintenance of memory CD8(+) T cells. Nat Cell Biol 20:21–27
Maddocks ODK, Berkers CR, Mason SM, Zheng L, Blyth K, Gottlieb E, Vousden KH (2013) Serine starvation induces stress and p53-dependent metabolic remodelling in cancer cells. Nature 493:542–546
Maddocks ODK, Labuschagne CF, Adams PD, Vousden KH (2016) Serine metabolism supports the Methionine cycle and DNA/RNA methylation through de novo ATP synthesis in cancer cells. Mol Cell 61:210–221
Maddocks ODK, Athineos D, Cheung EC, Lee P, Zhang T, van den Broek NJF, Mackay GM, Labuschagne CF, Gay D, Kruiswijk F et al (2017) Modulating the therapeutic response of tumours to dietary serine and glycine starvation. Nature 544:372–376
Madeo F, Pietrocola F, Eisenberg T, Kroemer G (2014) Caloric restriction mimetics: towards a molecular definition. Nat Rev Drug Discovery 13:727–740
Madsen AS, Andersen C, Daoud M, Anderson KA, Laursen JS, Chakladar S, Huynh FK, Colaco AR, Backos DS, Fristrup P et al (2016) Investigating the sensitivity of NAD+-dependent sirtuin deacylation activities to NADH. J Biol Chem 291:7128–7141
Maher EA, Marin-Valencia I, Bachoo RM, Mashimo T, Raisanen J, Hatanpaa KJ, Jindal A, Jeffrey FM, Choi C, Madden C et al (2012) Metabolism of [U-13 C]glucose in human brain tumors in vivo. NMR Biomed 25:1234–1244
Mahmood N, Rabbani SA (2019) DNA methylation readers and cancer: mechanistic and therapeutic applications. Front Oncol 9
Maile T, Kwoczynski S, Katzenberger RJ, Wassarman DA, Sauer F (2004) TAF1 activates transcription by phosphorylation of serine 33 in histone H2B. Science 304:1010–1014
Mann BS, Johnson JR, Cohen MH, Justice R, Pazdur R (2007) FDA approval summary: vorinostat for treatment of advanced primary cutaneous T-cell lymphoma. Oncologist 12:1247–1252
Manuyakorn A, Paulus R, Farrell J, Dawson NA, Tze S, Cheung-Lau G, Hines OJ, Reber H, Seligson DB, Horvath S et al (2010) Cellular histone modification patterns predict prognosis and treatment response in resectable pancreatic adenocarcinoma: results from RTOG 9704. J Clin Oncol 28:1358–1365
Marcucci G, Silverman L, Eller M, Lintz L, Beach CL (2005) Bioavailability of azacitidine subcutaneous versus intravenous in patients with the myelodysplastic syndromes. J Clin Pharmacol 45:597–602
Mardis ER, Ding L, Dooling DJ, Larson DE, McLellan MD, Chen K, Koboldt DC, Fulton RS, Delehaunty KD, McGrath SD et al (2009) Recurring mutations found by sequencing an acute myeloid leukemia genome. N Engl J Med 361:1058–1066
Marjon K, Cameron MJ, Quang P, Clasquin MF, Mandley E, Kunii K, Mcvay M, Choe S, Kernytsky A, Gross S et al (2016) MTAP deletions in cancer create vulnerability to targeting of the MAT2A/PRMT5/RIOK1 axis. Cell Rep 15:574–587
Markham GD, Pajares MA (2009) Structure-function relationships in methionine adenosyltransferases. Cell Mol Life Sci 66:636–648
Martinez Calejman C, Trefely S, Entwisle SW, Luciano A, Jung SM, Hsiao W, Torres A, Hung CM, Li H, Snyder NW et al (2020) mTORC2-AKT signaling to ATP-citrate lyase drives brown adipogenesis and de novo lipogenesis. Nat Commun 11:575
Martinez-Chantar ML, Vazquez-Chantada M, Ariz U, Martinez N, Varela M, Luka Z, Capdevila A, Rodriguez J, Aransay AM, Matthiesen R et al (2008) Loss of the glycine N-methyltransferase gene leads to steatosis and hepatocellular carcinoma in mice. Hepatology 47:1191–1199
Mashimo T, Pichumani K, Vemireddy V, Hatanpaa KJ, Singh DK, Sirasanagandla S, Nannepaga S, Piccirillo SG, Kovacs Z, Foong C et al (2014) Acetate is a bioenergetic substrate for human glioblastoma and brain metastases. Cell 159:1603–1614
Tanikawa C, Espinosa M, Suzuki A, Masuda K, Yamamoto K, Tsuchiya E, Ueda K, Daigo Y, Nakamura Y, Matsuda, K (2012) Regulation of histone modification and chromatin structure by the p53-PADI4 pathway. Nat Commun 3
Matsuda S, Adachi J, Ihara M, Tanuma N, Shima H, Kakizuka A, Ikura M, Ikura T, Matsuda T (2016) Nuclear pyruvate kinase M2 complex serves as a transcriptional coactivator of arylhydrocarbon receptor. Nucleic Acids Res 44:636–647
Mattson MP, Chan SL (2003) Calcium orchestrates apoptosis. Nat Cell Biol 5:1041–1043
Mauracher LM, Posch F, Martinod K, Grilz E, Daullary T, Hell L, Brostjan C, Zielinski C, Ay C, Wagner DD et al (2018) Citrullinated histone H3, a biomarker of neutrophil extracellular trap formation, predicts the risk of venous thromboembolism in cancer patients. J Thromb Haemost 16:508–518
Mavrakis KJ, McDonald ER, Schlabach MR, Billy E, Hoffman GR, deWeck A, Ruddy DA, Venkatesan K, Yu JJ, McAllister G et al (2016) Disordered methionine metabolism in MTAP/CDKN2A-deleted cancers leads to dependence on PRMT5. Science 351:1208–1213
Mayya V, Lundgren DH, Hwang SI, Rezaul K, Wu L, Eng JK, Rodionov V, Han DK (2009) Quantitative phosphoproteomic analysis of T cell receptor signaling reveals system-wide modulation of protein-protein interactions. Sci Signal 2:ra46
McBrian MA, Behbahan IS, Ferrari R, Su T, Huang TW, Li K, Hong CS, Christofk HR, Vogelauer M, Seligson DB et al (2013) Histone acetylation regulates intracellular pH. Mol Cell 49:310–321
McCabe MT, Ott HM, Ganji G, Korenchuk S, Thompson C, Van Aller GS, Liu Y, Graves AP, Della Pietra A, Diaz E et al (2012) EZH2 inhibition as a therapeutic strategy for lymphoma with EZH2-activating mutations. Nature 492:108–112
McCarthy N (2013) LEUKAEMIA knowing left from right. Nat Rev Cancer 13:220–220
McDonald OG, Li X, Saunders T, Tryggvadottir R, Mentch SJ, Warmoes MO, Word AE, Carrer A, Salz TH, Natsume S et al (2017) Epigenomic reprogramming during pancreatic cancer progression links anabolic glucose metabolism to distant metastasis. Nat Genet 49:367–376
McGrath J, Trojer P (2015) Targeting histone lysine methylation in cancer. Pharmacol Ther 150:1–22
Mentch SJ, Mehrmohamadi M, Huang L, Liu XJ, Gupta D, Mattocks D, Padilla PG, Ables G, Bamman MM, Thalacker-Mercer AE et al (2015) Histone methylation dynamics and gene regulation occur through the sensing of one-carbon metabolism. Cell Metab 22:861–873
Mertz JA, Conery AR, Bryant BM, Sandy P, Balasubramanian S, Mele DA, Bergeron L, Sims RJ (2011) Targeting MYC dependence in cancer by inhibiting BET bromodomains. Proc Natl Acad Sci USA 108:16669–16674
Merza M, Hartman H, Rahman M, Hwaiz R, Zhang EM, Renstrom E, Luo LT, Morgelin M, Regner S, Thorlacius H (2015) Neutrophil extracellular traps induce trypsin activation, inflammation, and tissue damage in mice with severe acute pancreatitis. Gastroenterology 149:1920–1931
Metallo CM, Gameiro PA, Bell EL, Mattaini KR, Yang JJ, Hiller K, Jewell CM, Johnson ZR, Irvine DJ, Guarente L et al (2012) Reductive glutamine metabolism by IDH1 mediates lipogenesis under hypoxia. Nature 481:380
Mews P, Donahue G, Drake AM, Luczak V, Abel T, Berger SL (2017) Acetyl-CoA synthetase regulates histone acetylation and hippocampal memory. Nature 546:381–386
Mi WY, Gu YC, Han CF, Liu HY, Fan QO, Zhang XL, Cong Q, Yu WG (2011) O-GlcNAcylation is a novel regulator of lung and colon cancer malignancy. Biochim Biophys Acta 1812:514–519
Michelakis ED, Sutendra G, Dromparis P, Webster L, Haromy A, Niven E, Maguire C, Gammer TL, Mackey JR, Fulton D et al (2010) Metabolic modulation of glioblastoma with dichloroacetate. Sci Transl Med 2:31ra34
Michelucci A, Cordes T, Ghelfi J, Pailot A, Reiling N, Goldmann O, Binz T, Wegner A, Tallam A, Rausell A et al (2013) Immune-responsive gene 1 protein links metabolism to immunity by catalyzing itaconic acid production. Proc Natl Acad Sci USA 110:7820–7825
Mills EL, Ryan DG, Prag HA, Dikovskaya D, Menon D, Zaslona Z, Jedrychowski MP, Costa ASH, Higgins M, Hams E et al (2018) Itaconate is an anti-inflammatory metabolite that activates Nrf2 via alkylation of KEAP1. Nature 556:113–117
Monti M, De Rosa V, Iommelli F, Carriero MV, Terlizzi C, Camerlingo R, Belli S, Fonti R, Di Minno G, and Del Vecchio S (2018) Neutrophil extracellular traps as an adhesion substrate for different tumor cells expressing RGD-binding integrins. Int J Mol Sci 19
