
Abstract
Obesity and aging are two important epidemic factors for metabolic syndrome and many other health issues, which contribute to devastating diseases such as cardiovascular diseases, stroke and cancers. The brain plays a central role in controlling metabolic physiology in that it integrates information from other metabolic organs, sends regulatory projections and orchestrates the whole-body function. Emerging studies suggest that brain dysfunction in sensing various internal cues or processing external cues may have profound effects on metabolic and other physiological functions. This review highlights brain dysfunction linked to genetic mutations, sex, brain inflammation, microbiota, stress as causes for whole-body pathophysiology, arguing brain dysfunction as a root cause for the epidemic of aging and obesity-related disorders. We also speculate key issues that need to be addressed on how to reveal relevant brain dysfunction that underlines the development of these disorders and diseases in order to develop new treatment strategies against these health problems.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
With the development of life quality, people eat better and live longer. Obesity and population aging have now become a great burden to people in modern society. According to the World Health Organization, more than 650 million adults worldwide are obese; till 2050, two billion people would be older than 65 years of age (Organization, 2015). Given the increasing prevalence of obesity and aging-associated dysfunctions in humans, it is of most importance to understand the mechanism underlying them.
Generally, metabolic dysfunction results from both intrinsic factors and environmental changes. Whole-genome sequencing and genome-wide association studies (GWAS) helped identify many susceptible loci associated with human obesity (Locke et al., 2015); the sexual dimorphism in metabolic regulation has long been emphasized in clinic research (Palmer and Clegg, 2015). As to environmental changes, gut-microbiota imbalance, chronic stress and micro-environmental inflammation have been associated with the obesity pandemic (Flier, 2004; Sominsky and Spencer, 2014; Razzoli et al., 2017; Tomiyama, 2018; Cani et al., 2019).
The brain plays a significant role in energy balance. As early as in 1849, Claude Bernard have observed that stimulation of the base of the fourth ventricle in rabbits caused a dramatic rise in blood glucose (Ruud et al., 2017). However, it was not until recent years that people began to discover neurobiological mechanisms in metabolic regulation with the help of powerful scientific tools. Now people have discovered multiple neural pathways participating in different aspects of metabolism. In addition, recent studies found that not only the key metabolic regulatory neurons, but also the previously largely neglected “supporting cells”, in particularly the microglia and astrocytes surrounding the neurons, are important players in the hypothalamic metabolic regulatory machinery (Thaler et al., 2012; Gao et al., 2014; García-Cáceres et al., 2016, 2019; Vicente-Gutierrez et al., 2019).
Metabolism and aging process influence each other. During aging, there is huge changes in body metabolism, in which the hypothalamus plays an important role. The food-regulatory neurons in the arcuate nucleus of hypothalamus (ARC), micro-inflammation and hypothalamic stem cells all influence aging process, indicating that the brain regulation, especially the hypothalamus, not only plays a significant role in metabolic pathology, but also in other metabolism-related physiology, such as aging.
In this review, we will list some of the most common factors that influence the regulatory functions of the brain on the whole-body metabolism, including gene mutations, sex, brain glial cells, gut microbiota and stress, and dissect molecular mechanisms and related neuronal circuits by which these factors interact with brain to regulate metabolism.
Genetic Basis for Human Obesity
Recent studies revealed genetic and epigenetic basis for variations in human body mass index (BMI) (Farooqi and O'Rahilly, 2006; Locke et al., 2015; Wahl et al., 2017), and strikingly the majority of BMI-associated genetic variants affect genes that are enriched in the brain (Locke et al., 2015; Turcot et al., 2018). While the roles of many of these genetic variants have not been illustrated yet, here we discussed some genes whose mutations are known to cause human obesity via dysregulations in the central nervous system (CNS) (Table 1).
Many obesity-related genes are involved in the regulation of leptin-melanocortin pathway. The leptin receptor (LEPR) mainly locates in the ARC (Nasrallah and Horvath, 2014). Loss of function mutation in the LEPR gene cause early-onset obesity in both humans (Clement et al., 1998) and mice (Cohen et al., 2001). Pro-opiomelanocortin (POMC) neurons are one of neuron types expressing the LEPR. Patients with POMC deficiency (Krude et al., 1998) or Pomc knockout mice (Yaswen et al., 1999; Shen et al., 2017) have severe metabolic disorders such as obesity and hyperphagia. Actually, not only homozygous POMC mutation, but also heterozygous point mutations, which lead to disrupted alpha-melanocyte-stimulating hormone/beta-melanocyte-stimulating hormone (α-MSH/β-MSH, products of the POMC gene), could also cause obesity (Farooqi, 2008). Steroid receptor co-activator-1 (SRC-1, encoded by nuclear receptor coactivator 1 gene, NCOA1) can enhance POMC expression by interacting with phosphorylated signal transducer and activator of transcription 3 (STAT3). When mutating Ncoa1 in POMC neurons in mice, Pomc expression decreased, which led to hyperphagia, increased fat mass and diet-induced obesity. To be noted, NCOA1 mutated variants are found in obese people with disrupted SRC-1 function, further revealing the functional role of SRC-1 in energy homeostasis (Yang et al., 2019). One of the downstream targets of α-MSH/β-MSH is the melanocortin 4 receptor (MC4R). MC4R mutations are closely associated with obesity in humans (Yeo et al., 1998), and about 1%–2.5% people with severe obesity contain a pathogenic MC4R mutation (Farooqi and O'Rahilly, 2006). MC4R inactivation in mice also caused obesity, hyperphagia and hyperglucemia (Huszar et al., 1997), suggesting that MC4R is one of the most common obesity-related genes.
Single-minded 1 (SIM1) is abundantly expressed in the paraventricular nucleus of the hypothalamus (PVH). SIM1 mutations cause of monogenic obesity (Holder et al., 2000), and 13 SIM1 gene variants are found in obese patients (Ramachandrappa et al., 2013). Studies in mice further confirmed that knocking out Sim1 gene induced obesity and hyperphagia, probably through melanocortin receptor (Tolson et al., 2010). Aryl hydrocarbon receptor nuclear translocator 2 (ARNT2) is a transcriptional factor which can form heterodimeric complex with SIM1. Arnt2 mutation in mice with either N-ethyl-N-nitrosourea (ENU) treatment or clustered regularly interspaced short palindromic repeats/Cas9 (CRISPR/Cas9) gene editing causes hyperphagia, abnormal glucose metabolism and excessive lipid storage (Turer et al., 2018). In humans, a research found that one common nsSNP (nonsynonymous single-nucleotide polymorphisms) Gly679Ser/rs4072568 in ARNT2 was closely associated with BMI and body fat percentage (Swarbrick et al., 2011). In 2013, six children were reported to have homozygous ARNT2 mutation. In addition to phenotypes such as hypopituitarism, microcephaly and central diabetes insipidus, some, but not all patients showed obese growth curve (Webb et al., 2013). Brain-derived neurotropic factor (BDNF) is initially known to promote neuron development and differentiation by binding to its receptor tropomyosin-related kinase B (TrkB, encoded by the neurotrophic receptor tyrosine kinase 2 gene, NTRK2) (Snider, 1994). Human BDNF gene polymorphism is proven closely associated with BMI (Hong et al., 2012), and NTRK2 mutation is also found in obese patients (Yeo et al., 2004). Bdnf is highly expressed in the ventromedial nucleus of hypothalamus (VMH) and PVH, regulating body weight and food intake downstream of MC4R signaling in mice (Xu et al., 2003). As to Ntrk2, a recent study revealed that Ntrk2 deletion in the dorsomedial hypothalamus (DMH) of mice caused obesity and hyperphagia (Liao et al., 2019).
In addition to the leptin-melanocortin pathway, some genes that normally build up basic cellular function are recently found to contribute to obesity-related phenotypes. Primary cilia locate on surface of multiple cell types, including neurons. Primary cilia have long been seen as cellular signaling transporter (Siljee et al., 2018). However, in recent years, it has been reported that primary cilia also involve in energy homeostasis. For example, ciliary protein centrosomal protein 19 (CEP19) was found mutated in people with obese symptom, and Cep19 knockout mice exhibited obesity, hyperphagia and other metabolic abnormalities, similar to human phenotypes (Shalata et al., 2013). In addition, GWAS of BMI speculated that single nucleotide polymorphism (SNP) in adenylate cyclase 3 (ADCY3) was strongly related to BMI (Stergiakouli et al., 2014). Indeed, Adcy3 gene mutated mice became fatter with immobile lifestyle and hyperphagia (Wang et al., 2009). More interestingly, it was reported that MC4R and ADCY3 co-localize in primary cilia of some neurons. When silencing ADCY3 in MC4R-expressing neurons, the regulation of body weight would be disrupted (Siljee et al., 2018). Whether ADCY3 participates in malenocortin pathway has not been clearly illustrated, but current results still pointed out that ADCY3 and other ciliary proteins may profoundly function in whole-body metabolism.
FTO alpha-ketoglutarate dependent dioxygenase (FTO, which is called fat mass and obesity associated in Mus musculus) gene was firstly identified for development-regulatory function. It was soon found that FTO SNPs are associated with BMI and link to more food intake in human. However, in the mouse model, knocking out the Fto gene in rodents reduced body fat mass, while overexpression of Fto cause obesity and increase of food intake, which is opposite to human studies (Speakman, 2015). Myelin transcription factor 1 like (MYT1L) only expresses in human brain. Patients with MYT1L single nucleotide variants (SNVs) suffer from intellectual disability and obesity (Loid et al., 2018; Al Tuwaijri and Alfadhel, 2019). Deletion of chromosome 16p11.2, which SH2B adaptor protein 1 (SH2B1) locates nearby, causes morbid obesity (Walters et al., 2010). Sh2b1 knocking out mice had disrupted metabolic phenotypes including obesity and leptin resistance, while restoring SH2B1β in neurons attenuates these metabolic disorders by improving leptin signaling pathways (Ren et al., 2007).
Although the evidence is compelling that the mutation of these genes causes obesity in both humans and rodents, obesity caused by these mutations is rare, which is in contrast to the ever-growing obesity epidemic. Thus, the major cause of the current human obesity is not driven by single gene mutations, but rather by a combination of environmental changes and overall genetic susceptibility.
Sex Differences in Metabolic Regulation
Sexual differences exist in the regulation of feeding behavior and energy homeostasis in rodents (Shi et al., 2009; Sugiyama and Agellon, 2012). For example, male rats consume higher energy than females even when corrected by their larger lean body mass and metabolic rate (Woodward and Emery, 1989). Further, male rodents are more susceptible to obesity and insulin resistance induced by high fat diet (HFD) feeding compared to female counterparts (Grove et al., 2010; Benz et al., 2012; Stubbins et al., 2012; Yang et al., 2014; Morselli et al., 2016; Dorfman et al., 2017). To understand biological mechanisms for these sex differences, much of effort has been focused on roles of sex chromosomes and gonadal hormones. Emerging evidence also indicates that other factors (encoded by autosome genes) also contribute to sexual dimorphism in energy balance (see Wang and Xu, 2019; Mauvais-Jarvis et al., 2020; Tramunt et al., 2020 for detailed reviews). Here we highlight a few such factors and signals.
Sex chromosomes
The biological sex is ultimately determined by the sex chromosomes. The sex determining region Y (Sry) gene in the Y chromosome in male animals initiates the differentiation of the testes, whereas the absence of the Sry gene in females will allow ovaries to develop (Goodfellow and Lovell-Badge, 1993). Using a “four core genotypes’’ mouse model to distinguish the effects of gonadal sex (testes or ovaries) and sex chromosomes (XX or XY) (De Vries et al., 2002), it has been demonstrated that both the gonads (or gonadal hormones) and sex chromosome independently contribute to the sex differences in body weight (Chen et al., 2012; Reue, 2017). A few X-linked genes have been implicated in the regulation of energy balance. One such example is 5-hydroxytryptamine (5-HT) 2C receptor (5-TH2CR), encoded by an X-linked gene (Ht2cr) (Tecott et al., 1995). Male mice with global deficiency of 5-TH2CR develop a late onset hyperphagic obesity (Tecott et al., 1995; Nonogaki et al., 1998). 5-HT and its analogs, e.g., d-fenfluramine and lorcaserin, suppress food intake largely through acting upon the 5-TH2CR expressed by POMC neurons (Xu et al., 2008; Berglund et al., 2013; D'Agostino et al., 2018) and dopamine neurons in male animals (Xu et al., 2017). Mechanisms for 5-TH2CR’s effects on appetite control involve its actions to activate the transient receptor potential channel 5 (TrpC5) which leads to depolarization of neurons (Gao et al., 2017c). Interestingly, TrpC5 is also encoded by an X-linked gene, and it has been shown to mediate actions of multiple hormones, including estrogen (Qiu et al., 2018), leptin (Qiu et al., 2010; Gao et al., 2017c) and insulin (Qiu et al., 2014). Importantly, deletion of TrpC5 in POMC neurons leads to obesity in male mice, which is associated with increased daylight feeding and decreased energy expenditure (Gao et al., 2017c). Another X-linked gene is O-GlcNAc transferase (OGT), although its functions on body weight balance appears to be site specific, with OGT in orexigenic agouti-related peptide (AgRP) neurons promoting weight gain (Ruan et al., 2014) while OGT in anorexigenic PVH neurons prevent overeating and obesity (Lagerlof et al., 2016). In summary, the Sry gene on the Y chromosome determines the development of male or female gonads, which influence the circulating gonadal hormones and therefore energy homeostasis (See below). The X-linked genes also influence the energy balance, but their contributions to the sex difference in body weight control remain to be further investigated.
Gonadal hormones
Estrogen
Estrogen is well known to play an essential role in preventing body weight gain in females, as the withdrawal of endogenous estrogen by ovariectomy (OVX) in female animals leads to body weight gain and hyperadiposity which can be prevented by estrogen treatment but not by progesterone (Drewett, 1973; Schwartz and Wade, 1981; Geary et al., 2001; Wallen et al., 2001; Rogers et al., 2009; Paladini and Roeper, 2014). The most studied estrogen receptor in the context of body weight balance is ERα. Humans or mice with mutations in the ERα (which is also called estrogen receptor 1, ESR1) gene are obese (Heine et al., 2000; Okura et al., 2003), and loss of ERα in mice blocks the anorexigenic effects of estrogen (Geary et al., 2001). Brain ERα has been implicated in body weight control (Palmer and Gray, 1986; Butera and Beikirch, 1989; Santollo et al., 2011). Consistently, female mice with Esr1 deletion in the brain develop obesity (Xu et al., 2011). In particular, ERα is expressed by some POMC neurons in the ARC (Miller et al., 1995; Attia, 2010; de Souza et al., 2011), which can be activated by estrogen (Gao et al., 2007; Malyala et al., 2008; Saito et al., 2015). Female (but not male) mice lacking ERα only in POMC neurons develop hyperphagia and modest body weight gain (Xu et al., 2011). Another hypothalamic ERα population with sexually dimorphic functions is that in the VMH, and multiple groups reported that ERα signals in the VMH in females (but not in males) prevent obesity primarily by stimulating energy expenditure (Musatov et al., 2007; Xu et al., 2011; Martinez de Morentin et al., 2014; Correa et al., 2015). Notably, a recent study used single-cell RNA sequencing to identify a sub-population of VMH neurons co-expressing ERα and reprimo (RPRM), which is only present in female mice and regulate thermogenesis (van Veen et al., 2019). ERα is also abundant in the hindbrain, including the nucleus of the solitary tract (NTS) and dorsal raphe nucleus (DRN) (Osterlund et al., 1998; Merchenthaler et al., 2004; Schlenker and Hansen, 2006), and estrogenic actions in these regions promote satiation (Geary et al., 2001; Asarian and Geary, 2007) and inhibit binge-like eating (Robichaud and Debonnel, 2005; Dalmasso et al., 2011; Santollo et al., 2011; Cao et al., 2014).
It is important to point out that ERα also prevents obesity in males. For example, ERα gene deficiency results in obesity in male mice (Heine et al., 2000; Callewaert et al., 2009) and in men (Smith et al., 1994; Grumbach and Auchus, 1999). Further, male mice lacking ERα in the brain develop modest obesity (Xu et al., 2011). In addition, administration of estrogen or its analogs reduces body weight in male mice (Gao et al., 2007; Finan et al., 2012). Notably, the male gonadal hormone, testosterone, can be converted to estrogen by aromatase (Jarvie and Hentges, 2012) and abundant aromatase is expressed in a few brain regions (Wu et al., 2009). Thus, it is possible that certain male brain regions with high aromatase activity could be exposed to high levels of estrogen despite the lack of circulating estrogen. Indeed, both male and female aromatase knockout mice develop obesity (Jones et al., 2000), highlighting the physiological function of aromatase and estrogen in both sexes. In particular, loss of ERα in the medial amygdala (MeA), a brain region with high aromatase expression (Wu et al., 2009), not only causes obesity in female mice, but also in male mice (Xu et al., 2015).