Morrish F, Noonan J, Perez-Olsen C, Gafken PR, Fitzgibbon M, Kelleher J, VanGilst M, Hockenbery D (2010) Myc-dependent mitochondrial generation of acetyl-CoA contributes to fatty acid biosynthesis and histone acetylation during cell cycle entry. J Biol Chem 285:36267–36274
Moscarello MA, Mastronardi FG, Wood DD (2007) The role of citrullinated proteins suggests a novel mechanism in the pathogenesis of multiple sclerosis. Neurochem Res 32:251–256
Moss TJ, Wallrath LL (2007) Connections between epigenetic gene silencing and human disease. Mutat Res 618:163–174
Mullen AR, Wheaton WW, Jin ES, Chen PH, Sullivan LB, Cheng T, Yang YF, Linehan WM, Chandel NS, DeBerardinis RJ (2012) Reductive carboxylation supports growth in tumour cells with defective mitochondria. Nature 481:385
Murphy MP, O’Neill LAJ (2018) Krebs cycle reimagined: the emerging roles of succinate and itaconate as signal transducers. Cell 174:780–784
Musselman CA, Khorasanizadeh S, Kutateladze TG (2014) Towards understanding methyllysine readout. Biochim Biophys Acta 1839:686–693
Muthusamy T, Cordes T, Handzlik MK, You L, Lim EW, Gengatharan J, Pinto AFM, Badur MG, Kolar MJ, Wallace M et al (2020) Serine restriction alters sphingolipid diversity to constrain tumour growth. Nature
Nair S, Huynh JP, Lampropoulou V, Loginicheva E, Esaulova E, Gounder AP, Boon ACM, Schwarzkopf EA, Bradstreet TR, Edelson BT et al (2018) Irg1 expression in myeloid cells prevents immunopathology during M. tuberculosis infection. J Exp Med 215:1035–1045
Namgaladze D, Zukunft S, Schnutgen F, Kurrle N, Fleming I, Fuhrmann D, Brune B (2018) Polarization of human macrophages by interleukin-4 does not require ATP-citrate lyase. Front Immunol 9
Nanduri J, Semenza GL, Prabhakar NR (2017) Epigenetic changes by DNA methylation in chronic and intermittent hypoxia. Am J Physiol 313:L1096–L1100
Nencioni A, Caffa I, Cortellino S, Longo VD (2018) Fasting and cancer: molecular mechanisms and clinical application. Nat Rev Cancer 18:707–719
Newman JC, Verdin E (2014a) beta-Hydroxybutyrate: much more than a metabolite. Diabetes Res Clin Pract 106:173–181
Newman JC, Verdin E (2014b) Ketone bodies as signaling metabolites. Trends Endocrinol Metab 25:42–52
Newman JC, Verdin E (2017) beta-Hydroxybutyrate: a signaling metabolite. Annu Rev Nutr 37(37):51–76
Nicodeme E, Jeffrey KL, Schaefer U, Beinke S, Dewell S, Chung CW, Chandwani R, Marazzi I, Wilson P, Coste H et al (2010) Suppression of inflammation by a synthetic histone mimic. Nature 468:1119–1123
Nicolas E, Roumillac C, Trouche D (2003) Balance between acetylation and methylation of histone H3 lysine 9 on the E2F-responsive dihydrofolate reductase promoter. Mol Cell Biol 23:1614–1622
Nonnenmacher Y, Hiller K (2018) Biochemistry of proinflammatory macrophage activation. Cell Mol Life Sci 75:2093–2109
Nowak SJ, Corces VG (2004) Phosphorylation of histone H3: a balancing act between chromosome condensation and transcriptional activation. Trends Genet 20:214–220
Obata F, Kuranaga E, Tomioka K, Ming M, Takeishi A, Chen CH, Soga T, Miura M (2014) Necrosis-driven systemic immune response alters SAM metabolism through the FOXO-GNMT axis. Cell Rep 7:821–833
Oermann EK, Wu J, Guan KL, Xiong Y (2012) Alterations of metabolic genes and metabolites in cancer. Semin Cell Dev Biol 23:370–380
Oldham WM, Clish CB, Yang Y, Loscalzo J (2015) Hypoxia-mediated Increases in L-2-hydroxyglutarate coordinate the metabolic response to reductive stress. Cell Metab 22:291–303
Olson LE, Tollkuhn J, Scafoglio C, Krones A, Zhang J, Ohgi KA, Wu W, Taketo MM, Kemler R, Grosschedl R et al (2006) Homeodomain-mediated beta-catenin-dependent switching events dictate cell-lineage determination. Cell 125:593–605
Onakpoya I, Hung SK, Perry R, Wider B, Ernst E (2011) The use of garcinia extract (hydroxycitric acid) as a weight loss supplement: a systematic review and meta-analysis of randomised clinical trials. J Obes 2011:
O’Neill LAJ, Artyomov MN (2019) Itaconate: the poster child of metabolic reprogramming in macrophage function. Nat Rev Immunol 19:273–281
O’Neill LAJ, Pearce EJ (2016) Immunometabolism governs dendritic cell and macrophage function. J Exp Med 213:15–23
Osinalde N, Mitxelena J, Sanchez-Quiles V, Akimov V, Aloria K, Arizmendi JM, Zubiaga AM, Blagoev B, Kratchmarova I (2016) Nuclear phosphoproteomic screen uncovers ACLY as mediator of IL-2-induced proliferation of CD4(+) T lymphocytes. Mol Cell Proteomics 15:2076–2092
Pan PW, Feldman JL, Devries MK, Dong A, Edwards AM, Denu JM (2011) Structure and biochemical functions of SIRT6. J Biol Chem 286:14575–14587
Pan J, Zhao XY, Lin CN, Xu HC, Yin ZL, Liu TZ, Zhang SZ (2014) Immune responsive gene 1, a novel oncogene, increases the growth and tumorigenicity of glioma. Oncol Rep 32:1957–1966
Pan M, Reid MA, Lowman XH, Kulkarni RP, Tran TQ, Liu XJ, Yang Y, Hernandez-Davies JE, Rosales KK, Li HQ et al (2016) Regional glutamine deficiency in tumours promotes dedifferentiation through inhibition of histone demethylation. Nat Cell Biol 18:1090–1101
Papandreou I, Cairns RA, Fontana L, Lim AL, Denko NC (2006) HIF-1 mediates adaptation to hypoxia by actively downregulating mitochondrial oxygen consumption. Cell Metab 3:187–197
Papathanassiu AE, Ko JH, Imprialou M, Bagnati M, Srivastava PK, Vu HA, Cucchi D, McAdoo SP, Ananieva EA, Mauro C et al (2017) BCAT1 controls metabolic reprogramming in activated human macrophages and is associated with inflammatory diseases. Nat Commun 8
Park J, Chen Y, Tishkoff DX, Peng C, Tan MJ, Dai LZ, Xie ZY, Zhang Y, Zwaans BMM, Skinner ME et al (2013) SIRT5-mediated lysine desuccinylation impacts diverse metabolic pathways. Mol Cell 50:919–930
Parker SJ, Metallo CM (2016) Chasing one-carbon units to understand the role of serine in epigenetics. Mol Cell 61:185–186
Parker CW, Kern M, Eisen HN (1962) Polyfunctional dinitrophenyl haptens as reagents for elicitation of immediate type allergic skin responses. J Exp Med 115:789–801
Parsa S, Ortega-Molina A, Ying H-Y, Jiang M, Teater M, Wang J, Zhao C, Reznik E, Pasion JP, Kuo D et al (2020) The serine hydroxymethyltransferase-2 (SHMT2) initiates lymphoma development through epigenetic tumor suppressor silencing. Nature Cancer 1:653–664
Parsons DW, Jones S, Zhang XS, Lin JCH, Leary RJ, Angenendt P, Mankoo P, Carter H, Siu IM, Gallia GL et al (2008) An integrated genomic analysis of human glioblastoma multiforme. Science 321:1807–1812
Pastor WA, Aravind L, Rao A (2013) TETonic shift: biological roles of TET proteins in DNA demethylation and transcription. Nat Rev Mol Cell Biol 14:341–356
Patel MS, Nemeria NS, Furey W, Jordan F (2014) The pyruvate dehydrogenase complexes: structure-based function and regulation. J Biol Chem 289:16615–16623
Pavlova NN, Thompson CB (2016) The emerging hallmarks of cancer metabolism. Cell Metab 23:27–47
Pearce NJ, Yates JW, Berkhout TA, Jackson B, Tew D, Boyd H, Camilleri P, Sweeney P, Gribble AD, Shaw A et al (1998) The role of ATP citrate-lyase in the metabolic regulation of plasma lipids–hypolipidaemic effects of SB-204990, a lactone prodrug of the potent ATP citrate-lyase inhibitor SB-201076. Biochem J 334:113–119
Pearce EL, Walsh MC, Cejas PJ, Harms GM, Shen H, Wang LS, Jones RG, Choi YW (2009) Enhancing CD8 T-cell memory by modulating fatty acid metabolism. Nature 460:103–U118
Perez-Chacon G, Astudillo AM, Balgoma D, Balboa MA, Balsinde J (2009) Control of free arachidonic acid levels by phospholipases A2 and lysophospholipid acyltransferases. Biochim Biophys Acta 1791:1103–1113
Pietrocola F, Galluzzi L, Bravo-San Pedro JM, Madeo F, Kroemer G (2015) Acetyl Coenzyme A: a central metabolite and second messenger. Cell Metab 21:805–821
Pineiro M, Gonzalez PJ, Hernandez F, Palacian E (1991) Interaction of RNA polymerase II with acetylated nucleosomal core particles. Biochem Biophys Res Commun 177:370–376
Pineiro M, Hernandez F, Palacian E (1992) Succinylation of histone amino groups facilitates transcription of nucleosomal cores. Biochim Biophys Acta 1129:183–187
Poillet-Perez L, Xie XQ, Zhan L, Yang Y, Sharp DW, Hu ZS, Su XY, Maganti A, Jiang C, Lu WY et al (2018) Autophagy maintains tumour growth through circulating arginine. Nature 563:569–573
Poli V, Fagnocchi L, Fasciani A, Cherubini A, Mazzoleni S, Ferrillo S, Miluzio A, Gaudioso G, Vaira V, Turdo A et al (2018) MYC-driven epigenetic reprogramming favors the onset of tumorigenesis by inducing a stem cell-like state (vol 9, 1024, 2018). Nat Commun 9
Polletta L, Vernucci E, Carnevale I, Arcangeli T, Rotili D, Palmerio S, Steegborn C, Nowak T, Schutkowski M, Pellegrini L et al (2015) SIRT5 regulation of ammonia-induced autophagy and mitophagy. Autophagy 11:253–270
Poole CJ, van Riggelen J (2017) MYC-master regulator of the cancer epigenome and transcriptome. Genes (Basel) 8
Prickaerts P, Adriaens ME, van den Beucken T, Koch E, Dubois L, Dahlmans VEH, Gits C, Evelo CTA, Chan-Seng-Yue M, Wouters BG et al (2016) Hypoxia increases genome-wide bivalent epigenetic marking by specific gain of H3K27me3. Epigenet Chromatin 9
Prigent C, Dimitrov S (2003) Phosphorylation of serine 10 in histone H3, what for? J Cell Sci 116:3677–3685
Pritzker LB, Joshi S, Gowan JJ, Harauz G, Moscarello MA (2000) Deimination of myelin basic protein. 1. Effect of deimination of arginyl residues of myelin basic protein on its structure and susceptibility to digestion by cathepsin D. Biochemistry 39:5374–5381
Pufulete M, Al-Ghnaniem R, Khushal A, Appleby P, Harris N, Gout S, Emery PW, Sanders TA (2005) Effect of folic acid supplementation on genomic DNA methylation in patients with colorectal adenoma. Gut 54:648–653
Qi H, Ning X, Yu C, Ji X, Jin Y, McNutt MA, Yin Y (2019) Succinylation-dependent mitochondrial translocation of PKM2 promotes cell survival in response to nutritional stress. Cell Death Dis 10:170
Qin W, Qin K, Zhang Y, Jia W, Chen Y, Cheng B, Peng L, Chen N, Liu Y, Zhou W et al (2019) S-glycosylation-based cysteine profiling reveals regulation of glycolysis by itaconate. Nat Chem Biol 15:983–991
Qin W, Zhang Y, Tang H, Liu D, Chen Y, Liu Y, Wang C (2020) Chemoproteomic profiling of itaconation by bioorthogonal probes in inflammatory macrophages. J Am Chem Soc 142:10894–10898
Rardin MJ, He WJ, Nishida Y, Newman JC, Carrico C, Danielson SR, Guo A, Gut P, Sahu AK, Li B et al (2013) SIRT5 regulates the mitochondrial lysine succinylome and metabolic networks. Cell Metab 18:920–933
Reid MA, Dai Z, Locasale JW (2017) The impact of cellular metabolism on chromatin dynamics and epigenetics. Nat Cell Biol 19:1298–1306
Ren J, Singh BN, Huang Q, Li ZF, Gao Y, Mishra P, Hwa YL, Li JP, Dowdy SC, Jiang SW (2011) DNA hypermethylation as a chemotherapy target. Cell Signal 23:1082–1093
Reytor E, Perez-Miguelsanz J, Alvarez L, Perez-Sala D, Pajares MA (2009) Conformational signals in the C-terminal domain of methionine adenosyltransferase I/III determine its nucleocytoplasmic distribution. FASEB J 23:3347–3360
Richie CT, Golden A (2005) Chromosome segregation: Aurora B gets tousled. Curr Biol 15:R379–R382
Richon VM, Emiliani S, Verdin E, Webb Y, Breslow R, Rifkind RA, Marks PA (1998) A class of hybrid polar inducers of transformed cell differentiation inhibits histone deacetylases. Proc Natl Acad Sci USA 95:3003–3007
Riggs MG, Whittaker RG, Neumann JR, Ingram VM (1977) n-Butyrate causes histone modification in HeLa and friend erythroleukaemia cells. Nature 268:462–464
Roediger WE (1982) Utilization of nutrients by isolated epithelial cells of the rat colon. Gastroenterology 83:424–429
Rohle D, Popovici-Muller J, Palaskas N, Turcan S, Grommes C, Campos C, Tsoi J, Clark O, Oldrini B, Komisopoulou E et al (2013) An inhibitor of mutant IDH1 delays growth and promotes differentiation of glioma cells. Science 340:626–630
Romano KA, Martinez-Del Campo A, Kasahara K, Chittim CL, Vivas EI, Amador-Noguez D, Balskus EP, Rey FE (2017) Metabolic, epigenetic, and transgenerational effects of gut bacterial choline consumption. Cell Host Microbe 22(279–290):
Ruan HB, Han XM, Li MD, Singh JP, Qian K, Azarhoush S, Zhao L, Bennett AM, Samuel VT, Wu J et al (2012) O-GlcNAc transferase/host cell factor C1 complex regulates gluconeogenesis by modulating PGC-1 alpha stability. Cell Metab 16:226–237
Ruan HB, Nie YZ, Yang XY (2013) Regulation of protein degradation by O-GlcNAcylation: crosstalk with ubiquitination. Mol Cell Proteomics 12:3489–3497
Ruderman NB, Xu XJ, Nelson L, Cacicedo JM, Saha AK, Lan F, Ido Y (2010) AMPK and SIRT1: a long-standing partnership? Am J Physiol-Endocrinol Metab 298:E751–E760
Ruetz M, Campanello GC, Purchal M, Shen HY, McDevitt L, Gouda H, Wakabayashi S, Zhu JH, Rubin EJ, Warncke K et al (2019) Itaconyl-CoA forms a stable biradical in methylmalonyl-CoA mutase and derails its activity and repair. Science 366:589–593
Rufer AC, Thoma R, Hennig M (2009) Structural insight into function and regulation of carnitine palmitoyltransferase. Cell Mol Life Sci 66:2489–2501
Rzem R, Vincent MF, Schaftingen E, Veiga-da-Cunha M (2007) L-2-Hydroxyglutaric aciduria, a defect of metabolite repair. J Inherit Metab Dis 30:681–689
Sabari BR, Zhang D, Allis CD, Zhao YM (2017) Metabolic regulation of gene expression through histone acylations. Nat Rev Mol Cell Biol 18:90–101
Sadakierska-Chudy A, Filip M (2015) A comprehensive view of the epigenetic landscape. Part II: Histone post-translational modification, nucleosome level, and chromatin regulation by ncRNAs. Neurotox Res 27:172–197
Sakabe K, Wang Z, Hart GW (2010) Beta-N-acetylglucosamine (O-GlcNAc) is part of the histone code. Proc Natl Acad Sci USA 107:19915–19920
Sakata SF, Shelly LL, Ruppert S, Schutz G, Chou JY (1993) Cloning and expression of murine S-adenosylmethionine synthetase. J Biol Chem 268:13978–13986
Saldana-Meyer R, Recillas-Targa F (2011) Transcriptional and epigenetic regulation of the p53 tumor suppressor gene. Epigenetics 6:1068–1077
Salvesen HB, MacDonald N, Ryan A, Jacobs IJ, Lynch ED, Akslen LA, Das S (2001) PTEN methylation is associated with advanced stage and microsatellite instability in endometrial carcinoma. Int J Cancer 91:22–26
Sassone-Corsi P, Mizzen CA, Cheung P, Crosio C, Monaco L, Jacquot S, Hanauer A, Allis CD (1999) Requirement of Rsk-2 for epidermal growth factor-activated phosphorylation of histone H3. Science 285:886–891
Scheppach W, Weiler F (2004) The butyrate story: old wine in new bottles? Curr Opin Clin Nutr Metab Care 7:563–567
Schmitt AM, Schmid S, Rudolph T, Anlauf M, Prinz C, Kloppel G, Moch H, Heitz PU, Komminoth P, Perren A (2009) VHL inactivation is an important pathway for the development of malignant sporadic pancreatic endocrine tumors. Endocr Relat Cancer 16:1219–1227
Schug ZT, Peck B, Jones DT, Zhang QF, Grosskurth S, Alam IS, Goodwin LM, Smethurst E, Mason S, Blyth K et al (2015) Acetyl-CoA synthetase 2 promotes acetate utilization and maintains cancer cell growth under metabolic stress. Cancer Cell 27:57–71
Sciacovelli M, Goncalves E, Johnson TI, Zecchini VR, da Costa ASH, Gaude E, Drubbel AV, Theobald SJ, Abbo SR, Tran MGB et al (2016) Fumarate is an epigenetic modifier that elicits epithelial-to-mesenchymal transition (vol 537, pg 544, 2016). Nature 540
Seligson DB, Horvath S, Shi T, Yu H, Tze S, Grunstein M, Kurdistani SK (2005) Global histone modification patterns predict risk of prostate cancer recurrence. Nature 435:1262–1266
Seligson DB, Horvath S, McBrian MA, Mah V, Yu H, Tze S, Wang Q, Chia D, Goodglick L, Kurdistani SK (2009) Global levels of histone modifications predict prognosis in different cancers. Am J Pathol 174:1619–1628
Serefidou M, Venkatasubramani AV, Imhof A (2019) The impact of one carbon metabolism on histone methylation. Front Genet 10
Shan CL, Elf S, Ji QJ, Kang HB, Zhou L, Hitosugi T, Jin LT, Lin RT, Zhang L, Seo JH et al (2014) Lysine acetylation activates 6-phosphogluconate dehydrogenase to promote tumor growth. Mol Cell 55:552–565