Collectively, high levels of circulating estrogen prevent obesity in female animals. Many of these estrogenic actions are mediated by multiple ERα populations, which therefore at least partly account for sex differences in body weight. However, ERα in certain brain regions with enriched aromatase activity, e.g., the MeA, also contributes to the maintenance of male energy balance. Notably, two other estrogen receptors, estrogen receptor beta (ERβ) and G protein-coupled receptor 30 (GPR30), are also implicated in the regulation of regulate body weight balance (Foryst-Ludwig et al., 2008; Haas et al., 2009; Sharma et al., 2013; Davis et al., 2014), but their sex-specific functions remain to be further investigated.
Testosterone
Testosterone can be converted to estrogen by aromatase (Jarvie and Hentges, 2012). Thus, testosterone actions can be mediated by either the androgen receptor (AR) or estrogen receptors. Notably, AR is encoded by an X-linked gene. Interestingly, while young Ar knockout male mice show decreased body weight, aged mutant male mice have increased body weight and adiposity (Sato et al., 2003; Fan et al., 2005; Lin et al., 2005). Consistently, brain-specific deletion of Ar leads to a late onset obesity in male mice (Yu et al., 2013), indicating that brain AR is required to prevent aging-associated obesity in males. Further, Fagman et al. reported that whole-body deletion of AR in female mice on an apolipoprotein E (apoE)-deficient background are prone to diet-induced obesity compared to apolipoprotein E (apoE)-deficient female mice (Fagman et al., 2015). While these results suggest potential anti-obesity actions of AR in females, these effects need to be further confirmed in animals with normal apoE functions. Future, testosterone, when exposed to neonatal animals can change structures and excitability of hypothalamic neurons in both males and females (Matsumoto and Arai, 1980), and cause obesity during the adulthood at least in females (Nohara et al., 2011, 2013). Importantly, neonatal administration of dihydrotestosterone, a non-aromatizable androgen, cannot induce similar obese phenotypes in females (Nohara et al., 2011), suggesting that the programming effects of testosterone may be mediated by estrogen receptors, rather than by AR.
Other factors
While the majority of the field focus on the functions of sex chromosomal genes and gonadal hormones, autosome genes may exist to regulate the sex differences in energy homeostasis.
First, some factors could regulate levels of gonadal hormones or receptors, and therefore contribute to sex differences in energy balance. For example, kisspeptin (Kiss1) and its receptor, G protein-coupled receptor 54 (GPR54), are key regulators of reproduction (Murphy, 2005), and are also required to maintain normal circulating levels of estrogen in females (Seminara et al., 2003; d'Anglemont de Tassigny et al., 2007). Thus, loss of GPR54 leads to massive obesity in female mice but to a lesser extent in males (Tolson et al., 2014, 2016). Similarly, deficiency in angiotensin II receptor (AT2R) renders female mice more prone to diet-induced obesity, associated with reduced estrogen levels, but the same mutation does not affect male mice (Samuel et al., 2013).
In addition, factors that function downstream of gonadal hormones can also contribute to sexual dimorphism in energy balance. One such example is SRC-1. SRC-1 can interact with ERα upon estrogen stimulation, and is required to mediate estrogenic actions to reduce food intake in female mice (Zhu et al., 2013; Yang et al., 2019). Another example is STAT3, which is required to mediate anorexigenic effects of estrogen (Gao et al., 2007). Loss of STAT3 in POMC neurons leads to modest obesity in female mice but does not affect male mice (Xu et al., 2007).
It is important to point out that some autosome gene-encoded factors may contribute to sex differences independent of gonadal hormones. For example, an autosome-encoded transcription factor, TAp63, is found to drive enhanced POMC mRNAs and neural activity in females compared to male counterparts (Wang et al., 2018), corresponding to lower food intake in female mice (Nohara et al., 2011). Deletion of Tap63 in POMC neurons does not affect male body weight but renders females more susceptible to diet-induced obesity (Wang et al., 2018). Importantly, loss of TAp63 does not influence anorexigenic effects of estrogen (Wang et al., 2018), suggesting that TAp63 and estrogenic actions are largely independent of each other. Interestingly, Sirtuin 1 (SIRT1), a transcriptional target of TAp63 (Su et al., 2012), plays a sex-specific role in POMC neurons to prevent obesity in females but not in males (Ramadori et al., 2010), similar to effects of TAp63. Microglial cells in the hypothalamus also contribute to the sex differences in body weight control. HFD feeding reduces hypothalamic levels of C-X3-C motif chemokine ligand 1 (CX3CL1, a neuron-released chemokine) and its receptor expressed by microglia, CX3CR1, specifically in male mice but not in female mice (Dorfman et al., 2017). Importantly, Cx3cr1 deletion in female mice causes obesity, associated with “male-like” hypothalamic microglial accumulation and activation (Dorfman et al., 2017).
In summary, the sex chromosomes are conceivably the fundamental contributors to the sexual dimorphism. The Sry gene in the Y chromosome determines the gonadal development, and therefore influence the circulating levels of estrogen and testosterone. Increasing numbers of the X-linked genes are shown to regulate energy balance, although their roles in sex differences remain elusive. Estrogen-ER and testosterone-AR signals in the brain contribute to the regulation of energy balance in females and males, respectively; however, since testosterone can be converted to estrogen in certain brain regions, ER can also partially mediate testosterone’s actions in males. Importantly, autosome-encoded genes also contribute to the sex differences in energy balance through gonadal hormone-dependent or independent mechanisms (Fig. 1).

Mechanisms underlying sex differences in body weight control. The Sry gene in the Y chromosome determines the gonadal development, and therefore influences the circulating levels of estrogen and testosterone. Testosterone can directly act upon the androgen receptor, and can also be converted to estrogen which acts upon the estrogen receptor. These gonadal hormones, together with the X-linked genes and other factors encoded by autosomes, all contribute to sex differences in energy balance partially through their actions in the brain. This figure was illustrated partially via the Biorender.com
Hypothalamic Glial Immune and Metabolic Changes in Obesity and Diabetes
Microglia innate immunity-driven inflammatory response at frontlines in response to HFD
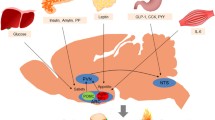
Brain microglia are long-surviving and self-renewing innate immune cells, that share the same developmental origin as peripheral tissue macrophages (Ginhoux et al., 2010; Prinz and Priller, 2014; Gomez Perdiguero et al., 2015; Goldmann et al., 2016; Tay et al., 2017). Microglia are crucial for scavenging cell debris and pathogens to maintain brain tissue homeostasis. Various neural disorders in the CNS are associated with microglial dysfunction (Perry et al., 2010; Prinz and Priller, 2014; Heneka et al., 2015). Recent genome-wide analyses showed that microglia have distinctive brain region-dependent transcriptional identities with differences in bioenergetic and immune-regulatory pathways (Grabert et al., 2016). The hypothalamus contains highly heterogeneous and condensed populations of neurons in different regions. The ventromedial arcuate nucleus and median eminence complex at the mediobasal hypothalamus (MBH) is an area lacking normal blood-brain barrier (BBB) (Broadwell et al., 1983; Yi et al., 2006), thus the MBH functions as an important “brain window” for different brain cells sensing blood-born substances (Horvath, 2005; Friedman, 2016; Timper and Bruning, 2017). It is also reasonable that in this region, neural synaptic rewiring events occurs frequently during different metabolic states (Horvath, 2005), and constantly produce cell debris and metabolic waste. In order to keep a healthy and clean microenvironment for the hypothalamic neurons to function, the microglial activity in the MBH needs to match the high demands for immune surveillances and debris phagocytosing/clearances (Fig. 2). This was demonstrated by the fact that microglia in the MBH showed a significantly higher reactivity than in other hypothalamic regions when experimental animals were exposed to HFD, and that this occurred rapidly within 3 days after receiving the HFD (Thaler et al., 2012; Kim et al., 2019). The exact factors that mediate the HFD effects on hypothalamic microglial reactivity is yet unclear. Intriguingly, it is unlikely directly through any major blood-borne components, since microglial cytokines production was not changed upon exposure to the serum derived from HFD-fed animals (Baufeld et al., 2016). Alternatively, the HFD stimulated advanced glycation end-products (AGEs) produced by MBH neurons was proposed to be one of the brain mediators, since AGEs drive microglial inflammatory response via their receptors highly expressed by microglial cells (Gao et al., 2017a), though how the HFD induces AGEs production in the hypothalamic neurons are unknown.

A schematic illustrates the difference of microglia and astrocytes in MBH between metabolic homeostasis or metabolic syndrome. In metabolic homeostasis, MBH neurons such as POMC neurons produce regular amount of cell debris and metabolic wastes, matches the phagocytic capacity of the microglia, whereas in metabolic syndrome induced by obesogenic HFD, the over-production of cell debris and wastes drives microglial activation and immunometabolic reprogramming, as evidenced by increased cytokines production (IL-1β, TNFα) and lipids utilization (LPL, UCP2), as well as downregulation of glycolysis and glutamate/glutamine utilization (HK2, GDH). This immunometabolic reprogramming is associated with impaired phagocytic capacity (CD68) in the reactive microglia and increased astrocytes activity, which might contribute to the increased IL-6 production in the local microenvironment. Furthermore, although the number of astrocytes is increased upon HFD, these reactive astrocytes display shortened high-order cell processes, which is an indication of a loss of process plasticity. All these changes in microglia and astrocytes are associated with hypothalamic neuronal dysfunction in controlling food intake and energy homeostasis
Immunometabolic reprogramming in proinflammatory microglia
To ensure efficient immune surveillance and phagocytosis, the microglia need to tightly modulate their intracellular metabolic processes to match the fuel demand and control their effector functions. This so-called immunometabolism is one of the key mechanisms for understanding microglial pathophysiology (O'Neill et al., 2016). Unlike the peripheral macrophages that can be defined into a pro-inflammatory (M1) and an anti-inflammatory/pro-resolving (M2) phenotypes, the microglial phenotypes are way more complex following their varied non-macrophage-like functions in the brain (Ransohoff, 2016). It is well-known that M1 macrophages rely mainly on glycolysis, whereas M2 cells are more dependent on oxidative phosphorylation (OXPHOS). However, in HFD-fed animals, the reactive microglia in the MBH likely carry both M1- and M2-like features, since these cells not only express more M1 phenotype-associated pro-inflammatory cytokines such as interleukin 1 beta (IL-1β) and tumor necrosis factor alpha (TNFα) (Thaler et al., 2012), but also more mitochondrial uncoupling protein 2 (UCP2) (Kim et al., 2019), more lipids’ uptake gate-keeper lipoprotein lipase (LPL) (Gao et al., 2017b) and less hexokinase 2 (HK2) (Milanova et al., 2019), indicating an increase of fatty-acid oxidation and OXPHOS and less glycolysis which belong to the M2 phenotype. This illustrates that in HFD-activated microglia, their immunometabolism is reprogrammed differently from the canonical switching process between peripheral M1 and M2 macrophages.
As mentioned above, one important metabolic factor is the mitochondrial UCP2 which is linked to cytokine production in the pro-inflammatory microglia (Cannon and Nedergaard, 2004). At early stage of HFD feeding, the hypothalamic microglia produce more UCP2, which is associated with increases of IL-1β, interleukin-6 (IL-6) and TNFα expression, as well as changes in the mitochondrial number and size (Kim et al., 2019). These HFD-induced changes in cytokine levels and mitochondrial morphology can be prevented when UCP2 is deleted in microglia. Importantly, deletion of UCP2 in microglia is also associated with more excitatory synaptic inputs onto the POMC neurons, and this leads to increased sensitivity to leptin in POMC neurons and resistance to HFD-induced obesity in mice (Kim et al., 2019). These data suggest that shutting down fatty-acid oxidation and OXPHOS in mitochondria is crucial to reprogram the microglial immunometabolism, reduce cytokine production in reactive microglia, and protect the POMC neurons from HFD-induced dysfunction.
An important issue regarding studies on microglial immunometabolism is that these cells are highly active with their motility and cellular metabolism, logically, none of the in vitro experimental approaches can closely mimic in vivo conditions (Timmerman et al., 2018). Thus, in vitro tools should be only applied when, for example, high cell number is critical for the study (Timmerman et al., 2018).
Dysregulation of phagocytosis in proinflammatory microglia
TNFα is a major cytokine produced by reactive microglia in obesogenic environments. On one hand, TNFα-activated nuclear factor-kappa B (NF-κB) pathway plays a critical role in the reactive microglia to enhance their pro-inflammatory responses by increased cytokine production and cell proliferation, thus boosting the microglial activity as an autocrine factor (Kuno et al., 2005; Block et al., 2007). On the other hand, TNFα secreted by the reactive microglia acts on neighboring hypothalamic POMC neurons and induces mitochondrial stress (Yi et al., 2017). The TNFα-impaired mitochondrial function might be one of the major causes of HFD-induced POMC neural dysfunction, which is not only observed in the long-term HFD-fed animals (Thaler et al., 2012; Yi et al., 2017; Kim et al., 2019), but also in postmortem hypothalamic tissue of type 2 diabetic patients (Alkemade et al., 2012; Kalsbeek et al., 2020). Correspondingly, restraining the TNFα/NF-κB pathway in microglia can effectively reduce microglial activation and prevent diet-induced obesity (Valdearcos et al., 2017), and blocking the TNFα/NF-κB pathway in POMC neurons can also reduce the diet-induced obesity. This highlights the TNFα/NF-κB pathway in the MBH as a potential brain target for treating obesity.
The reactive microglia in the MBH upon HFD is not only characterized by increased cytokine production, but also impaired phagocytic capacity, as shown by a downregulation of phagocytic indicator cluster of differentiation 68 (CD68) expression during day and night in HFD-fed rats (Milanova et al., 2019). Intriguingly, HFD feeding stimulates microglia to produce more LPL (Gao et al., 2017b). Besides governing lipid uptake for fueling, the LPL-gated phospholipid production is also crucial for phagolysosome formation and turnover (Boulais et al., 2010). Thus, HFD feeding might impair microglial phagocytosis in the MBH, whereas a compensatory mechanism drives more LPL expression and phospholipids production in order to maintain the phagocytic capacity. Not surprisingly, in HFD-fed mice, lacking LPL on microglia, there was a further down-regulation of CD68, and worsened phagocytic capacity, ultimately associated with less POMC-expressing neurons and more vulnerability to HFD-induced metabolic disorders (Gao et al., 2017b). This suggests that lacking a sufficient microglial phagocytosis has a detrimental effect on POMC neural survival upon HFD challenge (Fig. 2).
Consistent with the finds in animal studies, microglial functional changes are also found to be associated with human obesity. This is demonstrated by an increased area covered by microglial cell bodies in the infundibular nucleus (an area equivalents to the ARC in rodents) in obese individuals, accompanied by microglial dystrophy/cytorrhexis (Baufeld et al., 2016). Microglial cytorrhexic dysmorphology are often observed in neurodegenerative diseases, where microglia are over-challenged with immune or phagocytic tasks and eventually become “burn out” (Baron et al., 2014; Streit et al., 2014). Thus, after long-lasting activation, microglial phagocytic inability could occur late in the course of the metabolic disorders, which then eventually contribute to the loss of hypothalamic neurons in the type 2 diabetic patients.
As in HFD-activated microglia, the increased pro-inflammatory cytokines oppose phagocytic capacity, targeting microglial pathway in treating obesity needs to tackle both inflammatory and phagocytic pathways. Hypothalamic cell-specific therapeutic targeting is one of the most promising strategies to create an efficient anti-obesity treatment. The eventually goal is to identify intracellular signaling pathways in the hypothalamic microglia that can be targeted by cell-specific compounds. By normalizing the dysfunctional hypothalamic microglia and the associated innate immune response, as induced by our hypercaloric environment, these compounds should be able to treat obesity and prevent associated metabolic syndromes.
Hypothalamic astrocytes reactivity in obesity
Similar to microglia, astrocytes also get activated rapidly upon HFD feeding, characterized by increases in the cell number (Thaler et al., 2012; Buckman et al., 2015; Zhang et al., 2017b), and with the NF-κB pathway as an important player in astrogliosis (Buckman et al., 2015; Zhang et al., 2017b). Not surprisingly, the astrocytes-derived cytokine IL-6 (Van Wagoner and Benveniste, 1999) at the downstream of NF-κB pathway (Oeckinghaus et al., 2011) was also found to be elevated within 24 h of HFD exposure (Thaler et al., 2012). Intriguingly, the HFD-stimulated astrogliosis is associated with shortening of the high-order processes of individual astrocyte and impairment of astrocytic process plasticity, mediated by IKKb/NF-kB pathway (Zhang et al., 2017b). It is known that under various pathophysiological conditions, the coating of neurons by astrocytic processes is important for maintaining inter-neuronal communications (Syková, 2004) and rewiring the output of the melanocortin neurocircuit (Horvath et al., 2010). Indeed, the impaired astrocytic process plasticity was associated with dysregulation of extracellular gamma-aminobutyric acid (GABA) (Zhang et al., 2017b), which may affect neurocircuits in different brain regions that control feeding and energy metabolism (Kim et al., 2015; Wu et al., 2015). Two blood-borne metabolic hormones insulin and leptin, with their receptors highly expressed by astrocytes, are found to profoundly regulate astrocytes activity (Kim et al., 2014; García-Cáceres et al., 2016). Deletion of either receptor not only changed astrocytes morphology, but also impacted the activity of the hypothalamic melanocortin circuitry (Kim et al., 2014; García-Cáceres et al., 2016), indicating the essential role of astrocytes in metabolic sensing and regulation in the MBH.
It should be noted that both microglia and astrocytes constitute heterogeneous populations in the brain. The microglial heterogeneity is mainly related to their distinctive molecular markers or morphological diversities (Burns et al., 2020; Masuda et al., 2020; Olah et al., 2020). Moreover, besides the resident microglia, brain also contains macrophages under different pathophysiological conditions (Masuda et al., 2020). Nevertheless, all the microglia and macrophages in the brain share some common markers, such as Cx3cr1, that serve as the promotor for Cre line for genetically manipulation of microglia/macrophages function. Unlike microglia that share common markers, not all astrocytes in the brain can be recognized by a single common marker. The glial fibrillary acidic protein (GFAP) that is commonly used, can only recognized sub-population of astrocytes (Tatsumi et al., 2018). Thus, data obtained by GFAP immunohistology or using GFAP promotor (human or mouse) Cre lines need to be interpreted with caution. Alternatively, a combinational genetic manipulation targeting subpopulation of astrocytes that express 10-formyltetrahydrofolate dehydrogenase (Aldh1l1) (Srinivasan et al., 2016) or glutamate aspartate transporter (Glast) (Mori et al., 2006), might be essential for the complete understanding of astrocytes function in the context of obesity and metabolic disorders.
Gut Microbiota and Brain-Regulated Metabolism
Gut microbiota, brain and metabolism
Microbiota forms an ecological balance with human body during evolution. The symbiotic relationship between gut microbiota and humans provides beneficial effects to human health including: (1) defense against pathogen colonization by nutrient competition and production of antimicrobial substances; (2) fortification of intestinal epithelial barrier and induction of secretory immunoglobulin A synthesis to limit pathogenic bacteria penetration into tissues; (3) facilitation of nutrient absorption by metabolizing indigestible dietary compounds; and (4) participation in the maturation and functionality of the host immune system by providing diverse signals for “tuning” the host immune status (Round and Mazmanian, 2009; Adak and Khan, 2019). Importantly, emerging studies have demonstrated a profound involvement of gut microbiota in an array of diseases through modulating brain neuron function. Given the expanding literature involving the function of gut microbiota, this section will only focus on those that are closely related to brain control of metabolism and obesity.
The role of gut microbiota in influencing brain-regulated metabolism
Gut microbiota can be influenced by and in turn influence host metabolism. There is a huge change of microbiota community composition in obese and diabetic people (Clemente et al., 2012). Overall, obese people contain low microbiota richness with an increased Firmicutes to Bacteroidetes ratio, though this conclusion seems controversial in some studies (Patterson et al., 2016). Using metagenomics, researchers found that anti-inflammatory microbial species, like Faecalibacterium prausnitzii, are more abundant in lean people with high microbiota richness, while Bacteroides and R. gnavus enrichments are more closely related to obese individuals (Le Chatelier et al., 2013). As to animal models, HFD fed germ-free (GF) mice gain less fat mass compared to mice with normal gut microbiota (Bäckhed et al., 2004), and obese phenotypes can be mimicked on GF mice if they receive fecal microbiota transplantation (FMT) from obese mice (Turnbaugh et al., 2006).
The “gut-brain axis” concept describes a bidirectional interaction between the brain and gut. The “microbiota-gut-brain axis” is subsequently developed as gut microbiota is significantly involved in the gut-brain axis (Cryan et al., 2019). Now, studies have found that gut microbiota can produce metabolites and proteins to modulate neuron-regulated host metabolism, although the exact mechanisms are largely unknown. Here, we discuss current progress in exploring the role of gut microbiota in neural control of metabolism (Fig. 3), and raise unsolved questions for future directions (For techniques involved in microbiota researches, please see review Cryan et al., 2019 for details).

Gut microbiota involved in a complex interaction with host metabolism. The gut microbiota influences neural control of metabolism through multiple ways. SCFAs produced by gut microbiota can activate GPR41 and GPR43 receptors on intestine L cells. After being activated, L cells produce GLP-1 and PYY peptides, which act in the brain by either blood circulation or vagus nerve to reduce food intake and balance glucose metabolism. BAs are another kind of microbiota metabolites that promote GLP-1 production from intestine L cells. ClpB is the only known bacterial protein that can reduce food intake by gut-brain axis. HFD intake causes increased Gram-negative bacteria proportion, LPS concentration and systemic inflammation. As LPS can activate vagus nerve, gut microbiota may modulate systemic and brain inflammatory levels by LPS production. To be noted, when studying functional roles of specific microbiota clusters, antibodies/prebiotics supplements and FMT are the most common approaches to choose
Gut microbiota produce short-chain fatty acid (SCFA) by fermentation of organic compounds such as carbohydrates, lipids and dietary fibers. SCFAs influence gut-brain axis mostly based on endocrine-circulating system. Apart from crossing BBB directly (Mitchell et al., 2011), SCFAs are endogenous ligands of G-protein coupled receptor 41 and 43 (GPR41 and GPR43). Intestine L cells express these two receptors as well as glucagon-like peptide-1 (GLP-1) and peptide YY (PYY) (Karaki et al., 2006), so it is likely that some SCFAs promote GLP-1 and PYY production by binding to GPR41 and GPR43. Indeed, degeneration of dietary fibers by microbiota increased GLP-1 (Cani et al., 2004) and PYY (Delzenne et al., 2005) levels and modulate food intake by acting in the brain (Murphy and Bloom, 2006). Consistently, the feeding-suppressing effects of SCFAs require the intact vagal neurons, where GLP-1 and PYY are known to act (Baraboi et al., 2010; Krieger et al., 2015, 2016). For example, hepatic vagotomy or capsaicin treatment (which denervates vagal neurons) attenuates or abolishes effects of intraperitoneal (IP) injection of acetate, propionate and butyrate to decrease food intake (C et al., 2018). In addition, propionate supplement from diet also induces dramatic activation of the dorsal vagal complex (DVC), which can be reduced by capsaicin (De Vadder et al., 2014). Further, subdiaphragmatic vagotomy blunts food-suppression effect of butyrate (Li et al., 2018b), supporting that SCFAs act, at least in part, through vagal afferent neurons.
Bile acids (BAs) are another kind of metabolites associated with microbiota. BAs can not only activate intestine L cells for GLP-1 production (Brighton et al., 2015), but also induce fibroblast growth factor 19 (FGF19) production from enterocytes (Steven et al., 2015). GLP-1, FGF19 and BAs themselves can all cross the BBB to modulate feeding and glucose metabolism by binding to respective receptors on neurons (Jameson et al., 2020).
In addition to the microbiota-associated metabolites, bacterial proteins may also influence metabolism via the gut-brain axis. For example, the bacterial protein caseinolytic protease B (ClpB) was shown to mimic α-MSH and act in the brain to reduce food intake (Breton et al., 2016).
Further, gut microbiota themselves can influence brain function. For example, Lactobacillus rhamnosus supplement increases GABA levels in the brain and reduces anxiety controlled by the vagus nerve (Bravo et al., 2011), although it remains unclear whether such mechanisms also influence brain functions that are relevant to metabolic control.
Notably, obesity in HFD-fed animals is associated with increased Gram-negative bacteria proportion in gut microbiota, with subsequent increased lipopolysaccharide (LPS) plasma concentration, systemic inflammation and obesity (Cani et al., 2008). Since LSP can activate vagal afferent neurons (Klingbeil and de La Serre, 2018), such association suggest that dysregulated gut microbiota may contribute to diet-induced obesity, although this cause–effect relation remains to be definitively tested (Dalby et al., 2018).
In summary, current researches in the connection between gut microbiota, brain and metabolism have suggested that compositions of gut microbiota changes in association with BMI. Gut microbiota regulates neural control of metabolism, probably via endocrine system and vagus nerve.
Stress, Feeding and Obesity Development
Stress, anxiety and related emotional modalities
The mental and physiological processes involved in stress and related emotional modalities are complex and often difficult to precisely define. In general, stress is described as a state of mental or emotional strain or tension resulting from adverse or very demanding circumstances. Anxiety is the reaction to situations perceived as stressful and dangerous. Both stress and anxiety are a normal part of life and elicits changes with physical, mental, and emotional responses necessary for maintaining welfare and survival of individuals and species. However, a disproportional level of anxiety with excessive nervousness and fear without apparent triggers leads to anxiety disorders (Patriquin and Mathew, 2017). Depression is normally regarded as a generalized form of anxiety with symptoms of anhedonia and reduced motivation, a major form of psychiatric disorders and can result from chronic experience of stress and anxiety (Kessler, 1997; Pizzagalli, 2014). Despite potential difference in stress and related emotional modalities between humans and rodents (Tye, 2018), studies in rodent models on human-emotion like behaviors have provided important insights on neurobiology related to stress related behaviors. Given the vast literature on the topic of stress and related behaviors, this review only focuses on stress related to feeding and obesity development.
Stress and body weight regulation
Corticotropin releasing hormone (CRH) neurons, widely distributed in the brain, are thought to be one of the first responders in orchestrating behavioral and hormonal responses to stress (Deussing and Chen, 2018). Hypothalamic CRH neurons, a major subset of brain CRH neurons, are located in the PVH, a key brain region for feeding and body weight regulation, and therefore likely serve as a link between metabolism and stress responses. These neurons are known to elicit neuroendocrine responses through the autonomic nervous system and hypothalamus–pituitary–adrenal axis (HPA). Extensive studies in humans implicate the HPA axis in obesity development. Uncontrolled glucocorticoid levels are associated with hyperphagia, insulin resistance, obesity, and diabetes, as observed in human Cushing’s diseases (Pivonello et al., 2015). Heightened glucocorticoid levels have been suggested to increase feeding and obesity through insulin action and producing a bias toward increased motivation to eat and decreased executive function through altering brain cognitive function (Sinha and Jastreboff, 2013). Consistent with the observations in humans, a heightened HPA axis is also associated with obese animal models including leptin deficient ob/ob mouse models (Huang et al., 1998). Interestingly, manipulation of CRH expression in adult mice affects diet preference (Okamoto et al., 2018).
Given the consequence of chronic stress on emotional changes including anxiety and depression (Berry et al., 2012), it is not surprising that there is a strong association between psychiatric disorders including anxiety and depression, and obesity in humans (Siervo et al., 2009). It has been proposed that stress-induced emotional eating links anxiety/depression and obesity through alterations in cortisol and brain reward circuitry, leading to promotion of compulsive overeating (Dallman, 2010; Nederkoorn et al., 2015). Conversely, eating disorders are often accompanied with alterations in emotion including stress, anxiety and aggression (Sweeney and Yang, 2017). Importantly, treatments of psychological disorders are known to be associated with obesity development (Domecq et al., 2015). Collectively, these observations argue that chronic stress and consequent emotional derangements including anxiety and depression contribute to human obesity.
Given the substantial evidence in humans on chronic stress and obesity, it is surprising that results using animal stress models are inconsistent (Razzoli and Bartolomucci, 2016). Some studies conclude that stress reduces feeding and body weight (Michel et al., 2005; Liu et al., 2014; Harris, 2017; Qu et al., 2020), while others show the opposite that stress induces weight gain (Lutter et al., 2008; Melhorn et al., 2010; Razzoli et al., 2015). One important reason, among others, may lie in the nature of stressors imposed (Harris, 2015; Razzoli et al., 2017). Self-reported stress in humans, as normally conducted in human studies, is largely chronic and psychological in nature, likely resulting from socioeconomic pressure. Thus, it is rather difficult to mimic the same chronic stress situation in rodents. Those reports on chronic stress reducing body weight in rodents often use repeated environmental stresses, i.e., foot shock or restraint (Michel et al., 2005; Liu et al., 2014; Harris, 2017), which are essentially repeated acute stressors with periods of relieving phases. Acute stress is known to reduce feeding in short term (Maniscalco and Rinaman, 2017; Terrill et al., 2018), and acute brain action of CRH and its related urocortins reduces feeding (Stengel and Tache, 2014). In contrast, chronic stress in animal models with weight gain is normally induced by psychological social defeat (Lutter et al., 2008; Melhorn et al., 2010; Razzoli et al., 2015). Chronic psychological stress produces social avoidance and other signs common to anxiety and depression patients (Chiba et al., 2012; Venzala et al., 2013), and therefore may better mimic chronic stress in humans. Importantly, human genetic studies are beginning to identify candidate genes underlying the association between stress/anxiety and obesity, which can be confirmed in mice (Meier et al., 2019), supporting that the effect of chronic stress on obesity may be evolutionally conserved. A combination of mouse genetics and chronic psychological stress models may help to identify key brain neurons and pathways in mediating the effect of chronic stress on obesity.
HFD, stress and obesity
As human obesity is contributed by both HFD and stress, it is imperative to consider whether HFD and chronic stress interact to precipitate obesity development. HFD alters both basal and stress-induced HPA axis, reduces corticosterone rhythmicity (Auvinen et al., 2012; Kalyani et al., 2016), induces anxiety-like behaviors (Gomes et al., 2020), and suppresses stress responses (Appiakannan et al., 2019). Conversely, stress aggravates HFD-induced insulin resistance and heightened glucocorticoid action worsens obesity and hyperglycemia induced by HFD (Bartolomucci et al., 2009; Tsai et al., 2018; Garcia-Eguren et al., 2019). These observations strongly suggest that HFD and chronic stress synergistically promote obesity and associated metabolic syndrome.
Given the similar effect of HFD and chronic stress on obesity development, it is important to identify the underlying neural basis. Studies focusing on the PVH CRH neurons have suggested an importance of the responsiveness of these neurons in HFD-induced obesity. PVH CRH neurons exhibit a diurnal pattern of activity, which orchestrates a diurnal pattern of the HPA activity. HFD alters synaptic inputs to PVH neurons (Mazier et al., 2019), and blunts diurnal patterns of CRH expression (Auvinen et al., 2012), and therefore may alter the activity pattern of these neurons. Importantly, clamping the activity level of PVH CRH neurons either at high or low levels, which render these neurons not responsive to either stressors or nutritional challenges, mimicking HFD effects on obesity development (Zhu et al., 2020), which illustrates a potential importance of dynamic responses of PVH CRH neurons to environmental or nutritional challenges in HFD-induced obesity. Interestingly, HFD reduces responsiveness of CRH and AgRP neurons (Beutler et al., 2020; Zhu et al., 2020). It is conceivable that the responsiveness of neurons underlies their activity pattern by integrating dynamic changes in synaptic and hormonal inputs, and therefore shapes the diurnal pattern activity of these neurons (Ulrich-Lai and Herman, 2009). In addition, intrinsic neuron properties, for example, the core clock genes are known to drive an intricate gene network that ultimately orchestrates neuron responsiveness and activity patterns related to stress (Hood and Amir, 2018). Deletion of brain and muscle arnt-like 1 (Bmal1), a key core clock gene, in the PVH reduces the responsiveness of these neurons to external stressful stimuli, which blunts diurnal rhythms in energy expenditure and feeding, and causes obesity (Kim et al., 2020). HFD is known to disrupt diurnal rhythms in feeding and metabolism (Hatori et al., 2012). Thus, studies aiming to examine whether neuron responsiveness underlying the interplay between stress/HFD and circadian rhythms will provide important insights on the role of stress in obesity development.