Shen H, Campanello GC, Flicker D, Grabarek Z, Hu J, Luo C, Banerjee R, Mootha VK (2017) The human knockout gene CLYBL connects itaconate to vitamin B12. Cell 171(771–782):
Shi Y, Lan F, Matson C, Mulligan P, Whetstine JR, Cole PA, Casero RA, Shi Y (2004) Histone demethylation mediated by the nuclear amine oxidase homolog LSD1. Cell 119:941–953
Shi Y, Tomic J, Wen F, Shaha S, Bahlo A, Harrison R, Dennis JW, Williams R, Gross BJ, Walker S et al (2010) Aberrant O-GlcNAcylation characterizes chronic lymphocytic leukemia. Leukemia 24:1588–1598
Shi FT, Kim H, Lu W, He Q, Liu D, Goodell MA, Wan M, Songyang Z (2013) Ten-eleven translocation 1 (Tet1) is regulated by O-linked N-acetylglucosamine transferase (Ogt) for target gene repression in mouse embryonic stem cells. J Biol Chem 288:20776–20784
Shi J, Gu JH, Dai CL, Gu J, Jin X, Sun J, Iqbal K, Liu F, Gong CX (2015) O-GlcNAcylation regulates ischemia-induced neuronal apoptosis through AKT signaling. Sci Rep 5:14500
Shi WY, Yang X, Huang B, Shen WH, Liu L (2017) NOK mediates glycolysis and nuclear PDC associated histone acetylation. Front Biosci 22:1792–1804
Shim EH, Livi CB, Rakheja D, Tan J, Benson D, Parekh V, Kho EY, Ghosh AP, Kirkman R, Velu S et al (2014) L-2-hydroxyglutarate: an epigenetic modifier and putative oncometabolite in renal cancer. Cancer Discov 4:1290–1298
Shimazu T, Hirschey MD, Newman J, He W, Shirakawa K, Le Moan N, Grueter CA, Lim H, Saunders LR, Stevens RD et al (2013) Suppression of oxidative stress by beta-hydroxybutyrate, an endogenous histone deacetylase inhibitor. Science 339:211–214
Shin JH, Yang JY, Jeon BY, Yoon YJ, Cho SN, Kang YH, Ryu DH, Hwang GS (2011) (1)H NMR-based metabolomic profiling in mice infected with Mycobacterium tuberculosis. J Proteome Res 10:2238–2247
Shu LM, Khor TO, Lee JH, Boyanapalli SSS, Huang Y, Wu TY, Saw CLL, Cheung KL, Kong ANT (2011) Epigenetic CpG demethylation of the promoter and reactivation of the expression of neurog1 by curcumin in prostate LNCaP cells. Aaps J 13:606–614
Shyh-Chang N, Locasale JW, Lyssiotis CA, Zheng YX, Teo RY, Ratanasirintrawoot S, Zhang J, Onder T, Unternaehrer JJ, Zhu H et al (2013) Influence of threonine metabolism on S-adenosylmethionine and histone methylation. Science 339:222–226
Siedlecki P, Boy RG, Comagic S, Schirrmacher R, Wiessler M, Zielenkiewicz P, Suhai S, Lyko F (2003) Establishment and functional validation of a structural homology model for human DNA methyltransferase 1. Biochem Biophys Res Commun 306:558–563
Singer MS, Kahana A, Wolf AJ, Meisinger LL, Peterson SE, Goggin C, Mahowald M, Gottschling DE (1998) Identification of high-copy disruptors of telomeric silencing in Saccharomyces cerevisiae. Genetics 150:613–632
Singh N, Duenas-Gonzalez A, Lyko F, Medina-Franco JL (2009a) Molecular modeling and molecular dynamics studies of hydralazine with human DNA methyltransferase 1. ChemMedChem 4:792–799
Singh RK, Kabbaj MH, Paik J, Gunjan A (2009b) Histone levels are regulated by phosphorylation and ubiquitylation-dependent proteolysis. Nat Cell Biol 11:925–933
Singh BN, Shankar S, Srivastava RK (2011) Green tea catechin, epigallocatechin-3-gallate (EGCG): mechanisms, perspectives and clinical applications. Biochem Pharmacol 82:1807–1821
Singh JP, Zhang K, Wu J, Yang X (2015) O-GlcNAc signaling in cancer metabolism and epigenetics. Cancer Lett 356:244–250
Sivanand S, Rhoades S, Jiang Q, Lee JV, Benci J, Zhang J, Yuan S, Viney I, Zhao S, Carrer A et al (2017) Nuclear acetyl-CoA production by ACLY promotes homologous recombination. Mol Cell 67(252–265):
Slawson C, Hart GW (2011) O-GlcNAc signalling: implications for cancer cell biology. Nat Rev Cancer 11:678–684
Slawson C, Pidala J, Potter R (2001) Increased N-acetyl-beta-glucosaminidase activity in primary breast carcinomas corresponds to a decrease in N-acetylglucosamine containing proteins. Biochim Biophys Acta 1537:147–157
Smestad J, Erber L, Chen Y, Maher LJ (2018) Chromatin succinylation correlates with active gene expression and is perturbed by defective TCA cycle metabolism. Iscience 2:63–75
Soesanto YA, Luo B, Jones D, Taylor R, Gabrielsen JS, Parker G, McClain DA (2008) Regulation of Akt signaling by O-GlcNAc in euglycemia. Am J Physiol Endocrinol Metab 295:E974–980
Soloaga A, Thomson S, Wiggin GR, Rampersaud N, Dyson MH, Hazzalin CA, Mahadevan LC, Arthur JS (2003) MSK2 and MSK1 mediate the mitogen- and stress-induced phosphorylation of histone H3 and HMG-14. EMBO J 22:2788–2797
Song H, Ma J, Bian ZX, Chen SH, Zhu JB, Wang J, Huang N, Yin MZ, Sun FY, Xu M et al (2019) Global profiling of O-GlcNAcylated and/or phosphorylated proteins in hepatoblastoma. Signal Transduct Target Ther 4
Soria JC, Lee HY, Lee JI, Wang L, Issa JP, Kemp BL, Liu DD, Kurie JM, Mao L, Khuri FR (2002) Lack of PTEN expression in non-small cell lung cancer could be related to promoter methylation. Clin Cancer Res 8:1178–1184
Sorm F, Piskala A, Cihak A, Vesely J (1964) 5-Azacytidine, a new, highly effective cancerostatic. Experientia 20:202–203
Sreedhar A, Wiese EK, Hitosugi T (2020) Enzymatic and metabolic regulation of lysine succinylation. Genes Dis 7:166–171
Stafford JM, Lee CH, Voigt P, Descostes N, Saldana-Meyer R, Yu JR, Leroy G, Oksuz O, Chapman JR, Suarez F et al (2018) Multiple modes of PRC2 inhibition elicit global chromatin alterations in H3K27M pediatric glioma. Sci Adv 4
Steinberg GR, Carling D (2019) AMP-activated protein kinase: the current landscape for drug development. Nat Rev Drug Discov 18:527–551
Stine ZE, Walton ZE, Altman BJ, Hsieh AL, Dang CV (2015) MYC, metabolism, and cancer. Cancer Discov 5:1024–1039
Stram AR, Payne RM (2016) Post-translational modifications in mitochondria: protein signaling in the powerhouse. Cell Mol Life Sci 73:4063–4073
Strelko CL, Lu WY, Dufort FJ, Seyfried TN, Chiles TC, Rabinowitz JD, Roberts MF (2011) Itaconic acid is a mammalian metabolite induced during macrophage activation. J Am Chem Soc 133:16386–16389
Su CH, Shann YJ, Hsu MT (2009) p53 chromatin epigenetic domain organization and p53 transcription. Mol Cell Biol 29:93–103
Sugimoto M, Sakagami H, Yokote Y, Onuma H, Kaneko M, Mori M, Sakaguchi Y, Soga T, Tomita M (2011) Non-targeted metabolite profiling in activated macrophage secretion. Metabolomics 8:624–633
Sugimura T, Birnbaum SM, Winitz M, Greenstein JP (1959) Quantitative nutritional studies with water-soluble, chemically defined diets. VII. Nitrogen balance in normal and tumor-bearing rats following forced feeding. Arch Biochem Biophys 81:439–447
Sulkowski PL, Corso CD, Robinson ND, Scanlon SE, Purshouse KR, Bai H, Liu Y, Sundaram RK, Hegan DC, Fons NR et al (2017) 2-Hydroxyglutarate produced by neomorphic IDH mutations suppresses homologous recombination and induces PARP inhibitor sensitivity. Sci Transl Med 9
Sulkowski PL, Sundaram RK, Oeck S, Corso CD, Liu Y, Noorbakhsh S, Niger M, Boeke M, Ueno D, Kalathil AN et al (2018) Krebs-cycle-deficient hereditary cancer syndromes are defined by defects in homologous-recombination DNA repair. Nat Genet 50:1086–1092
Sulkowski PL, Oeck S, Dow J, Economos NG, Mirfakhraie L, Liu Y, Noronha K, Bao X, Li J, Shuch BM et al (2020) Oncometabolites suppress DNA repair by disrupting local chromatin signalling. Nature 582:586–591
Sun LC, Gao P (2017) Reproducibility in cancer biology: small molecules remain on target for c-Myc. Elife 6
Sun LC, Song LB, Wan QF, Wu GW, Li XH, Wang YH, Wang J, Liu ZJ, Zhong XY, He XP et al (2015) cMyc-mediated activation of serine biosynthesis pathway is critical for cancer progression under nutrient deprivation conditions. Cell Res 25:429–444
Sun LC, Suo CX, Li ST, Zhang HF, Gao P (2018) Metabolic reprogramming for cancer cells and their microenvironment: beyond the Warburg effect. Biochim Biophys Acta 1870:51–66
Sutendra G, Kinnaird A, Dromparis P, Paulin R, Stenson TH, Haromy A, Hashimoto K, Zhang N, Flaim E, Michelakis ED (2014) A nuclear pyruvate dehydrogenase complex is important for the generation of acetyl-CoA and histone acetylation. Cell 158:84–97