Common neural pathways in feeding and stress-related emotional modalities
Emerging studies suggest that feeding reciprocally affects emotion. AgRP neurons in the ARC, a key group of feeding-promoting neurons, are inhibited by stress stimuli while their activation promotes feeding and reduces behavioral signs of anxiety (Dietrich et al., 2015; Burnett et al., 2016; Padilla et al., 2016). GABAergic lateral hypothalamic area (LHA) neurons, while potently promoting feeding, produce place preference and rewarding effects and increases aggression and predatory behaviors (Jennings et al., 2015; Nieh et al., 2015; Li et al., 2018a; Mangieri et al., 2018; Cassidy et al., 2019), behavioral signs of reduced anxiety and stress perception. In contrast, glutamatergic LHA neurons reduce feeding through promoting behavior signs of stress and anxiety (Nieh et al., 2015; Stamatakis et al., 2016; Mangieri et al., 2018). PVH neurons, in addition to the well-studied CRH neurons, contain other types of neurons that send projections to the lateral septum (LS), a known brain region involved in aggression and anxiety, to inhibit feeding while promoting anxiety-like behaviors and reducing aggression (Xu et al., 2019). In particular, double knockout of synapse-associated protein 90/postsynaptic density-95-associated protein-3 (Sapap3), a key gene involved in anxiety-like behaviors, and Mc4r, one of the most frequent obesogenic genes in humans, completely rescues anxiety-like behaviors seen in Sapap3 knockouts and obesity in Mc4r knockouts (Xu et al., 2013), demonstrating an intimate interaction between the two seemingly unrelated genes. Thus, these and other findings from rodents argue that feeding and stress-related emotional modalities are regulated by a shared neuronal network widely distributed in the brain (Sweeney and Yang, 2017). Consistently, studies in humans suggest a profound effect of emotional feeding on obesity development, and aversive compulsivity and refusal to feed are co-existent in anorexia nervosa (Mattar et al., 2012; Godier and Park, 2014), suggesting that the co-regulation of feeding and stress-related emotional modalities is evolutionarily conserved. Thus, it is of importance to examine how these divergent brain regions respond to stress and HFD, and contribute to the current obesity epidemic.
Aging in the Central Nervous System of Organisms
Neuroendocrinal control of C. elegans aging from metabolic regulation
Caenorhabditis elegans (C. elegans) model was first introduced as a genetic model organism more than 50 years ago. In laboratory environment, the lifespan of C. elegans is approximately 3 weeks, and this nematode can also develop to a fertile adult in 3 days. Longevity studies using C. elegans have been conducted for decades, and among others, insulin/insulin-like growth factor-1 signaling (IIS) pathway has been best appreciated for the influence on longevity of this animal (Morris et al., 1996). There are three main components of the IIS pathway in C. elegans, including DAF-2 (C. elegans homolog of the insulin/insulin-like growth), AGE-1 (C. elegans homolog of phosphatidylinositol 3-kinase), and DAF-16 (C. elegans homolog of the forkhead box FoxO transcription factor). The mutants that regulate the activity of these signaling components have significant effects on increasing the lifespan of C. elegans (Friedman and Johnson, 1988a, b; Kenyon et al., 1993). The IIS pathway is also important for sensory perception involved in the regulation of the lifespan of C. elegans. While chemosensation-deficient C. elegans increased lifespan, chemosensory defects or elimination of gustatory neurons can dramatically decrease the lifespan effect of daf-16 mutation (Apfeld and Kenyon, 1999; Alcedo and Kenyon, 2004). The underlying mechanisms could be partially related to increased abilities of worms to handle various environmental and physiological stresses. In the long-lived strain of C. elegans, it was found that the increased stress resistance with reduced insulin signaling (Kenyon, 2010). Consistently, lifespan studies of C. elegans with genetic manipulation increasing stress resistance suggest that stress response is an important mechanism for modulating the lifespan (Durieux et al., 2011; Taylor and Dillin, 2013). Studies further showed that blocking of chemosensation can increase the nuclear translocation of DAF-16 in C. elegans, and decrease in DAF-16 nuclear localization can be induced through re-exposure to food in food-deprived C. elegans (Artan et al., 2016). DAF-9/P450, which is responsible for lipid hormone signaling in C. elegans, has also been shown to regulate larval development and aging (Gerisch et al., 2001; Jia et al., 2002; Rottiers et al., 2006). Temperature is one of the important factors in controlling lifespan by altering the metabolic rates (Jeong et al., 2012; Xiao et al., 2015). Ablation of thermosensory neurons can decrease the lifespan of C. elegans at 25 °C (Lee and Kenyon, 2009). Transient receptor potential cation channel subfamily V member 1 (TRPV1), a transient receptor potential (TRP) cation channel, is one of the major pain receptors that respond to noxious heat in vertebrates. Previous study showed that TRPV1-null mice increased lifespan compared to wild type (WT). Similarly, TRPV double-null mutant worms with deletion of both osm-9 and ocr-2 can prolong their lifespan (Riera et al., 2014). C. elegans uses three receptors, GCY-8, GCY-18 and GCY-23 as thermal sensors, and while triple mutant of these receptors did not detect an increasing temperature, it can lead to a shortened lifespan of worms at 25 °C (Inada et al., 2006; Chen et al., 2016). The response of organisms to various environmental and physiological stresses can determine their lifespan. In the long-lived strain of C. elegans, it was found that the increased stress resistance with reduced insulin signaling (Kenyon, 2010). In addition, lifespan studies of C. elegans with genetic manipulation increasing stress resistance suggest that the stress response pathway is an important mechanism for regulating lifespan (Durieux et al., 2011; Taylor and Dillin, 2013).
Hypothalamic control of aging in mammals: an emerging and exciting theme
One of the characteristics of physiological changes that occur during aging is the change in energy homeostasis (Hildrum et al., 2007). Despite being a healthy old man without degenerative disease, changes in metabolic activity can include sarcopenic obesity, a problem of fat gain but muscle loss in body composition. The loss of appetite in the elderly also leads to an imbalance between energy metabolism and nutrition (Wolden-Hanson et al., 2004). The ARC, located in the bottom of the hypothalamus, play important roles in regulating food uptake to maintain energy homeostasis. The ARC consists of orexigenic AgRP/neuropeptide Y (NPY) neurons and anorexigenic POMC/cocaine- and amphetamine-regulated transcript (CART) neurons, and notably POMC activity in aged mice was significantly reduced (Yang et al., 2012), but Pomc mRNA levels in ARC were not changed in young and middle-aged mice (Burke et al., 2014). Age-dependent metabolic alteration in aged rats was alleviated by overexpressing POMC gene in the hypothalamic ARC, including improved glucose metabolism, insulin sensitivity and reduced food consumption in aged rats (Li et al., 2005). NPY protein and NPY receptor levels also decrease in hypothalamic area of aged rats (Kowalski et al., 1992; Veyrat-Durebex et al., 2013). Through studies during the recent decade, it was discovered that the hypothalamus plays causal roles in systematic aging (Satoh et al., 2013; Zhang et al., 2013, 2017a). Previously, hypothalamic atrophy and signaling defects have been found in aging-related neurodegenerative diseases (Loskutova et al., 2010; Ishii and Iadecola, 2015; Vercruysse et al., 2018). The concept of NF-κB microinflammation in aging is clearly in alignment with its general role in metabolic syndrome which has been previously appreciated (Cai and Khor, 2019). Through experimental models, mid-aged rodents with an increase of hypothalamic NF-κB activity were reported to present aging acceleration and lifespan loss, while inhibition of NF-κB signaling in the hypothalamus was sufficient to lead to aging slowdown and a lifespan increase (Zhang et al., 2013). Caloric restriction, an effective dietary approach for aging intervention, has been shown to reduce hypothalamic gliosis and inflammatory changes (Yan et al., 2014; Sadagurski et al., 2017). Also, it should be mentioned that anti-aging drugs such as acarbose (Kothari et al., 2016), aspirin (Ong et al., 2007), rapamycin (Thomson et al., 2009), and metformin (Barzilai et al., 2016) all seem to be counter inflammatory. Thus, future research is warranted to investigate if hypothalamic inflammation is a common mechanism shared by these medical and non-medical anti-aging approaches.
Hypothalamic control of aging: targeting hypothalamic stem cells
In aging brains, one of the physiological features is a decreased production of new functional neurons from adult neural stem cells (Seib and Martin-Villalba, 2015). The derogatory effects of NF-κB signaling in the hypothalamus on aging seem to be causally related to its inhibition on gonadotropin-stimulating hormone (GnRH), a hypothalamic peptide which is classically known for its regulation on reproductive physiology but was found to important for promoting neurogenesis in this study (Zhang et al., 2013). Previous studies have found that hypothalamic neural stem/progenitor cells (htNSCs) are present in adult mice and physiologically important for hypothalamic long-term regulation over metabolic balance (Li et al., 2012). In 2017, it was discovered that htNSCs can function to supervise the process of aging while age-dependent loss of these cells is responsible for aging acceleration, and the mechanism is critically related to a novel endocrine function of these cells in secreting miRNA exosomes (Zhang et al., 2017a). This finding was consistent with the appreciation that decreased neurogenesis correlates with aging and the development of aging-related disorders (Greenberg and Jin, 2006; Molofsky et al., 2006; Encinas et al., 2011; Villeda et al., 2011; Sun et al., 2013; Baruch et al., 2014). As htNSC transplantation was tested for anti-aging effects, poor survival of implanted htNSCs was directly correlated with inflammatory status, but NF-κB inhibition in htNSCs was required for their survival (Li et al., 2012; Zhang et al., 2017a). A recent study have revealed a mechanism that seems important for aging-associated loss of htNSCs due to a noncoding long RNA named Hnscr (Xiao et al., 2020). In this study, it was reported that specific depletion of Hnscr in htNSCs accelerated the senescence of htNSCs and promoted aging-related physiological symptoms. The underlying molecular basis is related to the function of Hnscr in stabilizing Y-box binding protein 1 (YB-1) which is important for anti-apoptosis. Overall, the role of hypothalamic stem cells in systemic aging is not only consistent with neurodegenerative paradigm of aging and but has added a new perspective of aging mechanism from neuroendocrine regulation and dysregulation.
The Next Decade of Neural Control of Metabolism and Aging
It has become increasingly clear that human obesity is driven by both genetics and environmental changes. Research in last decades, especially recent emerging techniques including sophisticated mouse genetics, optogenetic manipulation, calcium indicator and single-cell sequencing have revealed ever-growing brain sites and neurons that are implicated in feeding and likely body weight regulation. However, it remains to be established which of these brain regions are relevant to human obesity development and therefore can be used as therapeutic targets against human obesity. This is thus critical to identify dysfunctions in brain neurons and pathways that underlie human obesity. Given the high palatable food environment as a strong driver for the current obesity epidemic, special emphasis should be put on the action of HFD on brain neurons. Recent studies have suggested a defective neuron responsiveness in AgRP neurons and CRH neurons by HFD (Beutler et al., 2020; Mazzone et al., 2020; Zhu et al., 2020), which may play a critical role in diet-induced obesity. Here we briefly speculate on a few major directions that we think are critical to unravel key brain neurons and neural pathways that are most relevant to human obesity. It is important to point out that these directions may represent a few of many potential important ones on obesity research. To go further in this field, several directions would be expected as below:
Single-cell sequencing
Single-cell sequencing is now a useful tool precisely combining neuron sub-types to metabolic functions. However, much efforts should be paid before completing single-cell sequencing. Subtle division of neuronal subtypes and brain regions are required for appropriate sample sites. Different sequencing methods (e.g., single-cell sequencing and translating ribosome affinity purification sequencing) should be combined to map the exact function-related neuron population. If possible, improved cell immunofluorescence with computational remodeling could provide anatomical evidence in parallel. After neuronal subdivision, how to label and manipulate those neurons is still a question to be solved.
Human relevance
Through decades of research using cells and/or experimental animals as model systems, a great deal of knowledge has been obtained regarding the molecular, cellular and systemic mechanisms for the regulation of whole-body metabolism in the studied models. However, a major question in the field still remains unsolved: what cause metabolic disorders in humans? Since most of the obesity-associated human variants affect genes that are enriched in the brain (Locke et al., 2015), we suggest to bring together the diverse expertise in human metabolic research and basic neuroendocrinology, to reveal the fundamental genetic basis and the neuroendocrine mechanisms that regulate metabolic homeostasis not only in animals but also in humans.
Sex difference
While the sex difference in energy homeostasis and other physiological systems are being increasingly recognized (Arnold, 2014), the underlying mechanisms remain elusive. In addition to the genes encoded by the sex chromosomes and the gonadal hormones (and their receptors), we predict that a large number of autosome-encoded genes also contribute to the sex differences in energy balance. However, a lot of such genes have been missed simply because female animals are purposely omitted in most studies to reduce costs and efforts. Therefore we suggest that all pharmacological and genetic investigations into energy balance in animal models should include both males and females, as required by the funding agencies, e.g., the National Institutes of Health (NIH) (Tannenbaum et al., 2016).
Immunometabolism
The impact of immune system on brain-regulated metabolism should be emphasized in future. As to the brain, the role of microglia and astrocytes in regulating metabolic disorder has long been ignored. With bulk of data accumulated in the recent years, the essential roles played by these glial cells become clearer than ever. Yet, whether and how microglia in the hypothalamus precisely detect diet-induced metabolic stresses, how microglia interact with astrocytes, and to which extent the heterogeneity is when different hypothalamic neurons interact with different glial cells are remain to be investigated to demonstrate this multi-system collaboration. Nevertheless, as we have discussed above, the ultimate role of glial population is to serves as the “supporting unit” for neurons to function, it is plausibly to prospect that glial cells are readily to adapt their immune and metabolic functions to match the demands of neuroprotection in the context of metabolic stress.
Gut microbiota
It is necessary to comprehend CNS-peripheral tissue network. We have known that gut microbiota produces different compounds and participates in brain control of metabolism. However, many details remain to be explored.
First, this field lack comprehensive understandings from microbiota to brain. Compared with compounds gut microbiota produces, evidence of how microbiota responds to environment, produces those compounds and finally mediate CNS is elusive. Second, the role of vagus nerve in innervating gut microbiota and brain-regulated metabolism need more clarification in future. Finally, further studies should consider human-subject reproduction, establishing causal link of microbiota and brain-metabolic diseases, and finally promote related therapeutic methods.
Stress
It has become increasingly clear from extensive human behavioral studies that both chronic stress and HFD contribute to the current obesity epidemic. In contrast to HFD, the mechanism underlying the effect of stress on body weight has received much less attention in rodent research. One major reason is that it is difficult to model chronic stress-induced emotional modalities in rodents. Identification of a reliable chronic stress model that recapitulates the effect of human chronic stress is a critical next step to unravel the underlying mechanism for chronic stress induced obesity in humans. It appears that chronic social stress shows some promises in mimicking the effect on feeding and body weight of human chronic stress. Given the availability of imaging data from humans with chronic stress, future studies using rodent brain imaging may help reveal whether there are similar changes in brain imaging. Given the complex nature of underlying neurocircuitry for both feeding and stress-related emotional modalities (Yau and Potenza, 2013; Herzog, 2020), many brain neurons and neural pathways including traditionally viewed as feeding and emotion brain centers are likely to be involved. Emerging observations demonstrate that feeding brain centers are deeply involved in stress-related emotional modalities and emotional brain centers inherently regulate feeding behaviors. The cellular mechanism underlying the effect of HFD and chronic stress is largely unknown. Notably, HFD blunts PVH CRH neuron responsiveness and the diurnal rhythms of downstream HPA axis, suggesting disruptive neuron responsiveness as a potential cellular mechanism. It is critical to examine whether chronic stress also induce a similar effect on these neurons. It is clear that many brain neurons and neural pathways are involved in chronic stress responses, feeding and body weight, it is thus imperative to understand the relative contribution of these brain regions and pathways in mediating chronic stress and HFD on obesity development, especially those brain regions that mediate the synergistic effect of HFD and chronic stress on weight gain.
Aging
Recent research has led to appreciation the hypothalamic basis for developing metabolic syndrome and aging involving some overlapping cellular and signaling pathways through NF-κB-dependent microinflammatory induction in neurons, glial cells and hypothalamic neural stem/progenitor cells. With these findings which have begun to shed a light on this important topic, there are many remarkable questions which can be asked, and the following is to briefly list a few perspectives for researchers to consider for future studies.
First of all, given the close relationships among hypothalamic dysfunction, aging and metabolic syndrome (Fig. 4), it is necessary to deeply understand how these various aspects of general pathological and pathophysiological domains are biologically and programmatically integrated. Secondly, regarding the underlying molecular operation, given the critical role of hypothalamic NF-κB as revealed in a good body of literature, it will be informative to investigate how NF-κB might interact with other aging-regulating molecular events such as SIRTs, Adenosine 5′-monophosphate (AMP)-activated protein kinase (AMPK) and mammalian target of rapamycin (mTOR) signaling and if there are NF-κB-unrelated molecular pathways to be critically accountable. Thirdly, while animal models have revealed the causal contribution of hypothalamic microinflammation to the hypothalamic mechanism of aging and related metabolic syndrome, it should be highly valuable to profile if these inflammatory changes occur in human conditions and address the importance of this hypothalamic microinflammatory condition in terms of human aging and related disease, Finally, as there has been a great deal of interest to develop anti-inflammatory therapeutical approaches, current clinical anti-inflammatory options have limited features and are more suitable for solving classical inflammation due to conditions such as infection rather than non-classical microinflammation in aging and related disorders. Thus, new anti-inflammatory approaches should be explored, for example, hypothalamic stem cell exosome-based intervention, which could be more effective in targeting aging-related microinflammation and neuroinflammation. Besides, to incorporate aging-related genes and neural pathways from animal studies into applications will be worthy of future clinical research.

Hypothalamic scheme for connecting aging with metabolic disorders. The diagram depicts the backbone relationships among the hypothalamus, aging and metabolic syndrome, suggesting that hypothalamic dysfunctions represent a key cause for initiating the onset of aging and metabolic syndromes although also individually depending on the context of age and chronic nutritional status but with some overlapping processes through the changes in neuroendocrine system and autonomic nervous system (ANS) which are important for hypothalamic and central control of whole-body physiological balance and homeostasis. Over a chronic term, aging and metabolic syndrome physiological changes can not only enhance each other but also reinforce hypothalamic dysfunctions, thus keeping the mechanisms and outcomes in a self-sustaining loop
Abbreviations
5-HT, 5-hydroxytryptamine; 5-HT2CR, 5-hydroxytryptamine 2C receptor; ADCY3, adenylate cyclase 3; AGEs, advanced glycation end-products; AgRP, agouti-related peptide; ALDH1L1, 10-formyltetrahydrofolate dehydrogenase; AMP, adenosine 5‘-monophosphate; AMPK, adenosine 5′-monophosphate-activated protein kinase; ANKRD26, ankyrin repeat domain 26; ANS, autonomic nervous system, APOE, apolipoprotein E; AR, androgen receptor; ARC, arcuate nucleus of hypothalamus; ARNT2, Aryl hydrocarbon receptor nuclear translocator 2; AT2R, angiotensin II receptor; BAs, bile acids; BBB, blood-brain barrier; BDNF, brain-derived neurotropic factor; BMAL1, brain and muscle arnt-like 1; BMI, body mass index; C. elegans, Caenorhabditis elegans; CART, cocaine- and amphetamine-regulated transcript; CD68, cluster of differentiation 68; CEP19, centrosomal protein 19; ClpB, caseinolytic protease B; CNS, central nervous system; CRH, corticotropin releasing hormone; CRISPR, clustered regularly interspaced short palindromic repeats; CX3CL1, C-X3-C motif chemokine ligand 1; dLS, dorsal lateral septal nuclei; DMH, dorsomedial hypothalamus; DRN, dorsal raphe nucleus; DVC, dorsal vagal complex; ENU, N-ethyl-N-nitrosourea; ERα, estrogen receptor alpha (also called estrogen receptor 1, ESR1); ERβ, estrogen receptor beta; FGF19, fibroblast growth factor 19; FMT, fecal microbiota transplantation; FTO, FTO alpha-ketoglutarate dependent dioxygenase in Homo sapiens and fat mass and obesity associated in Mus musculus; GABA, gamma-aminobutyric acid; GF, germ-free; GLAST, glutamate aspartate transporter; GLP-1, glucagon-like peptide-1; Glp1R, glucagon-like peptide-1 receptor; GnRH, gonadotropin-stimulating hormone; GPR30, G protein-coupled receptor 30; GPR41, G protein-coupled receptor 41; GPR43, G protein-coupled receptor 43; GPR54, G protein-coupled receptor 54; GWAS, genome-wide association studies; HFD, high-fat diet; HK2, hexokinase 2; HPA, hypothalamus-pituitary-adrenal axis; htNSCs, hypothalamic neural stem/progenitor cells; IIS, insulin/insulin-like growth factor-1 signaling; IL-1β, interleukin 1 beta; IL-6, interleukin- 6;IP, intraperitoneal; Kiss1, kisspeptin; LEPR, leptin receptor; LHA, lateral hypothalamic area; LPL, lipoprotein lipase; LPS, lipopolysaccharide; LS, lateral septum; MBH, mediobasal hypothalamus; MC4R, melanocortin 4 receptor; ME, median eminence; MeA, medial amygdala; mTOR, mammalian target of rapamycin; MYT1L, myelin transcription factor 1 like; NCOA1, nuclear receptor coactivator 1; NF-κB, nuclear factor-kappa B; NIH, National Institutes of Health; NPY, neuropeptide Y; NREM, non-rapid eye movement; nsSNP, nonsynonymous single‐nucleotide polymorphisms; NTRK2, neurotrophic receptor tyrosine kinase 2; NTS, nucleus of the solitary tract; OGT, O-GlcNAc transferase; OVX, ovariectomy; OXPHOS, oxidative phosphorylation; Oxtr, oxytocin receptor; POA, preoptic area; POMC, pro-opiomelanocortin; PVH, paraventricular nucleus of the hypothalamus; PYY, peptide YY; ROS, oxygen species; Rprm, reprimo; Sapap3, synapse-associated protein 90/postsynaptic density-95-associated protein-3; SCFA, short-chain fatty acid; SCN, suprachiasmatic nucleus; SH2B1, SH2B adaptor protein 1; SIM1, single-minded 1; SIRT1, Sirtuin 1; SNP, single nucleotide polymorphism; SNV, single nucleotide variants; SRC-1, steroid receptor co-activator-1; SRY, sex determining region Y; SST, somatostatin; STAT3, signal transducer and activator of transcription 3; Tac1, tachykinin 1; TLR4. Toll-like receptor-4; TNFα, tumor necrosis factor alpha; TrkB, tropomyosin-related kinase B; TRP, transient receptor potential; TrpC5, transient receptor potential channel 5; TRPV1, transient receptor potential cation channel subfamily V member 1; UCP2, uncoupling protein 2; Vglut2, vesicular glutamate transporter2; VMH, ventromedial hypothalamus; WT, wide type; YB-1, Y-box binding protein 1; α-MSH, alpha-melanocyte-stimulating hormone; β-MSH, beta-melanocyte-stimulating hormone
REFERENCES
Adak A, Khan MR (2019) An insight into gut microbiota and its functionalities. Cell Mol Life Sci 76:473–493
Al Tuwaijri A, Alfadhel M (2019) MYT1L mutation in a patient causes intellectual disability and early onset of obesity: a case report and review of the literature. J Pediatr Endocrinol Metab 32:409–413
Alcedo J, Kenyon C (2004) Regulation of C elegans longevity by specific gustatory and olfactory neurons. Neuron 41:45–55
Alkemade A, Yi CX, Pei L, Harakalova M, Swaab DF, la Fleur SE, Fliers E, Kalsbeek A (2012) AgRP and NPY expression in the human hypothalamic infundibular nucleus correlate with body mass index, whereas changes in alphaMSH are related to type 2 diabetes. J Clin Endocrinol Metab 97:E925–E933
Apfeld J, Kenyon C (1999) Regulation of lifespan by sensory perception in Caenorhabditis elegans. Nature 402:804–809
Appiakannan HS, Kestyus DR, Weber ET (2019) Effects of high fat diet and chronic circadian challenge on glucocorticoid regulation in C57BL/6J mice. Physiol Behav 204:100–105
Arnold AP (2014) Conceptual frameworks and mouse models for studying sex differences in physiology and disease: why compensation changes the game. Exp Neurol 259:2–9
Artan M, Jeong DE, Lee D, Kim YI, Son HG, Husain Z, Kim J, Altintas O, Kim K, Alcedo J et al (2016) Food-derived sensory cues modulate longevity via distinct neuroendocrine insulin-like peptides. Genes Dev 30:1047–1057
Asarian L, Geary N (2007) Estradiol enhances cholecystokinin-dependent lipid-induced satiation and activates estrogen receptor-alpha-expressing cells in the nucleus tractus solitarius of ovariectomized rats. Endocrinology 148:5656–5666
Attia E (2010) Anorexia nervosa: current status and future directions. Annu Rev Med 61:425–435
Auvinen HE, Romijn JA, Biermasz NR, Pijl H, Havekes LM, Smit JW, Rensen PC, Pereira AM (2012) The effects of high fat diet on the basal activity of the hypothalamus–pituitary–adrenal axis in mice. J Endocrinol 214:191–197
Bäckhed F, Ding H, Wang T, Hooper LV, Koh GY, Nagy A, Semenkovich CF, Gordon JI (2004) The gut microbiota as an environmental factor that regulates fat storage. Proc Natl Acad Sci USA 101:15718–15723
Baraboi ED, Michel C, Smith P, Thibaudeau K, Ferguson AV, Richard D (2010) Effects of albumin-conjugated PYY on food intake: the respective roles of the circumventricular organs and vagus nerve. Eur J Neurosci 32:826–839
Baron R, Babcock AA, Nemirovsky A, Finsen B, Monsonego A (2014) Accelerated microglial pathology is associated with Aβ plaques in mouse models of Alzheimer’s disease. Aging Cell 13:584–595
Bartolomucci A, Cabassi A, Govoni P, Ceresini G, Cero C, Berra D, Dadomo H, Franceschini P, Dell’Omo G, Parmigiani S et al (2009) Metabolic consequences and vulnerability to diet-induced obesity in male mice under chronic social stress. PLoS ONE 4:e4331
Baruch K, Deczkowska A, David E, Castellano JM, Miller O, Kertser A, Berkutzki T, Barnett-Itzhaki Z, Bezalel D, Wyss-Coray T et al (2014) Aging. Aging-induced type I interferon response at the choroid plexus negatively affects brain function. Science 346:89–93
Barzilai N, Crandall JP, Kritchevsky SB, Espeland MA (2016) Metformin as a tool to target aging. Cell Metab 23:1060–1065
Baufeld C, Osterloh A, Prokop S, Miller KR, Heppner FL (2016) High-fat diet-induced brain region-specific phenotypic spectrum of CNS resident microglia. Acta Neuropathol 132:361–375
Benz V, Bloch M, Wardat S, Bohm C, Maurer L, Mahmoodzadeh S, Wiedmer P, Spranger J, Foryst-Ludwig A, Kintscher U (2012) Sexual dimorphic regulation of body weight dynamics and adipose tissue lipolysis. PLoS ONE 7:e37794
Berglund ED, Liu C, Sohn JW, Liu T, Kim MH, Lee CE, Vianna CR, Williams KW, Xu Y, Elmquist JK (2013) Serotonin 2C receptors in pro-opiomelanocortin neurons regulate energy and glucose homeostasis. J Clin Investig 123:5061–5070
Berry A, Bellisario V, Capoccia S, Tirassa P, Calza A, Alleva E, Cirulli F (2012) Social deprivation stress is a triggering factor for the emergence of anxiety- and depression-like behaviours and leads to reduced brain BDNF levels in C57BL/6J mice. Psychoneuroendocrinology 37:762–772
Beutler LR, Corpuz TV, Ahn JS, Kosar S, Song W, Chen Y, Knight ZA (2020) Obesity causes selective and long-lasting desensitization of AgRP neurons to dietary fat. Elife 9:e55909
Block ML, Zecca L, Hong JS (2007) Microglia-mediated neurotoxicity: uncovering the molecular mechanisms. Nat Rev Neurosci 8:57–69
Boulais J, Trost M, Landry CR, Dieckmann R, Levy ED, Soldati T, Michnick SW, Thibault P, Desjardins M (2010) Molecular characterization of the evolution of phagosomes. Mol Syst Biol 6:423
Bravo JA, Forsythe P, Chew MV, Escaravage E, Savignac HM, Dinan TG, Bienenstock J, Cryan JF (2011) Ingestion of Lactobacillus strain regulates emotional behavior and central GABA receptor expression in a mouse via the vagus nerve. Proc Natl Acad Sci USA 108:16050–16055
Breton J, Tennoune N, Lucas N, Francois M, Legrand R, Jacquemot J, Goichon A, Guerin C, Peltier J, Pestel-Caron M et al (2016) Gut commensal E. coli proteins activate host satiety pathways following nutrient-induced bacterial growth. Cell Metab 23:324–334
Brighton CA, Rievaj J, Kuhre RE, Glass LL, Schoonjans K, Holst JJ, Gribble FM, Reimann F (2015) Bile acids trigger GLP-1 release predominantly by accessing basolaterally located G protein-coupled bile acid receptors. Endocrinology 156:3961–3970
Broadwell RD, Balin BJ, Salcman M, Kaplan RS (1983) Brain–blood barrier? Yes and no. Proc Natl Acad Sci USA 80:7352–7356
Buckman LB, Thompson MM, Lippert RN, Blackwell TS, Yull FE, Ellacott KL (2015) Evidence for a novel functional role of astrocytes in the acute homeostatic response to high-fat diet intake in mice. Mol Metab 4:58–63
Burke LK, Doslikova B, D’Agostino G, Garfield AS, Farooq G, Burdakov D, Low MJ, Rubinstein M, Evans ML, Billups B et al (2014) 5-HT obesity medication efficacy via POMC activation is maintained during aging. Endocrinology 155:3732–3738
Burnett CJ, Li C, Webber E, Tsaousidou E, Xue SY, Bruning JC, Krashes MJ (2016) Hunger-driven motivational state competition. Neuron 92:187–201
Burns JC, Cotleur B, Walther DM, Bajrami B, Rubino SJ, Wei R, Franchimont N, Cotman SL, Ransohoff RM, Mingueneau M (2020) Differential accumulation of storage bodies with aging defines discrete subsets of microglia in the healthy brain. Elife 9:e57495
Butera PC, Beikirch RJ (1989) Central implants of diluted estradiol: independent effects on ingestive and reproductive behaviors of ovariectomized rats. Brain Res 491:266–273
Goswami C, Iwasaki Y, Yada T (2018) Short-chain fatty acids suppress food intake by activating vagal afferent neurons. J Nutr Biochem 57:130–135
Cai D, Khor S (2019) “Hypothalamic Microinflammation” paradigm in aging and metabolic diseases. Cell Metab 30:19–35