Takahashi H, McCaffery JM, Irizarry RA, Boeke JD (2006) Nucleocytosolic acetyl-coenzyme A synthetase is required for histone acetylation and global transcription. Mol Cell 23:207–217
Takawa M, Masuda K, Kunizaki M, Daigo Y, Takagi K, Iwai Y, Cho HS, Toyokawa G, Yamane Y, Maejima K et al (2011) Validation of the histone methyltransferase EZH2 as a therapeutic target for various types of human cancer and as a prognostic marker. Cancer Sci 102:1298–1305
Takusagawa F, Kamitori S, Markham GD (1996) Structure and function of S-adenosylmethionine synthetase: crystal structures of S-adenosylmethionine synthetase with ADP, BrADP, and PPi at 28 angstroms resolution. Biochemistry 35:2586–2596
Tallam A, Perumal TM, Antony PM, Jager C, Fritz JV, Vallar L, Balling R, del Sol A, Michelucci A (2016) Gene regulatory network inference of immunoresponsive gene 1 (IRG1) identifies interferon regulatory factor 1 (IRF1) as its transcriptional regulator in mammalian macrophages. PLoS ONE 11
Tannahill GM, Curtis AM, Adamik J, Palsson-McDermott EM, McGettrick AF, Goel G, Frezza C, Bernard NJ, Kelly B, Foley NH et al (2013) Succinate is an inflammatory signal that induces IL-1 beta through HIF-1 alpha. Nature 496:238–242
Taplin ME, Hussain A, Shore ND, Bradley B, Trojer P, Lebedinsky C, Senderowicz AM, Antonarakis ES (2018) A phase 1b/2 study of CPI-1205, a small molecule inhibitor of EZH2, combined with enzalutamide (E) or abiraterone/prednisone (A/P) in patients with metastatic castration resistant prostate cancer (mCRPC). J Clin Oncol 36
Thalin C, Lundstrom S, Seignez C, Daleskog M, Lundstrom A, Henriksson P, Helleday T, Phillipson M, Wallen H, Demers M (2018). Citrullinated histone H3 as a novel prognostic blood marker in patients with advanced cancer. PLoS ONE 13
Thompson CB (2019) Cancer cell metabolism: reexamining the regulation of anabolic growth in health and disease. Faseb J 33
Tohme S, Yazdani HO, Al-Khafaji AB, Chidi AP, Loughran P, Mowen K, Wang YM, Simmons RL, Huang H, Tsung A (2016) Neutrophil extracellular traps promote the development and progression of liver metastases after surgical stress. Cancer Res 76:1367–1380
Topper MJ, Vaz M, Chiappinelli KB, DeStefano Shields CE, Niknafs N, Yen RC, Wenzel A, Hicks J, Ballew M, Stone M et al (2017) Epigenetic therapy ties MYC depletion to reversing immune evasion and treating lung cancer. Cell 171(1284–1300):
Trojan J, Brieger A, Raedle J, Esteller M, Zeuzem S (2000) 5 ‘-CpG island methylation of the LKB1/STK11 promoter and allelic loss at chromosome 19p13.3 in sporadic colorectal cancer. Gut 47:272–276
Tyrakis PA, Palazon A, Macias D, Lee KL, Phan AT, Velica P, You J, Chia GS, Sim J, Doedens A et al (2016) S-2-hydroxyglutarate regulates CD8(+) T-lymphocyte fate. Nature 540:236–241
Uddin MJ, Joe Y, Kim SK, Jeong SO, Ryter SW, Pae HO, Chung HT (2016) IRG1 induced by heme oxygenase-1/carbon monoxide inhibits LPS-mediated sepsis and pro-inflammatory cytokine production. Cell Mol Immunol 13:170–179
Ulivi P, Mercatali L, Casoni GL, Scarpi E, Bucchi L, Silvestrini R, Sanna S, Monteverde M, Amadori D, Poletti V et al (2013) Multiple marker detection in peripheral blood for NSCLC diagnosis. PLoS ONE 8
van den Berg MA, de Jong-Gubbels P, Kortland CJ, van Dijken JP, Pronk JT, Steensma HY (1996) The two acetyl-coenzyme A synthetases of Saccharomyces cerevisiae differ with respect to kinetic properties and transcriptional regulation. J Biol Chem 271:28953–28959
Van Quickelberghe E, Martens A, Goeminne LJE, Clement L, van Loo G, Gevaert K (2018) Identification of immune-responsive gene 1 (IRG1) as a target of A20. J Proteome Res 17:2182–2191
Vanharanta S, Shu W, Brenet F, Hakimi AA, Heguy A, Viale A, Reuter VE, Hsieh JJ, Scandura JM, Massague J (2013) Epigenetic expansion of VHL-HIF signal output drives multiorgan metastasis in renal cancer. Nat Med 19:50–56
Varambally S, Dhanasekaran SM, Zhou M, Barrette TR, Kumar-Sinha C, Sanda MG, Ghosh D, Pienta KJ, Sewalt RGAB, Otte AP et al (2002) The polycomb group protein EZH2 is involved in progression of prostate cancer. Nature 419:624–629
Varambally S, Cao Q, Mani RS, Shankar S, Wang XS, Ateeq B, Laxman B, Cao XH, Jing XJ, Ramnarayanan K et al (2008) Genomic loss of microRNA-101 leads to overexpression of histone methyltransferase EZH2 in Cancer. Science 322:1695–1699
Vella P, Scelfo A, Jammula S, Chiacchiera F, Williams K, Cuomo A, Roberto A, Christensen J, Bonaldi T, Helin K et al (2013) Tet proteins connect the O-linked N-acetylglucosamine transferase Ogt to chromatin in embryonic stem cells. Mol Cell 49:645–656
Ventura M, Mateo F, Serratosa J, Salaet I, Carujo S, Bachs O, Pujol MJ (2010) Nuclear translocation of glyceraldehyde-3-phosphate dehydrogenase is regulated by acetylation. Int J Biochem Cell Biol 42:1672–1680
Verdin E (2015) NAD(+) in aging, metabolism, and neurodegeneration. Science 350:1208–1213
Verschueren KHG, Blanchet C, Felix J, Dansercoer A, De Vos D, Bloch Y, Van Beeumen J, Svergun D, Gutsche I, Savvides SN et al (2019) Structure of ATP citrate lyase and the origin of citrate synthase in the Krebs cycle. Nature 568:571–575
Vidali G, Boffa LC, Bradbury EM, Allfrey VG (1978) Butyrate suppression of histone deacetylation leads to accumulation of multiacetylated forms of histones H3 and H4 and increased DNase I sensitivity of the associated DNA sequences. Proc Natl Acad Sci USA 75:2239–2243
Volkel P, Dupret B, Le Bourhis X, Angrand PO (2015) Diverse involvement of EZH2 in cancer epigenetics. Am J Transl Res 7:175–193
Vrba L, Junk DJ, Novak P, Futscher BW (2008) p53 induces distinct epigenetic states at its direct target promoters. Bmc Genomics 9
Wagener N, Macher-Goeppinger S, Pritsch M, Husing J, Hoppe-Seyler K, Schirmacher P, Pfitzenmaier J, Haferkamp A, Hoppe-Seyler F, Hohenfellner M (2010) Enhancer of zeste homolog 2 (EZH2) expression is an independent prognostic factor in renal cell carcinoma. Bmc Cancer 10
Wang YP, Lei QY (2018) Metabolic recoding of epigenetics in cancer. Cancer Commun (Lond) 38:25
Wang Y, Wysocka J, Sayegh J, Lee YH, Perlin JR, Leonelli L, Sonbuchner LS, McDonald CH, Cook RG, Dou Y et al (2004) Human PAD4 regulates histone arginine methylation levels via demethylimination. Science 306:279–283
Wang J, Alexander P, Wu L, Hammer R, Cleaver O, McKnight SL (2009) Dependence of mouse embryonic stem cells on threonine catabolism. Science 325:435–439
Wang YJ, Li PX, Wang S, Hu J, Chen XA, Wu JH, Fisher M, Oshaben K, Zhao N, Gu Y et al (2012) Anticancer peptidylarginine deiminase (PAD) inhibitors regulate the autophagy flux and the mammalian target of rapamycin complex 1 activity. J Biol Chem 287:25941–25953
Wang F, Travins J, DeLaBarre B, Penard-Lacronique V, Schalm S, Hansen E, Straley K, Kernytsky A, Liu W, Gliser C et al (2013) Targeted inhibition of mutant IDH2 in leukemia cells induces cellular differentiation. Science 340:622–626
Wang F, Wang K, Xu W, Zhao SM, Ye D, Wang Y, Xu Y, Zhou LS, Chu YW, Zhang CP et al (2017a) SIRT5 desuccinylates and activates pyruvate kinase M2 to block macrophage IL-1 beta production and to prevent DSS-induced colitis in mice. Cell Rep 19:2331–2344
Wang Y, Guo YR, Liu K, Yin Z, Liu R, Xia Y, Tan L, Yang P, Lee JH, Li XJ et al (2017b) KAT2A coupled with the alpha-KGDH complex acts as a histone H3 succinyltransferase. Nature 552:273–277
Wang Y, Jin J, Chung MWH, Feng L, Sun HY, Hao Q (2018a) Identification of the YEATS domain of GAS41 as a pH-dependent reader of histone succinylation. Proc Natl Acad Sci USA 115:2365–2370
Wang YG, Guo YR, Xing DM, Tao YJ, Lu ZM (2018b) Supramolecular assembly of KAT2A with succinyl-CoA for histone succinylation. Cell Discov 4
Wang C, Zhang C, Li X, Shen J, Xu Y, Shi H, Mu X, Pan J, Zhao T, Li M et al (2019a) CPT1A-mediated succinylation of S100A10 increases human gastric cancer invasion. J Cell Mol Med 23:293–305