Callewaert F, Venken K, Ophoff J, De Gendt K, Torcasio A, van Lenthe GH, Van Oosterwyck H, Boonen S, Bouillon R, Verhoeven G et al (2009) Differential regulation of bone and body composition in male mice with combined inactivation of androgen and estrogen receptor-alpha. FASEB J 23:232–240
Cani PD, Bibiloni R, Knauf C, Waget A, Neyrinck AM, Delzenne NM, Burcelin RJD (2008) Changes in gut microbiota control metabolic endotoxemia-induced inflammation in high-fat diet-induced obesity and diabetes in mice. Diabetes 57:1470–1481
Cani PD, Dewever C, Delzenne NM (2004) Inulin-type fructans modulate gastrointestinal peptides involved in appetite regulation (glucagon-like peptide-1 and ghrelin) in rats. Br J Nutr 92:521–526
Cani PD, Van Hul M, Lefort C, Depommier C, Rastelli M, Everard A (2019) Microbial regulation of organismal energy homeostasis. Nat Metab 1:34–46
Cannon B, Nedergaard J (2004) Brown adipose tissue: function and physiological significance. Physiol Rev 84:277–359
Cao X, Xu P, Oyola MG, Xia Y, Yan X, Saito K, Zou F, Wang C, Yang Y, Hinton A Jr et al (2014) Estrogens stimulate serotonin neurons to inhibit binge-like eating in mice. J Clin Investig 124:4351–4362
Cassidy RM, Lu Y, Jere M, Tian JB, Xu Y, Mangieri LR, Felix-Okoroji B, Selever J, Xu Y, Arenkiel BR et al (2019) A lateral hypothalamus to basal forebrain neurocircuit promotes feeding by suppressing responses to anxiogenic environmental cues. Sci Adv 5:eaav1640
Chen X, McClusky R, Chen J, Beaven SW, Tontonoz P, Arnold AP, Reue K (2012) The number of x chromosomes causes sex differences in adiposity in mice. PLoS Genet 8:e1002709
Chen YC, Chen HJ, Tseng WC, Hsu JM, Huang TT, Chen CH, Pan CL (2016) A C. elegans thermosensory circuit regulates longevity through crh-1/CREB-dependent flp-6 neuropeptide signaling. Dev Cell 39:209–223
Chiba S, Numakawa T, Ninomiya M, Richards MC, Wakabayashi C, Kunugi H (2012) Chronic restraint stress causes anxiety- and depression-like behaviors, downregulates glucocorticoid receptor expression, and attenuates glutamate release induced by brain-derived neurotrophic factor in the prefrontal cortex. Prog Neuropsychopharmacol Biol Psychiatry 39:112–119
Clement K, Vaisse C, Lahlou N, Cabrol S, Pelloux V, Cassuto D, Gourmelen M, Dina C, Chambaz J, Lacorte JM et al (1998) A mutation in the human leptin receptor gene causes obesity and pituitary dysfunction. Nature 392:398–401
Clemente JC, Ursell LK, Parfrey LW, Knight RJC (2012) The impact of the gut microbiota on human health: an integrative view. Cell 148:1258–1270
Cohen P, Zhao C, Cai X, Montez JM, Rohani SC, Feinstein P, Mombaerts P, Friedman JM (2001) Selective deletion of leptin receptor in neurons leads to obesity. J Clin Investig 108:1113–1121
Correa SM, Newstrom DW, Warne JP, Flandin P, Cheung CC, Lin-Moore AT, Pierce AA, Xu AW, Rubenstein JL, Ingraham HA (2015) An estrogen-responsive module in the ventromedial hypothalamus selectively drives sex-specific activity in females. Cell Rep 10:62–74
Cryan JF, O’Riordan KJ, Cowan CSM, Sandhu KV, Bastiaanssen TFS, Boehme M, Codagnone MG, Cussotto S, Fulling C, Golubeva AV et al (2019) The microbiota-gut-brain axis. Physiol Rev 99:1877–2013
D’Agostino G, Lyons D, Cristiano C, Lettieri M, Olarte-Sanchez C, Burke LK, Greenwald-Yarnell M, Cansell C, Doslikova B, Georgescu T et al (2018) Nucleus of the solitary tract serotonin 5-HT2C receptors modulate food intake. Cell Metab 28:619-630.e615
d’Anglemont de Tassigny X, Fagg LA, Dixon JP, Day K, Leitch HG, Hendrick AG, Zahn D, Franceschini I, Caraty A, Carlton MB et al (2007) Hypogonadotropic hypogonadism in mice lacking a functional Kiss1 gene. Proc Natl Acad Sci USA 104:10714–10719
Dalby MJ, Aviello G, Ross AW, Walker AW, Barrett P, Morgan PJ (2018) Diet induced obesity is independent of metabolic endotoxemia and TLR4 signalling, but markedly increases hypothalamic expression of the acute phase protein, SerpinA3N. Sci Rep 8:15648
Dallman MF (2010) Stress-induced obesity and the emotional nervous system. Trends Endocrinol Metab 21:159–165
Dalmasso C, Amigone JL, Vivas L (2011) Serotonergic system involvement in the inhibitory action of estrogen on induced sodium appetite in female rats. Physiol Behav 104:398–407
Davis KE, Carstens EJ, Irani BG, Gent LM, Hahner LM, Clegg DJ (2014) Sexually dimorphic role of G protein-coupled estrogen receptor (GPER) in modulating energy homeostasis. Horm Behav 66:196–207
de Souza FS, Nasif S, Lopez-Leal R, Levi DH, Low MJ, Rubinsten M (2011) The estrogen receptor alpha colocalizes with proopiomelanocortin in hypothalamic neurons and binds to a conserved motif present in the neuron-specific enhancer nPE2. Eur J Pharmacol 660:181–187
De Vadder F, Kovatcheva-Datchary P, Goncalves D, Vinera J, Zitoun C, Duchampt A, Backhed F, Mithieux G (2014) Microbiota-generated metabolites promote metabolic benefits via gut-brain neural circuits. Cell 156:84–96
De Vries GJ, Rissman EF, Simerly RB, Yang LY, Scordalakes EM, Auger CJ, Swain A, Lovell-Badge R, Burgoyne PS, Arnold AP (2002) A model system for study of sex chromosome effects on sexually dimorphic neural and behavioral traits. J Neurosci 22:9005–9014
Delzenne NM, Cani PD, Daubioul C, Neyrinck AM (2005) Impact of inulin and oligofructose on gastrointestinal peptides. Br J Nutr 93(Suppl 1):S157–S161
Deussing JM, Chen A (2018) The corticotropin-releasing factor family: physiology of the stress response. Physiol Rev 98:2225–2286
Dietrich MO, Zimmer MR, Bober J, Horvath TL (2015) Hypothalamic Agrp neurons drive stereotypic behaviors beyond feeding. Cell 160:1222–1232
Domecq JP, Prutsky G, Leppin A, Sonbol MB, Altayar O, Undavalli C, Wang Z, Elraiyah T, Brito JP, Mauck KF et al (2015) Clinical review: drugs commonly associated with weight change: a systematic review and meta-analysis. J Clin Endocrinol Metab 100:363–370
Dorfman MD, Krull JE, Douglass JD, Fasnacht R, Lara-Lince F, Meek TH, Shi X, Damian V, Nguyen HT, Matsen ME et al (2017) Sex differences in microglial CX3CR1 signalling determine obesity susceptibility in mice. Nat Commun 8:14556
Drewett RF (1973) Sexual behaviour and sexual motivation in the female rat. Nature 242:476–477
Durieux J, Wolff S, Dillin A (2011) The cell-non-autonomous nature of electron transport chain-mediated longevity. Cell 144:79–91
Encinas JM, Michurina TV, Peunova N, Park JH, Tordo J, Peterson DA, Fishell G, Koulakov A, Enikolopov G (2011) Division-coupled astrocytic differentiation and age-related depletion of neural stem cells in the adult hippocampus. Cell Stem Cell 8:566–579
Fagman JB, Wilhelmson AS, Motta BM, Pirazzi C, Alexanderson C, De Gendt K, Verhoeven G, Holmang A, Anesten F, Jansson JO et al (2015) The androgen receptor confers protection against diet-induced atherosclerosis, obesity, and dyslipidemia in female mice. FASEB J 29:1540–1550
Fan W, Yanase T, Nomura M, Okabe T, Goto K, Sato T, Kawano H, Kato S, Nawata H (2005) Androgen receptor null male mice develop late-onset obesity caused by decreased energy expenditure and lipolytic activity but show normal insulin sensitivity with high adiponectin secretion. Diabetes 54:1000–1008
Farooqi IS (2008) Monogenic human obesity. Front Horm Res 36:1–11
Farooqi S, O’Rahilly S (2006) Genetics of obesity in humans. Endocr Rev 27:710–718
Finan B, Yang B, Ottaway N, Stemmer K, Muller TD, Yi CX, Habegger K, Schriever SC, Garcia-Caceres C, Kabra DG et al (2012) Targeted estrogen delivery reverses the metabolic syndrome. Nat Med 18:1847–1856
Flier JS (2004) Obesity wars: molecular progress confronts an expanding epidemic. Cell 116:337–350
Foryst-Ludwig A, Clemenz M, Hohmann S, Hartge M, Sprang C, Frost N, Krikov M, Bhanot S, Barros R, Morani A et al (2008) Metabolic actions of estrogen receptor beta (ERbeta) are mediated by a negative cross-talk with PPARgamma. PLoS Genet 4:e1000108
Friedman DB, Johnson TE (1988a) 3 Mutants that extend both mean and maximum life-span of the nematode, Caenorhabditis elegans, define the age-1 gene. J Gerontol 43:B102–B109
Friedman DB, Johnson TE (1988b) A mutation in the age-1 gene in Caenorhabditis elegans lengthens life and reduces hermaphrodite fertility. Genetics 118:75–86
Friedman J (2016) The long road to leptin. J Clin Investig 126:4727–4734
Gao Q, Mezei G, Nie Y, Rao Y, Choi CS, Bechmann I, Leranth C, Toran-Allerand D, Priest CA, Roberts JL et al (2007) Anorectic estrogen mimics leptin’s effect on the rewiring of melanocortin cells and Stat3 signaling in obese animals. Nat Med 13:89–94
Gao Y, Bielohuby M, Fleming T, Grabner GF, Foppen E, Bernhard W, Guzmán-Ruiz M, Layritz C, Legutko B, Zinser E et al (2017a) Dietary sugars, not lipids, drive hypothalamic inflammation. Mol Metab 6:897–908
Gao Y, Ottaway N, Schriever SC, Legutko B, García-Cáceres C, de la Fuente E, Mergen C, Bour S, Thaler JP, Seeley RJ et al (2014) Hormones and diet, but not body weight, control hypothalamic microglial activity. Glia 62:17–25
Gao Y, Vidal-Itriago A, Kalsbeek MJ, Layritz C, Garcia-Caceres C, Tom RZ, Eichmann TO, Vaz FM, Houtkooper RH, van der Wel N et al (2017b) Lipoprotein lipase maintains microglial innate immunity in obesity. Cell Rep 20:3034–3042
Gao Y, Yao T, Deng Z, Sohn JW, Sun J, Huang Y, Kong X, Yu KJ, Wang RT, Chen H et al (2017c) TrpC5 mediates acute leptin and serotonin effects via Pomc neurons. Cell Rep 18:583–592
García-Cáceres C, Balland E, Prevot V, Luquet S, Woods SC, Koch M, Horvath TL, Yi CX, Chowen JA, Verkhratsky A et al (2019) Role of astrocytes, microglia, and tanycytes in brain control of systemic metabolism. Nat Neurosci 22:7–14
García-Cáceres C, Quarta C, Varela L, Gao Y, Gruber T, Legutko B, Jastroch M, Johansson P, Ninkovic J, Yi CX et al (2016) Astrocytic insulin signaling couples brain glucose uptake with nutrient availability. Cell 166:867–880
Garcia-Eguren G, Giro O, Romero MDM, Grasa M, Hanzu FA (2019) Chronic hypercortisolism causes more persistent visceral adiposity than HFD-induced obesity. J Endocrinol 242:65–77
Geary N, Asarian L, Korach KS, Pfaff DW, Ogawa S (2001) Deficits in E2-dependent control of feeding, weight gain, and cholecystokinin satiation in ER-alpha null mice. Endocrinology 142:4751–4757
Gerisch B, Weitzel C, Kober-Eisermann C, Rottiers V, Antebi A (2001) A hormonal signaling pathway influencing C. elegans metabolism, reproductive development, and life span. Dev Cell 1:841–851
Ginhoux F, Greter M, Leboeuf M, Nandi S, See P, Gokhan S, Mehler MF, Conway SJ, Ng LG, Stanley ER et al (2010) Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science 330:841–845
Godier LR, Park RJ (2014) Compulsivity in anorexia nervosa: a transdiagnostic concept. Front Psychol 5:778
Goldmann T, Wieghofer P, Jordao MJ, Prutek F, Hagemeyer N, Frenzel K, Amann L, Staszewski O, Kierdorf K, Krueger M et al (2016) Origin, fate and dynamics of macrophages at central nervous system interfaces. Nat Immunol 17:797–805
Gomes JAS, Silva JF, Marcal AP, Silva GC, Gomes GF, de Oliveira ACP, Soares VL, Oliveira MC, Ferreira AVM, Aguiar DC (2020) High-refined carbohydrate diet consumption induces neuroinflammation and anxiety-like behavior in mice. J Nutr Biochem 77:108317
Gomez Perdiguero E, Klapproth K, Schulz C, Busch K, Azzoni E, Crozet L, Garner H, Trouillet C, de Bruijn MF, Geissmann F et al (2015) Tissue-resident macrophages originate from yolk-sac-derived erythro-myeloid progenitors. Nature 518:547–551
Goodfellow PN, Lovell-Badge R (1993) SRY and sex determination in mammals. Annu Rev Genet 27:71–92
Grabert K, Michoel T, Karavolos MH, Clohisey S, Baillie JK, Stevens MP, Freeman TC, Summers KM, McColl BW (2016) Microglial brain region-dependent diversity and selective regional sensitivities to aging. Nat Neurosci 19:504–516
Greenberg DA, Jin K (2006) Turning neurogenesis up a Notch. Nat Med 12:884–885
Grove KL, Fried SK, Greenberg AS, Xiao XQ, Clegg DJ (2010) A microarray analysis of sexual dimorphism of adipose tissues in high-fat-diet-induced obese mice. Int J Obes (Lond) 34:989–1000
Grumbach MM, Auchus RJ (1999) Estrogen: consequences and implications of human mutations in synthesis and action. J Clin Endocrinol Metab 84:4677–4694
Haas E, Bhattacharya I, Brailoiu E, Damjanovic M, Brailoiu GC, Gao X, Mueller-Guerre L, Marjon NA, Gut A, Minotti R et al (2009) Regulatory role of G protein-coupled estrogen receptor for vascular function and obesity. Circ Res 104:288–291
Harris RB (2015) Chronic and acute effects of stress on energy balance: are there appropriate animal models? Am J Physiol Regul Integr Comp Physiol 308:R250–R265
Harris RB (2017) Repeated restraint stress lowers the threshold for response to third ventricle CRF administration. Horm Behav 89:64–68
Hatori M, Vollmers C, Zarrinpar A, DiTacchio L, Bushong EA, Gill S, Leblanc M, Chaix A, Joens M, Fitzpatrick JA et al (2012) Time-restricted feeding without reducing caloric intake prevents metabolic diseases in mice fed a high-fat diet. Cell Metab 15:848–860
Heine PA, Taylor JA, Iwamoto GA, Lubahn DB, Cooke PS (2000) Increased adipose tissue in male and female estrogen receptor-alpha knockout mice. Proc Natl Acad Sci USA 97:12729–12734
Heneka MT, Carson MJ, El Khoury J, Landreth GE, Brosseron F, Feinstein DL, Jacobs AH, Wyss-Coray T, Vitorica J, Ransohoff RM et al (2015) Neuroinflammation in Alzheimer’s disease. Lancet Neurol 14:388–405
Herzog H (2020) Integrated pathways that control stress and energy homeostasis. Nat Rev Endocrinol 16:75–76
Hildrum B, Mykletun A, Hole T, Midthjell K, Dahl AA (2007) Age-specific prevalence of the metabolic syndrome defined by the International Diabetes Federation and the National Cholesterol Education Program: the Norwegian HUNT 2 study. BMC Public Health 7:220
Holder JL Jr, Butte NF, Zinn AR (2000) Profound obesity associated with a balanced translocation that disrupts the SIM1 gene. Hum Mol Genet 9:101–108
Hong KW, Lim JE, Go MJ, Shin Cho Y, Ahn Y, Han BG, Oh B (2012) Recapitulation of the association of the Val66Met polymorphism of BDNF gene with BMI in Koreans. Obesity (Silver Spring) 20:1871–1875
Hood S, Amir S (2018) Biological clocks and rhythms of anger and aggression. Front Behav Neurosci 12:4