Wang GX, Meyer JG, Cai WK, Softic S, Li ME, Verdin E, Newgard C, Schilling B, Kahn CR (2019b) Regulation of UCP1 and mitochondrial metabolism in brown adipose tissue by reversible succinylation. Mol Cell 74:844–857
Ward PS, Patel J, Wise DR, Abdel-Wahab O, Bennett BD, Coller HA, Cross JR, Fantin VR, Hedvat CV, Perl AE et al (2010) The common feature of leukemia-associated IDH1 and IDH2 mutations is a neomorphic enzyme activity converting alpha-ketoglutarate to 2-hydroxyglutarate. Cancer Cell 17:225–234
Watson JA, Watson CJ, McCann A, Baugh J (2010) Epigenetics, the epicenter of the hypoxic response. Epigenetics 5:293–296
Weinert BT, Scholz C, Wagner SA, Iesmantavicius V, Su D, Daniel JA, Choudhary C (2013) Lysine succinylation is a frequently occurring modification in prokaryotes and eukaryotes and extensively overlaps with acetylation. Cell Rep 4:842–851
Weiss JM, Davies LC, Karwan M, Ileva L, Ozaki MK, Cheng RYS, Ridnour LA, Annunziata CM, Wink DA, McVicar DW (2018) Itaconic acid mediates crosstalk between macrophage metabolism and peritoneal tumors. J Clin Investig 128:3794–3805
Wellen KE, Hatzivassiliou G, Sachdeva UM, Bui TV, Cross JR, Thompson CB (2009) ATP-citrate lyase links cellular metabolism to histone acetylation. Science 324:1076–1080
Williams NC, O’Neill LAJ (2018) A Role for the Krebs Cycle Intermediate Citrate in Metabolic Reprogramming in Innate Immunity and Inflammation. Front Immunol 9:141
Williams NC, O’Neill LA (2020) ACLY-matizing macrophages to histone modification during immunometabolic reprogramming. Trends Immunol 41:93–94
Williams SC, Karajannis MA, Chiriboga L, Golfinos JG, von Deimling A, Zagzag D (2011) R132H-mutation of isocitrate dehydrogenase-1 is not sufficient for HIF-1 alpha upregulation in adult glioma. Acta Neuropathol 121:279–281
Wise DR, Ward PS, Shay JES, Cross JR, Gruber JJ, Sachdeva UM, Platt JM, DeMatteo RG, Simon MC, Thompson CB (2011) Hypoxia promotes isocitrate dehydrogenase-dependent carboxylation of alpha-ketoglutarate to citrate to support cell growth and viability. Proc Natl Acad Sci USA 108:19611–19616
Witalison EE, Thompson PR, Hofseth LJ (2015) Protein arginine deiminases and associated citrullination: physiological functions and diseases associated with dysregulation. Curr Drug Targets 16:700–710
Wong CC, Qian Y, Yu J (2017) Interplay between epigenetics and metabolism in oncogenesis: mechanisms and therapeutic approaches. Oncogene 36:3359–3374
Wood JG, Rogina B, Lavu S, Howitz K, Helfand SL, Tatar M, Sinclair D (2004) Sirtuin activators mimic caloric restriction and delay ageing in metazoans. Nature 430:686–689
Wu GW, Yuan MQ, Shen SQ, Ma XY, Fang JW, Zhu LB, Sun LC, Liu ZJ, He XP, Huang D et al (2017) Menin enhances c-Myc-mediated transcription to promote cancer progression. Nat Commun 8
Xia S, Lin R, Jin L, Zhao L, Kang HB, Pan Y, Liu S, Qian G, Qian Z, Konstantakou E et al (2017) Prevention of dietary-fat-fueled ketogenesis attenuates BRAF V600E tumor growth. Cell Metab 25:358–373
Xiangyun Y, Xiaomin N, Linping G, Yunhua X, Ziming L, Yongfeng Y, Zhiwei C, Shun L (2017) Desuccinylation of pyruvate kinase M2 by SIRT5 contributes to antioxidant response and tumor growth. Oncotarget 8:6984–6993
Xiao HB, Cao WX, Yin HR, Lin YZ, Ye SH (2001) Influence of L-methionine-deprived total parenteral nutrition with 5-fluorouracil on gastric cancer and host metabolism. World J Gastroenterol 7:698–701
Xiao A, Li H, Shechter D, Ahn SH, Fabrizio LA, Erdjument-Bromage H, Ishibe-Murakami S, Wang B, Tempst P, Hofmann K et al (2009) WSTF regulates the H2A.X DNA damage response via a novel tyrosine kinase activity. Nature 457:57–62
Xiao M, Yang H, Xu W, Ma S, Lin H, Zhu H, Liu L, Liu Y, Yang C, Xu Y et al (2012) Inhibition of alpha-KG-dependent histone and DNA demethylases by fumarate and succinate that are accumulated in mutations of FH and SDH tumor suppressors. Genes Dev 26:1326–1338
Xie Z, Dai J, Dai L, Tan M, Cheng Z, Wu Y, Boeke JD, Zhao Y (2012) Lysine succinylation and lysine malonylation in histones. Mol Cell Proteomics 11:100–107
Xie ZY, Zhang D, Chung DJ, Tang ZY, Huang H, Dai LZ, Qi SK, Li JY, Colak G, Chen Y et al (2016) Metabolic regulation of gene expression by histone lysine beta-hydroxybutyrylation. Mol Cell 62:194–206
Xu Q, Li Y, Gao X, Kang K, Williams JG, Tong L, Liu J, Ji M, Deterding LJ, Tong X et al (2020) HNF4alpha regulates sulfur amino acid metabolism and confers sensitivity to methionine restriction in liver cancer. Nat Commun 11:3978
Xu D, Shao F, Bian X, Meng Y, Liang T, Lu Z (2021) The evolving landscape of noncanonical functions of metabolic enzymes in cancer and other pathologies. Cell Metab 33:33–50
Yadav T, Quivy JP, Almouzni G (2018) Chromatin plasticity: a versatile landscape that underlies cell fate and identity. Science 361:1332–1336
Yan H, Parsons DW, Jin GL, McLendon R, Rasheed BA, Yuan WS, Kos I, Batinic-Haberle I, Jones S, Riggins GJ et al (2009) IDH1 and IDH2 mutations in gliomas. N Engl J Med 360:765–773
Yang J (2019) Sweetly profiling the cysteinome. Nat Chem Biol 15:935–936
Yang X, Qian K (2017) Protein O-GlcNAcylation: emerging mechanisms and functions. Nat Rev Mol Cell Biol 18:452–465
Yang Y, Sauve AA (2016) NAD(+) metabolism: bioenergetics, signaling and manipulation for therapy. Biochim Biophys Acta 1864:1787–1800
Yang M, Vousden KH (2016) Serine and one-carbon metabolism in cancer. Nat Rev Cancer 16:650–662
Yang XY, Zhang FX, Kudlow JE (2002) Recruitment of O-GlcNAc transferase to promoters by corepressor mSin3A: coupling protein O-GlcNAcylation to transcriptional repression. Cell 110:69–80
Yang HY, Yang T, Baur JA, Perez E, Matsui T, Carmona JJ, Lamming DW, Souza-Pinto NC, Bohr VA, Rosenzweig A et al (2007) Nutrient-sensitive mitochondrial NAD(+) levels dictate cell survival. Cell 130:1095–1107
Yang CS, Wang X, Lu G, Picinich SC (2009) Cancer prevention by tea: animal studies, molecular mechanisms and human relevance. Nat Rev Cancer 9:429–439
Yang CF, Sun WY, Cui W, Li XK, Yao J, Jia XY, Li CJ, Wu HJ, Hu ZY, Zou XM (2015) Procoagulant role of neutrophil extracellular traps in patients with gastric cancer. Int J Clin Exp Pathol 8:14075–14086
Yao HJ, Li PX, Venters BJ, Zheng S, Thompson PR, Pugh BF, Wang YM (2008) Histone Arg modifications and p53 regulate the expression of OKL38, a mediator of apoptosis. J Biol Chem 283:20060–20068
Ye D, Ma SH, Xiong Y, Guan KL (2013) R-2-hydroxyglutarate as the key effector of IDH mutations promoting oncogenesis. Cancer Cell 23:274–276
Ye D, Guan KL, Xiong Y (2018) Metabolism, activity, and targeting of D- and L-2-hydroxyglutarates. Trends Cancer 4:151–165
Yen K, Travins J, Wang F, David MD, Artin E, Straley K, Padyana A, Gross S, DeLaBarre B, Tobin E et al (2017) AG-221, a first-in-class therapy targeting acute myeloid leukemia harboring oncogenic IDH2 mutations. Cancer Discov 7:478–493
Yi CH, Pan H, Seebacher J, Jang IH, Hyberts SG, Heffron GJ, Vander Heiden MG, Yang R, Li F, Locasale JW et al (2011) Metabolic regulation of protein N-alpha-acetylation by Bcl-xL promotes cell survival. Cell 146:607–620
Yoo J, Medina-Franco JL (2011) Homology modeling, docking and structure-based pharmacophore of inhibitors of DNA methyltransferase. J Comput Aided Mol Des 25:555–567
Yoo CB, Jeong S, Egger G, Liang GN, Phiasivongsa P, Tang CL, Redkar S, Jones PA (2007) Delivery of 5-aza-2 ‘-deoxycytidine to cells using oligodeoxynucleotides. Cancer Res 67:6400–6408
Yu J, Yu J, Rhodes DR, Tomlins SA, Cao X, Chen G, Mehra R, Wang X, Ghosh D, Shah RB et al (2007) A polycomb repression signature in metastatic prostate cancer predicts cancer outcome. Cancer Res 67:10657–10663
Yu XH, Zhang DW, Zheng XL, Tang CK (2019) Itaconate: an emerging determinant of inflammation in activated macrophages. Immunol Cell Biol 97:134–141
Yuan H, Han Y, Wang X, Li N, Liu Q, Yin Y, Wang H, Pan L, Li L, Song K et al (2020) SETD2 Restricts Prostate Cancer Metastasis by Integrating EZH2 and AMPK Signaling Pathways. Cancer Cell.
Yun MY, Wu J, Workman JL, Li B (2011) Readers of histone modifications. Cell Res 21:564–578
Yuzhalin AE (2019) Citrullination in cancer. Cancer Res 79:1274–1284
Yuzhalin AE, Gordon-Weeks AN, Tognoli ML, Jones K, Markelc B, Konietzny R, Fischer R, Muth A, O’Neill E, Thompson PR et al (2018) Colorectal cancer liver metastatic growth depends on PAD4-driven citrullination of the extracellular matrix. Nat Commun 9
Zaidi N, Swinnen JV, Smans K (2012) ATP-citrate lyase: a key player in cancer metabolism. Cancer Res 72:3709–3714
Zamierowski MM, Wagner C (1977) Identification of folate binding proteins in rat liver. J Biol Chem 252:933–938
Zeng JD, Wu WKK, Wang HY, Li XX (2019) Serine and one-carbon metabolism, a bridge that links mTOR signaling and DNA methylation in cancer. Pharmacol Res 149
Zhang ZH, Tan MJ, Xie ZY, Dai LZ, Chen Y, Zhao YM (2011) Identification of lysine succinylation as a new post-translational modification. Nat Chem Biol 7:58–63
Zhang Q, Liu X, Gao W, Li P, Hou J, Li J, Wong J (2014) Differential regulation of the ten-eleven translocation (TET) family of dioxygenases by O-linked beta-N-acetylglucosamine transferase (OGT). J Biol Chem 289:5986–5996
Zhang W, Zhang SL, Hu X, Tam KY (2015) Targeting tumor metabolism for cancer treatment: is pyruvate dehydrogenase kinases (PDKs) a viable anticancer target? Int J Biol Sci 11:1390–1400
Zhang D, Tang ZY, Huang H, Zhou GL, Cui C, Weng YJ, Liu WC, Kim S, Lee S, Perez-Neut M et al (2019a) Metabolic regulation of gene expression by histone lactylation. Nature 574:575–580
Zhang XR, Cao RL, Niu JR, Yang SM, Ma HD, Zhao S, Li HT (2019b) Molecular basis for hierarchical histone de-beta-hydroxybutmlation by SIRT3. Cell Discov 5
Zhang HF, Tang K, Ma JW, Zhou L, Liu JC, Zeng LP, Zhu LY, Xu PW, Chen J, Wei KK et al (2020) Ketogenesis-generated beta-hydroxybutyrate is an epigenetic regulator of CD8(+) T-cell memory development. Nat Cell Biol 22:18–25
Zhao K, Miao H (2020) Targeting metabolic/epigenetic pathways: a potential strategy for cancer therapy in diffuse intrinsic pontine gliomas. Signal Transduct Target Ther 5:226
Zhao G, Winkler ME (1996) A novel alpha-ketoglutarate reductase activity of the serA-encoded 3-phosphoglycerate dehydrogenase of Escherichia coli K-12 and its possible implications for human 2-hydroxyglutaric aciduria. J Bacteriol 178:232–239
Zhao S, Lin Y, Xu W, Jiang W, Zha Z, Wang P, Yu W, Li Z, Gong L, Peng Y et al (2009) Glioma-derived mutations in IDH1 dominantly inhibit IDH1 catalytic activity and induce HIF-1alpha. Science 324:261–265
Zhao S, Xu W, Jiang W, Yu W, Lin Y, Zhang T, Yao J, Zhou L, Zeng Y, Li H et al (2010) Regulation of cellular metabolism by protein lysine acetylation. Science 327:1000–1004
Zhao S, Torres A, Henry RA, Trefely S, Wallace M, Lee JV, Carrer A, Sengupta A, Campbell SL, Kuo YM et al (2016) ATP-citrate lyase controls a glucose-to-acetate metabolic switch. Cell Rep 17:1037–1052
Zhao D, Li YY, Xiong XZ, Chen ZL, Li HT (2017) YEATS domain-A histone acylation reader in health and disease. J Mol Biol 429:1994–2002
Zhao S, Zhang X, Li H (2018) Beyond histone acetylation-writing and erasing histone acylations. Curr Opin Struct Biol 53:169–177
Zhao S, Jang C, Liu J, Uehara K, Gilbert M, Izzo L, Zeng XF, Trefely S, Fernandez S, Carrer A et al (2020) Dietary fructose feeds hepatic lipogenesis via microbiota-derived acetate. Nature 579:586–591
Zheng QF, Maksimovic I, Upad A, David Y (2020) Non-enzymatic covalent modifications: a new link between metabolism and epigenetics. Protein Cell 11:401–416
Zhou LS, Wang F, Sun RQ, Chen XF, Zhang ML, Xu Q, Wang Y, Wang SW, Xiong Y, Guan KL et al (2016) SIRT5 promotes IDH2 desuccinylation and G6PD deglutarylation to enhance cellular antioxidant defense. EMBO Rep 17:811–822
Zippo A, De Robertis A, Serafini R, Oliviero S (2007) PIM1-dependent phosphorylation of histone H3 at serine 10 is required for MYC-dependent transcriptional activation and oncogenic transformation. Nat Cell Biol 9:932
Zuber J, Shi JW, Wang E, Rappaport AR, Herrmann H, Sison EA, Magoon D, Qi J, Blatt K, Wunderlich M et al (2011) RNAi screen identifies Brd4 as a therapeutic target in acute myeloid leukaemia. Nature 478:524
ACKNOWLEDGEMENTS
This work was supported by National Natural Science Foundation of China (91957203, 81930083, 81874060, 81821001), National Key R&D Program of China (2018YFA0107103, 2018YFA0800300, 2017YFA0205600), the Chinese Academy of Sciences (XDB39000000), the Program for Guangdong Introducing Innovative and Entrepreneurial Teams (2017ZT07S054), Outstanding Scholar Program of Guangzhou Regenerative Medicine and Health Guangdong Laboratory (2018GZR110102001), and the Fundamental Research Funds for the Central Universities (YD2070002008, 2020ZYGXZR038).