Horvath TL (2005) The hardship of obesity: a soft-wired hypothalamus. Nat Neurosci 8:561–565
Horvath TL, Sarman B, García-Cáceres C, Enriori PJ, Sotonyi P, Shanabrough M, Borok E, Argente J, Chowen JA, Perez-Tilve D et al (2010) Synaptic input organization of the melanocortin system predicts diet-induced hypothalamic reactive gliosis and obesity. Proc Natl Acad Sci USA 107:14875–14880
Huang Q, Rivest R, Richard D (1998) Effects of leptin on corticotropin-releasing factor (CRF) synthesis and CRF neuron activation in the paraventricular hypothalamic nucleus of obese (ob/ob) mice. Endocrinology 139:1524–1532
Huszar D, Lynch CA, Fairchild-Huntress V, Dunmore JH, Fang Q, Berkemeier LR, Gu W, Kesterson RA, Boston BA, Cone RD et al (1997) Targeted disruption of the melanocortin-4 receptor results in obesity in mice. Cell 88:131–141
Inada H, Ito H, Satterlee J, Sengupta P, Matsumoto K, Mori I (2006) Identification of guanylyl cyclases that function in thermosensory neurons of Caenorhabditis elegans. Genetics 172:2239–2252
Ishii M, Iadecola C (2015) Metabolic and non-cognitive manifestations of Alzheimer’s disease: the hypothalamus as both culprit and target of pathology. Cell Metab 22:761–776
Jameson KG, Olson CA, Kazmi SA, Hsiao EY (2020) Toward understanding microbiome-neuronal signaling. Mol Cell 78(4):577–583
Jarvie BC, Hentges ST (2012) Expression of GABAergic and glutamatergic phenotypic markers in hypothalamic proopiomelanocortin neurons. J Comp Neurol 520:3863–3876
Jennings JH, Ung RL, Resendez SL, Stamatakis AM, Taylor JG, Huang J, Veleta K, Kantak PA, Aita M, Shilling-Scrivo K et al (2015) Visualizing hypothalamic network dynamics for appetitive and consummatory behaviors. Cell 160:516–527
Jeong DE, Artan M, Seo K, Lee SJ (2012) Regulation of lifespan by chemosensory and thermosensory systems: findings in invertebrates and their implications in mammalian aging. Front Genet 3:218
Jia KL, Albert PS, Riddle DL (2002) DAF-9, a cytochrome P450 regulating C. elegans larval development and adult longevity. Development 129:221–231
Jones ME, Thorburn AW, Britt KL, Hewitt KN, Wreford NG, Proietto J, Oz OK, Leury BJ, Robertson KM, Yao S et al (2000) Aromatase-deficient (ArKO) mice have a phenotype of increased adiposity. Proc Natl Acad Sci USA 97:12735–12740
Kalsbeek MJ, Wolff SE, Korpel NL, la Fleur SE, Romijn JA, Fliers E, Kalsbeek A, Swaab DF, Huitinga I, Hol EM et al (2020) The impact of antidiabetic treatment on human hypothalamic infundibular neurons and microglia. JCI Insight 5(16):e133868
Kalyani M, Hasselfeld K, Janik JM, Callahan P, Shi H (2016) Effects of high-fat diet on stress response in male and female wildtype and prolactin knockout mice. PLoS ONE 11:e0166416
Karaki S, Mitsui R, Hayashi H, Kato I, Sugiya H, Iwanaga T, Furness JB, Kuwahara A (2006) Short-chain fatty acid receptor, GPR43, is expressed by enteroendocrine cells and mucosal mast cells in rat intestine. Cell Tissue Res 324:353–360
Kenyon C, Chang J, Gensch E, Rudner A, Tabtiang R (1993) A C. elegans mutant that lives twice as long as wild-type. Nature 366:461–464
Kenyon CJ (2010) The genetics of ageing. Nature 464:504–512
Kessler RC (1997) The effects of stressful life events on depression. Annu Rev Psychol 48:191–214
Kim ER, Wu Z, Sun H, Xu Y, Mangieri LR, Xu Y, Tong Q (2015) Hypothalamic non-AgRP, non-POMC GABAergic neurons are required for postweaning feeding and NPY hyperphagia. J Neurosci 35:10440–10450
Kim ER, Xu Y, Cassidy RM, Lu Y, Yang Y, Tian J, Li DP, Van Drunen R, Ribas-Latre A, Cai ZL et al (2020) Paraventricular hypothalamus mediates diurnal rhythm of metabolism. Nat Commun 11:3794
Kim JD, Yoon NA, Jin S, Diano S (2019) Microglial UCP2 mediates inflammation and obesity induced by high-fat feeding. Cell Metab 30:952-962.e955
Kim JG, Suyama S, Koch M, Jin S, Argente-Arizon P, Argente J, Liu ZW, Zimmer MR, Jeong JK, Szigeti-Buck K et al (2014) Leptin signaling in astrocytes regulates hypothalamic neuronal circuits and feeding. Nat Neurosci 17:908–910
Klingbeil E, de La Serre CB (2018) Microbiota modulation by eating patterns and diet composition: impact on food intake. Am J Physiol Regul Integr Comp Physiol 315:R1254–R1260
Kothari V, Galdo JA, Mathews ST (2016) Hypoglycemic agents and potential anti-inflammatory activity. J Inflamm Res 9:27–38
Kowalski C, Micheau J, Corder R, Gaillard R, Contedevolx B (1992) Age-related-changes in cortico-releasing factor, somatostatin, neuropeptide-Y, methionine enkephalin and beta-endorphin in specific rat-brain areas. Brain Res 582:38–46
Krieger JP, Arnold M, Pettersen KG, Lossel P, Langhans W, Lee SJ (2016) Knockdown of GLP-1 receptors in vagal afferents affects normal food intake and glycemia. Diabetes 65:34–43
Krieger JP, Langhans W, Lee SJ (2015) Vagal mediation of GLP-1’s effects on food intake and glycemia. Physiol Behav 152:372–380
Krude H, Biebermann H, Luck W, Horn R, Brabant G, Gruters A (1998) Severe early-onset obesity, adrenal insufficiency and red hair pigmentation caused by POMC mutations in humans. Nat Genet 19:155–157
Kuno R, Wang J, Kawanokuchi J, Takeuchi H, Mizuno T, Suzumura A (2005) Autocrine activation of microglia by tumor necrosis factor-alpha. J Neuroimmunol 162:89–96
Lagerlof O, Slocomb JE, Hong I, Aponte Y, Blackshaw S, Hart GW, Huganir RL (2016) The nutrient sensor OGT in PVN neurons regulates feeding. Science 351:1293–1296
Le Chatelier E, Nielsen T, Qin J, Prifti E, Hildebrand F, Falony G, Almeida M, Arumugam M, Batto JM, Kennedy S et al (2013) Richness of human gut microbiome correlates with metabolic markers. Nature 500:541–546
Lee SJ, Kenyon C (2009) Regulation of the longevity response to temperature by thermosensory neurons in Caenorhabditis elegans (vol 19, pg 715, 2009). Curr Biol 19:798–798
Li G, Zhang Y, Wilsey JT, Scarpace PJ (2005) Hypothalamic pro-opiomelanocortin gene delivery ameliorates obesity and glucose intolerance in aged rats. Diabetologia 48:2376–2385
Li J, Tang Y, Cai D (2012) IKKbeta/NF-kappaB disrupts adult hypothalamic neural stem cells to mediate a neurodegenerative mechanism of dietary obesity and pre-diabetes. Nat Cell Biol 14:999–1012
Li Y, Zeng J, Zhang J, Yue C, Zhong W, Liu Z, Feng Q, Luo M (2018a) Hypothalamic circuits for predation and evasion. Neuron 97:911-924.e915
Li Z, Yi CX, Katiraei S, Kooijman S, Zhou E, Chung CK, Gao Y, van den Heuvel JK, Meijer OC, Berbée JFP et al (2018b) Butyrate reduces appetite and activates brown adipose tissue via the gut-brain neural circuit. Gut 67:1269–1279
Liao GY, Kinney CE, An JJ, Xu B (2019) TrkB-expressing neurons in the dorsomedial hypothalamus are necessary and sufficient to suppress homeostatic feeding. Proc Natl Acad Sci USA 116:3256–3261
Lin HY, Xu Q, Yeh S, Wang RS, Sparks JD, Chang C (2005) Insulin and leptin resistance with hyperleptinemia in mice lacking androgen receptor. Diabetes 54:1717–1725
Liu YZ, Chen JK, Zhang Y, Wang X, Qu S, Jiang CL (2014) Chronic stress induces steatohepatitis while decreases visceral fat mass in mice. BMC Gastroenterol 14:106
Locke AE, Kahali B, Berndt SI, Justice AE, Pers TH, Day FR, Powell C, Vedantam S, Buchkovich ML, Yang J et al (2015) Genetic studies of body mass index yield new insights for obesity biology. Nature 518:197–206
Loid P, Mäkitie R, Costantini A, Viljakainen H, Pekkinen M, Mäkitie O (2018) A novel MYT1L mutation in a patient with severe early-onset obesity and intellectual disability. Am J Med Genet A 176:1972–1975
Loskutova N, Honea RA, Brooks WM, Burns JM (2010) Reduced limbic and hypothalamic volumes correlate with bone density in early Alzheimer’s disease. J Alzheimers Dis 20:313–322
Lutter M, Sakata I, Osborne-Lawrence S, Rovinsky SA, Anderson JG, Jung S, Birnbaum S, Yanagisawa M, Elmquist JK, Nestler EJ et al (2008) The orexigenic hormone ghrelin defends against depressive symptoms of chronic stress. Nat Neurosci 11:752–753
Malyala A, Zhang C, Bryant DN, Kelly MJ, Ronnekleiv OK (2008) PI3K signaling effects in hypothalamic neurons mediated by estrogen. J Comp Neurol 506:895–911
Mangieri LR, Lu Y, Xu Y, Cassidy RM, Xu Y, Arenkiel BR, Tong Q (2018) A neural basis for antagonistic control of feeding and compulsive behaviors. Nat Commun 9:52
Maniscalco JW, Rinaman L (2017) Interoceptive modulation of neuroendocrine, emotional, and hypophagic responses to stress. Physiol Behav 176:195–206
Martinez de Morentin PB, Gonzalez-Garcia I, Martins L, Lage R, Fernandez-Mallo D, Martinez-Sanchez N, Ruiz-Pino F, Liu J, Morgan DA, Pinilla L et al (2014) Estradiol regulates brown adipose tissue thermogenesis via hypothalamic AMPK. Cell Metab 20:41–53
Masuda T, Sankowski R, Staszewski O, Prinz M (2020) Microglia heterogeneity in the single-cell era. Cell Rep 30:1271–1281
Matsumoto A, Arai Y (1980) Sexual dimorphism in ‘wiring pattern’ in the hypothalamic arcuate nucleus and its modification by neonatal hormonal environment. Brain Res 190:238–242
Mattar L, Thiebaud MR, Huas C, Cebula C, Godart N (2012) Depression, anxiety and obsessive-compulsive symptoms in relation to nutritional status and outcome in severe anorexia nervosa. Psychiatry Res 200:513–517
Mauvais-Jarvis F, Bairey Merz N, Barnes PJ, Brinton RD, Carrero JJ, DeMeo DL, De Vries GJ, Epperson CN, Govindan R, Klein SL et al (2020) Sex and gender: modifiers of health, disease, and medicine. Lancet 396:565–582
Mazier W, Saucisse N, Simon V, Cannich A, Marsicano G, Massa F, Cota D (2019) mTORC1 and CB1 receptor signaling regulate excitatory glutamatergic inputs onto the hypothalamic paraventricular nucleus in response to energy availability. Mol Metab 28:151–159
Mazzone CM, Liang-Guallpa J, Li C, Wolcott NS, Boone MH, Southern M, Kobzar NP, Salgado IA, Reddy DM, Sun F et al (2020) High-fat food biases hypothalamic and mesolimbic expression of consummatory drives. Nat Neurosci 23:1253–1266
Meier SM, Trontti K, Purves KL, Als TD, Grove J, Laine M, Pedersen MG, Bybjerg-Grauholm J, Baekved-Hansen M, Sokolowska E et al (2019) Genetic variants associated with anxiety and stress-related disorders: a genome-wide association study and mouse-model study. JAMA Psychiat 76(9):924–932
Melhorn SJ, Krause EG, Scott KA, Mooney MR, Johnson JD, Woods SC, Sakai RR (2010) Meal patterns and hypothalamic NPY expression during chronic social stress and recovery. Am J Physiol Regul Integr Comp Physiol 299:R813–R822
Merchenthaler I, Lane MV, Numan S, Dellovade TL (2004) Distribution of estrogen receptor alpha and beta in the mouse central nervous system: in vivo autoradiographic and immunocytochemical analyses. J Comp Neurol 473:270–291
Michel C, Duclos M, Cabanac M, Richard D (2005) Chronic stress reduces body fat content in both obesity-prone and obesity-resistant strains of mice. Horm Behav 48:172–179
Milanova IV, Kalsbeek MJT, Wang XL, Korpel NL, Stenvers DJ, Wolff SEC, de Goede P, Heijboer AC, Fliers E, la Fleur SE et al (2019) Diet-induced obesity disturbs microglial immunometabolism in a time-of-day manner. Front Endocrinol (Lausanne) 10:424
Miller MM, Tousignant P, Yang U, Pedvis S, Billiar RB (1995) Effects of age and long-term ovariectomy on the estrogen-receptor containing subpopulations of beta-endorphin-immunoreactive neurons in the arcuate nucleus of female C57BL/6J mice. Neuroendocrinology 61:542–551
Mitchell RW, On NH, Del Bigio MR, Miller DW, Hatch GM (2011) Fatty acid transport protein expression in human brain and potential role in fatty acid transport across human brain microvessel endothelial cells. J Neurochem 117:735–746
Molofsky AV, Slutsky SG, Joseph NM, He S, Pardal R, Krishnamurthy J, Sharpless NE, Morrison SJ (2006) Increasing p16INK4a expression decreases forebrain progenitors and neurogenesis during ageing. Nature 443:448–452
Mori T, Tanaka K, Buffo A, Wurst W, Kühn R, Götz M (2006) Inducible gene deletion in astroglia and radial glia—a valuable tool for functional and lineage analysis. Glia 54:21–34
Morris JZ, Tissenbaum HA, Ruvkun G (1996) A phosphatidylinositol-3-OH kinase family member regulating longevity and diapause in Caenorhabditis elegans. Nature 382:536–539
Morselli E, Frank AP, Palmer BF, Rodriguez-Navas C, Criollo A, Clegg DJ (2016) A sexually dimorphic hypothalamic response to chronic high-fat diet consumption. Int J Obes (Lond) 40:206–209
Murphy KG (2005) Kisspeptins: regulators of metastasis and the hypothalamic–pituitary–gonadal axis. J Neuroendocrinol 17:519–525
Murphy KG, Bloom SR (2006) Gut hormones and the regulation of energy homeostasis. Nature 444:854–859
Musatov S, Chen W, Pfaff DW, Mobbs CV, Yang XJ, Clegg DJ, Kaplitt MG, Ogawa S (2007) Silencing of estrogen receptor alpha in the ventromedial nucleus of hypothalamus leads to metabolic syndrome. Proc Natl Acad Sci USA 104:2501–2506
Nasrallah CM, Horvath TL (2014) Mitochondrial dynamics in the central regulation of metabolism. Nat Rev Endocrinol 10:650–658
Nederkoorn C, Dassen FC, Franken L, Resch C, Houben K (2015) Impulsivity and overeating in children in the absence and presence of hunger. Appetite 93:57–61
Nieh EH, Matthews GA, Allsop SA, Presbrey KN, Leppla CA, Wichmann R, Neve R, Wildes CP, Tye KM (2015) Decoding neural circuits that control compulsive sucrose seeking. Cell 160:528–541
Nohara K, Waraich RS, Liu S, Ferron M, Waget A, Meyers MS, Karsenty G, Burcelin R, Mauvais-Jarvis F (2013) Developmental androgen excess programs sympathetic tone and adipose tissue dysfunction and predisposes to a cardiometabolic syndrome in female mice. Am J Physiol Endocrinol Metab 304:E1321–E1330
Nohara K, Zhang Y, Waraich RS, Laque A, Tiano JP, Tong J, Munzberg H, Mauvais-Jarvis F (2011) Early-life exposure to testosterone programs the hypothalamic melanocortin system. Endocrinology 152:1661–1669
Nonogaki K, Strack AM, Dallman MF, Tecott LH (1998) Leptin-independent hyperphagia and type 2 diabetes in mice with a mutated serotonin 5-HT2C receptor gene. Nat Med 4:1152–1156
O’Neill LA, Kishton RJ, Rathmell J (2016) A guide to immunometabolism for immunologists. Nat Rev Immunol 16:553–565
Oeckinghaus A, Hayden MS, Ghosh S (2011) Crosstalk in NF-κB signaling pathways. Nat Immunol 12:695–708