ABBREVIATIONS
AcAc, acetoacetate; ACACA, acetyl-CoA carboxylase 1; Ac-CoA, acetyl-CoA or acetyl coenzyme A; ACLY, ATP citrate lyase; ACOD1/CAD, aconitate decarboxylase 1; ACOX1, acyl-CoA oxidase 1; ACSSs, acyl-CoA synthetase short-chain family members; ADD, ATRX-DNMT3-DNMT3L; ADM, acinar-to-ductal metaplasia; α-KG, α-ketoglutarate; α-KGDH, α-ketoglutarate dehydrogenase complex; AKT, AKT serine/threonine kinase; AML, acute myeloid leukemia; AMPK, AMP-activated protein kinase; ATM, ataxia telangiectasia mutated; BAH, bromo adjacent homology; BAZ2A/B, bromodomain adjacent to zinc finger domain 2A/B; BCAAs, branched amino acids; BCAT2, branched-chain amino acid transaminase 2; Bcl-xL, B-cell lymphoma-2-like 1; BMDM, bone marrow-derived macrophage; CBP, CREB-binding protein; CD, chromodomain; ChIP-qPCR, chromatin immunoprecipitation (ChIP) coupled with quantitative PCR; COX-2, cyclooxygenase 2; CPT1A, carnitine palmitoyltransferase 1A; CRTC2, CREB-regulated transcription coactivator 2; CS, citrate synthase; CTCL, cutaneous T-cell lymphoma; CYP1A1, cytochrome P450 1A1; DCA, dichloroacetate; DCD, double chromodomain; D2HGDH, D-2-hydroxyglutarate dehydrogenase; DIPGs, diffuse intrinsic pontine gliomas; DLAT, dihydrolipoamide S-acetyltransferase; DLBCL, diffuse large B-cell lymphoma; DLD, dihydrolipoamide dehydrogenase; DNMTs, DNA methyltransferases; DOT1L, disruptor of telomeric silencing 1-like; DPF, double PHD finger; DSS, dextran sodium sulfate; EMT, epithelial-to-mesenchymal-transition; ESCs, embryonic stem cells; EZH2, enhancer of zeste homolog 2; FAD, flavin adenine dinucleotide; FASN, fatty acid synthase; FDA, US Food and Drug Administration; FH, fumarate hydratase; FIH, factor inhibiting HIF; FL, follicular lymphoma; FOXO1, forkhead box O1; FOXO3a, forkhead box O3A; Fum, fumarate; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; GBM, glioblastomas; GC/MS, gas chromatography mass spectrometry; GCN5, general control of amino acid synthesis 5-like 2 (Yeast); GDH, glutamate dehydrogenase; GLDC, glycine dehydrogenase; GLS, glutaminase; GNAT, G protein subunit alpha transducin; GNMT, Glycine N-methyltransferase; GSH, glutathione; HAT-like domain, histone acetyltransferase-like domain; HATs, histone acetylatransferases; HBP, hexosamine biosynthetic pathway; H2BpS36, phosphorylation of histone H2B at serine 36; HDACs, histone deacetylases; HDMs, histone demethylases; HDR, DNA homology-dependent repair; 2-HG, 2-hydroxyglutarate; HIF-1α, hypoxia-inducible factor 1α; HIFs, hypoxia-inducible factors; H3K4, histone H3 lysine 4; H3.3K27M, histone H3.3 lysine 27-to-methionine; H3K79, histone H3 lysine 79; HLRCC, hereditary leiomyomatosis and renal cell cancer; 5’-hmC, DNA 5’-hydroxymethylcytosine; HMGCS2, 3-hydroxy-3-methylglutaryl-CoA synthase 2; HMTs, histone methytransferases; HO-1, heme oxygenase-1; H3pS10, phosphorylation of histone H3 at serine 10; HR, homologous recombination; hTERT, human telomerase reverse transcriptase; IDH, isocitrate dehydrogenase; IFN-γ, interferon-γ; IGF-1, insulin-like growth factor-1; IHC, immunohistochemistry; IKK-α, IκB kinase-α; IL-1β, interleukin-1β; IL-4, interleukin-4; iMEF, immortalized mouse embryonic fibroblasts; IRF1, interferon regulatory factor 1; IRG1, immune-responsive gene 1; ITalk, itaconate-alkyne; JHDM/Jmj-KDM, Jumonji C (JmjC) domain-containing demethyalses; Kacetyl, lysine acetylation; KAT2A, lysine acetyl transferase 2A; KATs, lysine acetylatransferases; Kbhb, lysine β-hydroxybutyrylation; Kla, lysine lactylation; KRAS, Kirsten rat sarcoma 2 viral oncogene homolog; Ksucc, lysine succinylation; LCAD, long-chain acyl-CoA dehydrogenase; LC-MS, liquid chromatography-mass spectrometry; LDHA: Lactate dehydrogenase A; L2HGDH, L-2-hydroxyglutarate dehydrogenase; LKB1, liver kinase B1; LPS, lipopolysaccharide; LSD1, lysine-specific histone demethylase 1; MAL, MyD88-adaptor-like protein; MATIα, methionine adenosyltransferase Iα; MATIIα, methionine adenosyltransferase IIα; MBPs, methyl-binding proteins; MBT, malignant brain tumor; 5mC, 5-methylcytosine; MCM, methylmalonyl-CoA mutase; MDH, malate dehydrogenase; MDS, myelodysplastic syndrome; MeCP2, methyl-CpG-binding protein 2; mESCs, mouse embryonic stem cells; MET, methionine; me-THF, 5,10-methylene-THF; MLL, mixed-lineage leukemia; MPC, mitochondrial pyruvate carrier; MPO, myeloperoxidase; MSK1 and MSK2, mitogen- and stress-activated protein kinase 1 and 2; MTAP, methylthioadenosine phosphorylase; MTB, Mycobacterium tuberculosis; mTHF, 5-methyltetrahydrofolate; mTOR, mechanistic target of rapamycin kinase; MyD88, myeloid differentiation primary-response protein 88; MYST, Moz, Ybf2/Sas3, Sas2, and Tip60; NAD+, nicotinamide adenine dinucleotide; NADPH, nicotinamide adenine dinucleotide phosphate, reduced form; NAM, nicotinamide; NAMPT, NAM phosphoribosyltransferase; Ndufs4, NADH dehydrogenase [ubiquinone] iron-sulfur protein 4; NE, neutrophil elastase; NETs, neutrophil extracellular traps; NFATC1, nuclear factor of activated T cells cytoplasmic 1; NF-κB, nuclear factor-κB; NLS, nuclear localization signal; NMN, nicotinamide mononucleotide; NOK, novel oncogene with kinase-domain; OAA, oxaloacetate; 2-OG. 2-oxoglutarate; OGA, O-GlcNAcase; O-GlcNAcylation, O-linked β-N-acetylglucosamine glycosylation; OGT, O-GlcNAc transferase; β-OHB, β-hydroxybutyrate; OKL38, oxidative stress-induced growth inhibitor 1; oxPPP, oxidative pentose phosphate pathway; PAD or PADI, peptidyl-arginine deiminase; PARP, poly (ADP-ribose) polymerase; PCAF, p300/CBP-associated factor; p300/CBP, E1A-binding protein p300/CREB-binding protein; PCK1, phosphoenolpyruvate carboxykinase 1; PDAC, pancreatic ductal adenocarcinoma; PDC, pyruvate dehydrogenase complex; PDH, pyruvate dehydrogenase; PDHX, pyruvate dehydrogenase complex, component X; PDK, pyruvate dehydrogenase kinase; PDP, pyruvate dehydrogenase phosphatase; PEP, phosphoenolpyruvate; PEPCK, phosphoenolpyruvate carboxykinase; 3-PG, 3-phosphoglycerate; PGC1α, PPARG coactivator 1α; 6PGD, 6-phosphogluconate dehydrogenase; PHD2, prolyl hydroxylase domain-containing proteins, also known as Egl nine homologs (EGLN2); PI3K, phosphoinositide 3-kinase; PIP2, phosphatidylinositol-(4,5)-bisphosphate; PIP3, phosphatidylinositol-(3,4,5)-trisphosphate; PKM2, pyruvate kinase isozyme M2; PLK1, polo-like kinase 1; Pol II, RNA polymerase II; PPAR, peroxisome proliferator-activated receptor; pResMϕ, peritoneal tissue–resident macrophages; PRMT5, protein arginine N-methyltransferase 5; PSAT1, phosphoserine aminotransferase 1; PTCL, peripheral T-cell lymphoma; P-TEFb, positive transcription elongation factor b; PTEN, phosphatase and tensin homolog; PTMs, posttranslational modifications; PTPRM, protein tyrosine phosphatase receptor type M; RCC, renal cell carcinoma; RSK2, ribosomal S6 kinase 2; SAH, S-adenosylhomocysteine; SAHA, suberanilohydroxamic acid; SAHH, S-adenosyl homocysteinehydrolase; SAM, S-adenosyl methionine; SASH1, SAM and SH3 domain-containing protein 1; SDH, succinate dehydrogenase; SDHA, succinate dehydrogenase complex, subunit A; SDH PGL/PCC, succinate dehydrogenase-related hereditary paraganglioma and pheochromocytoma; SESAME, Serine-responsive SAM-containing metabolic enzyme complex; SETD2, SET domain-containing 2; SETDB1/2, SET domain bifurcated 1/2; SFK, Src-family kinase; SHMTs, serine hydroxymethyltransferases; SKP2, S-phase kinase-associated protein 2; SREBF, sterol regulatory element-binding transcription factor; SQLE, squalene epoxidase; STAC, sirtuin-activating compound; TAD domain, transcription activation domain; TAMs, tumor-associated macrophages; TDH, threonine dehydrogenase; Teff cell, effector T cell; TETs, Ten-eleven translocation enzymes; TFEB, transcription factor EB; THF, tetrahydrofolate; TIP60, tat-interacting protein, also known as histone acetyltransferase KAT5; TIRAP, TIR domain-containing adaptor protein; TLR4, Toll-like receptor 4; Tmem cell, memory T cell; Tn cell, Naïve T cell; TRAM, TRIF-related adaptor molecule; TRIF, TIR-domain-containing adaptor protein-inducing IFNB; TSA, trichostatin A; TTD, tandem Tudor domain; UCP1, Mitochondrial uncoupling protein 1; UDP, uridine diphosphate glucose; UDP-GlcNAc, uridine diphosphate GlcNAc; VDAC3, voltage-dependent anion channel 3; VHL, von Hippel-Lindau tumor suppressor; VTE, venous thromboembolism; zn-CW, zinc finger CW.
COMPLIANCE WITH ETHICS GUIDELINES
Linchong Sun, Huafeng Zhang, and Ping Gao declare that they have no conflict of interest. This article does not contain any studies with human or animal subjects performed by any of the authors.
Author information
Authors and Affiliations
Corresponding authors
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Sun, L., Zhang, H. & Gao, P. Metabolic reprogramming and epigenetic modifications on the path to cancer. Protein Cell 13, 877–919 (2022). https://doi.org/10.1007/s13238-021-00846-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13238-021-00846-7