Okamoto S, Sato T, Tateyama M, Kageyama H, Maejima Y, Nakata M, Hirako S, Matsuo T, Kyaw S, Shiuchi T et al (2018) Activation of AMPK-regulated CRH neurons in the PVH is sufficient and necessary to induce dietary preference for carbohydrate over fat. Cell Rep 22:706–721
Okura T, Koda M, Ando F, Niino N, Ohta S, Shimokata H (2003) Association of polymorphisms in the estrogen receptor alpha gene with body fat distribution. Int J Obes Relat Metab Disord 27:1020–1027
Olah M, Menon V, Habib N, Taga MF, Ma Y, Yung CJ, Cimpean M, Khairallah A, Coronas-Samano G, Sankowski R et al (2020) Single cell RNA sequencing of human microglia uncovers a subset associated with Alzheimer’s disease. Nat Commun 11:6129
Ong CK, Lirk P, Tan CH, Seymour RA (2007) An evidence-based update on nonsteroidal anti-inflammatory drugs. Clin Med Res 5:19–34
World Health Organization (2015) World report on ageing and health, p260
Osterlund M, Kuiper GG, Gustafsson JA, Hurd YL (1998) Differential distribution and regulation of estrogen receptor-alpha and -beta mRNA within the female rat brain. Brain Res Mol Brain Res 54:175–180
Padilla SL, Qiu J, Soden ME, Sanz E, Nestor CC, Barker FD, Quintana A, Zweifel LS, Ronnekleiv OK, Kelly MJ et al (2016) Agouti-related peptide neural circuits mediate adaptive behaviors in the starved state. Nat Neurosci 19:734–741
Paladini CA, Roeper J (2014) Generating bursts (and pauses) in the dopamine midbrain neurons. Neuroscience 282:109–121
Palmer BF, Clegg DJ (2015) The sexual dimorphism of obesity. Mol Cell Endocrinol 402:113–119
Palmer K, Gray JM (1986) Central vs. peripheral effects of estrogen on food intake and lipoprotein lipase activity in ovariectomized rats. Physiol Behav 37:187–189
Patriquin MA, Mathew SJ (2017) The neurobiological mechanisms of generalized anxiety disorder and chronic stress. Chronic Stress (Thousand Oaks). https://doi.org/10.1177/2470547017703993
Patterson E, Ryan PM, Cryan JF, Dinan TG, Ross RP, Fitzgerald GF, Stanton C (2016) Gut microbiota, obesity and diabetes. Postgrad Med J 92:286–300
Perry VH, Nicoll JA, Holmes C (2010) Microglia in neurodegenerative disease. Nat Rev Neurol 6:193–201
Pivonello R, De Leo M, Cozzolino A, Colao A (2015) The treatment of Cushing’s disease. Endocr Rev 36:385–486
Pizzagalli DA (2014) Depression, stress, and anhedonia: toward a synthesis and integrated model. Annu Rev Clin Psychol 10:393–423
Prinz M, Priller J (2014) Microglia and brain macrophages in the molecular age: from origin to neuropsychiatric disease. Nat Rev Neurosci 15:300–312
Qiu J, Bosch MA, Meza C, Navarro UV, Nestor CC, Wagner EJ, Ronnekleiv OK, Kelly MJ (2018) Estradiol protects proopiomelanocortin neurons against insulin resistance. Endocrinology 159:647–664
Qiu J, Fang Y, Ronnekleiv OK, Kelly MJ (2010) Leptin excites proopiomelanocortin neurons via activation of TRPC channels. J Neurosci 30:1560–1565
Qiu J, Zhang C, Borgquist A, Nestor CC, Smith AW, Bosch MA, Ku S, Wagner EJ, Ronnekleiv OK, Kelly MJ (2014) Insulin excites anorexigenic proopiomelanocortin neurons via activation of canonical transient receptor potential channels. Cell Metab 19:682–693
Qu N, He Y, Wang C, Xu P, Yang Y, Cai X, Liu H, Yu K, Pei Z, Hyseni I et al (2020) A POMC-originated circuit regulates stress-induced hypophagia, depression, and anhedonia. Mol Psychiatry 25:1006–1021
Ramachandrappa S, Raimondo A, Cali AM, Keogh JM, Henning E, Saeed S, Thompson A, Garg S, Bochukova EG, Brage S et al (2013) Rare variants in single-minded 1 (SIM1) are associated with severe obesity. J Clin Investig 123:3042–3050
Ramadori G, Fujikawa T, Fukuda M, Anderson J, Morgan DA, Mostoslavsky R, Stuart RC, Perello M, Vianna CR, Nillni EA et al (2010) SIRT1 deacetylase in POMC neurons is required for homeostatic defenses against diet-induced obesity. Cell Metab 12:78–87
Ransohoff RM (2016) A polarizing question: do M1 and M2 microglia exist? Nat Neurosci 19:987–991
Razzoli M, Bartolomucci A (2016) The dichotomous effect of chronic stress on obesity. Trends Endocrinol Metab 27:504–515
Razzoli M, Pearson C, Crow S, Bartolomucci A (2017) Stress, overeating, and obesity: insights from human studies and preclinical models. Neurosci Biobehav Rev 76:154–162
Razzoli M, Sanghez V, Bartolomucci A (2015) Chronic subordination stress induces hyperphagia and disrupts eating behavior in mice modeling binge-eating-like disorder. Front Nutr 1(30):30
Ren D, Zhou Y, Morris D, Li M, Li Z, Rui L (2007) Neuronal SH2B1 is essential for controlling energy and glucose homeostasis. J Clin Investig 117:397–406
Reue K (2017) Sex differences in obesity: X chromosome dosage as a risk factor for increased food intake, adiposity and co-morbidities. Physiol Behav 176:174–182
Riera CE, Huising MO, Follett P, Leblanc M, Halloran J, Van Andel R, de Magalhaes CD, Merkwirth C, Dillin A (2014) TRPV1 pain receptors regulate longevity and metabolism by neuropeptide signaling. Cell 157:1023–1036
Robichaud M, Debonnel G (2005) Oestrogen and testosterone modulate the firing activity of dorsal raphe nucleus serotonergic neurones in both male and female rats. J Neuroendocrinol 17:179–185
Rogers NH, Perfield JW II, Strissel KJ, Obin MS, Greenberg AS (2009) Reduced energy expenditure and increased inflammation are early events in the development of ovariectomy-induced obesity. Endocrinology 150(5):2161–2168
Rottiers V, Motola DL, Gerisch B, Cummins CL, Nishiwaki K, Mangelsdorf DJ, Antebi A (2006) Hormonal control of C. elegans Dauer formation and life span by a Rieske-like oxygenase. Dev Cell 10:473–482
Round JL, Mazmanian SK (2009) The gut microbiota shapes intestinal immune responses during health and disease. Nat Rev Immunol 9:313–323
Ruan HB, Dietrich MO, Liu ZW, Zimmer MR, Li MD, Singh JP, Zhang K, Yin R, Wu J, Horvath TL et al (2014) O-GlcNAc transferase enables AgRP neurons to suppress browning of white fat. Cell 159:306–317
Ruud J, Steculorum SM, Bruning JC (2017) Neuronal control of peripheral insulin sensitivity and glucose metabolism. Nat Commun 8:15259
Sadagurski M, Cady G, Miller RA (2017) Anti-aging drugs reduce hypothalamic inflammation in a sex-specific manner. Aging Cell 16:652–660
Saito K, Cao X, He Y, Xu Y (2015) Progress in the molecular understanding of central regulation of body weight by estrogens. Obesity (Silver Spring) 23:919–926
Samuel P, Khan MA, Nag S, Inagami T, Hussain T (2013) Angiotensin AT(2) receptor contributes towards gender bias in weight gain. PLoS ONE 8:e48425
Santollo J, Torregrossa AM, Eckel LA (2011) Estradiol acts in the medial preoptic area, arcuate nucleus, and dorsal raphe nucleus to reduce food intake in ovariectomized rats. Horm Behav 60:86–93
Sato T, Matsumoto T, Yamada T, Watanabe T, Kawano H, Kato S (2003) Late onset of obesity in male androgen receptor-deficient (AR KO) mice. Biochem Biophys Res Commun 300:167–171
Satoh A, Brace CS, Rensing N, Cliften P, Wozniak DF, Herzog ED, Yamada KA, Imai S (2013) Sirt1 extends life span and delays aging in mice through the regulation of Nk2 homeobox 1 in the DMH and LH. Cell Metab 18:416–430
Schlenker EH, Hansen SN (2006) Sex-specific densities of estrogen receptors alpha and beta in the subnuclei of the nucleus tractus solitarius, hypoglossal nucleus and dorsal vagal motor nucleus weanling rats. Brain Res 1123:89–100
Schwartz SM, Wade GN (1981) Effects of estradiol and progesterone on food intake, body weight, and carcass adiposity in weanling rats. Am J Physiol 240:E499–E503
Seib DRM, Martin-Villalba A (2015) Neurogenesis in the normal ageing hippocampus: a mini-review. Gerontology 61:327–335
Seminara SB, Messager S, Chatzidaki EE, Thresher RR, Acierno JS Jr, Shagoury JK, Bo-Abbas Y, Kuohung W, Schwinof KM, Hendrick AG et al (2003) The GPR54 gene as a regulator of puberty. N Engl J Med 349:1614–1627
Shalata A, Ramirez MC, Desnick RJ, Priedigkeit N, Buettner C, Lindtner C, Mahroum M, Abdul-Ghani M, Dong F, Arar N et al (2013) Morbid obesity resulting from inactivation of the ciliary protein CEP19 in humans and mice. Am J Hum Genet 93:1061–1071
Sharma G, Hu C, Brigman JL, Zhu G, Hathaway HJ, Prossnitz ER (2013) GPER deficiency in male mice results in insulin resistance, dyslipidemia, and a proinflammatory state. Endocrinology 154:4136–4145
Shen WJ, Yao T, Kong X, Williams KW, Liu T (2017) Melanocortin neurons: multiple routes to regulation of metabolism. Biochim Biophys Acta Mol Basis Dis 1863:2477–2485
Shi H, Seeley RJ, Clegg DJ (2009) Sexual differences in the control of energy homeostasis. Front Neuroendocrinol 30:396–404
Siervo M, Wells JC, Cizza G (2009) The contribution of psychosocial stress to the obesity epidemic: an evolutionary approach. Horm Metab Res 41:261–270
Siljee JE, Wang Y, Bernard AA, Ersoy BA, Zhang S, Marley A, Von Zastrow M, Reiter JF, Vaisse C (2018) Subcellular localization of MC4R with ADCY3 at neuronal primary cilia underlies a common pathway for genetic predisposition to obesity. Nat Genet 50:180–185
Sinha R, Jastreboff AM (2013) Stress as a common risk factor for obesity and addiction. Biol Psychiatry 73:827–835
Smith EP, Boyd J, Frank GR, Takahashi H, Cohen RM, Specker B, Williams TC, Lubahn DB, Korach KS (1994) Estrogen resistance caused by a mutation in the estrogen-receptor gene in a man. N Engl J Med 331:1056–1061
Snider WD (1994) Functions of the neurotrophins during nervous system development: what the knockouts are teaching us. Cell 77:627–638
Sominsky L, Spencer SJ (2014) Eating behavior and stress: a pathway to obesity. Front Psychol 5:434
Speakman JR (2015) The ‘Fat Mass and Obesity Related’ (FTO) gene: mechanisms of impact on obesity and energy balance. Curr Obes Rep 4:73–91
Srinivasan R, Lu TY, Chai H, Xu J, Huang BS, Golshani P, Coppola G, Khakh BS (2016) New transgenic mouse lines for selectively targeting astrocytes and studying calcium signals in astrocyte processes in situ and in vivo. Neuron 92:1181–1195
Stamatakis AM, Van Swieten M, Basiri ML, Blair GA, Kantak P, Stuber GD (2016) Lateral hypothalamic area glutamatergic neurons and their projections to the lateral habenula regulate feeding and reward. J Neurosci 36:302–311
Stengel A, Tache Y (2014) CRF and urocortin peptides as modulators of energy balance and feeding behavior during stress. Front Neurosci 8:52
Stergiakouli E, Gaillard R, Tavaré JM, Balthasar N, Loos RJ, Taal HR, Evans DM, Rivadeneira F, St Pourcain B, Uitterlinden AG et al (2014) Genome-wide association study of height-adjusted BMI in childhood identifies functional variant in ADCY3. Obesity (Silver Spring) 22:2252–2259
Kliewer SA, Mangelsdorf DJ (2015) Bile acids as hormones: the FXR-FGF15/19 pathway. Dig Dis 33(3):327–331
Streit WJ, Xue QS, Tischer J, Bechmann I (2014) Microglial pathology. Acta Neuropathol Commun 2:142
Stubbins RE, Holcomb VB, Hong J, Nunez NP (2012) Estrogen modulates abdominal adiposity and protects female mice from obesity and impaired glucose tolerance. Eur J Nutr 51:861–870
Su X, Gi YJ, Chakravarti D, Chan IL, Zhang A, Xia X, Tsai KY, Flores ER (2012) TAp63 is a master transcriptional regulator of lipid and glucose metabolism. Cell Metab 16:511–525
Sugiyama MG, Agellon LB (2012) Sex differences in lipid metabolism and metabolic disease risk. Biochem Cell Biol 90:124–141
Sun F, Mao X, Xie L, Ding M, Shao B, Jin K (2013) Notch1 signaling modulates neuronal progenitor activity in the subventricular zone in response to aging and focal ischemia. Aging Cell 12:978–987
Swarbrick MM, Evans DS, Valle MI, Favre H, Wu SH, Njajou OT, Li R, Zmuda JM, Miljkovic I, Harris TB et al (2011) Replication and extension of association between common genetic variants in SIM1 and human adiposity. Obesity (Silver Spring) 19:2394–2403
Sweeney P, Yang Y (2017) Neural circuit mechanisms underlying emotional regulation of homeostatic feeding. Trends Endocrinol Metab 28:437–448
Syková E (2004) Extrasynaptic volume transmission and diffusion parameters of the extracellular space. Neuroscience 129:861–876
Tannenbaum C, Schwarz JM, Clayton JA, de Vries GJ, Sullivan C (2016) Evaluating sex as a biological variable in preclinical research: the devil in the details. Biol Sex Differ 7:13
Tatsumi K, Isonishi A, Yamasaki M, Kawabe Y, Morita-Takemura S, Nakahara K, Terada Y, Shinjo T, Okuda H, Tanaka T et al (2018) Olig2-lineage astrocytes: a distinct subtype of astrocytes that differs from GFAP astrocytes. Front Neuroanat 12:8
Tay TL, Mai D, Dautzenberg J, Fernandez-Klett F, Lin G, Sagar DM, Drougard A, Stempfl T, Ardura-Fabregat A et al (2017) A new fate mapping system reveals context-dependent random or clonal expansion of microglia. Nat Neurosci 20:793–803
Taylor RC, Dillin A (2013) XBP-1 Is a cell-nonautonomous regulator of stress resistance and longevity. Cell 153:1435–1447
Tecott LH, Sun LM, Akana SF, Strack AM, Lowenstein DH, Dallman MF, Julius D (1995) Eating disorder and epilepsy in mice lacking 5-HT2c serotonin receptors. Nature 374:542–546
Terrill SJ, Maske CB, Williams DL (2018) Endogenous GLP-1 in lateral septum contributes to stress-induced hypophagia. Physiol Behav 192:17–22
Thaler JP, Yi CX, Schur EA, Guyenet SJ, Hwang BH, Dietrich MO, Zhao X, Sarruf DA, Izgur V, Maravilla KR et al (2012) Obesity is associated with hypothalamic injury in rodents and humans. J Clin Investig 122:153–162
Thomson AW, Turnquist HR, Raimondi G (2009) Immunoregulatory functions of mTOR inhibition. Nat Rev Immunol 9:324–337
Timmerman R, Burm SM, Bajramovic JJ (2018) An overview of in vitro methods to study microglia. Front Cell Neurosci 12:242
Timper K, Bruning JC (2017) Hypothalamic circuits regulating appetite and energy homeostasis: pathways to obesity. Dis Model Mech 10:679–689
Tolson KP, Garcia C, Delgado I, Marooki N, Kauffman AS (2016) Metabolism and energy expenditure, but not feeding or glucose tolerance, are impaired in young Kiss1r KO female mice. Endocrinology 157:4192–4199
Tolson KP, Garcia C, Yen S, Simonds S, Stefanidis A, Lawrence A, Smith JT, Kauffman AS (2014) Impaired kisspeptin signaling decreases metabolism and promotes glucose intolerance and obesity. J Clin Investig 124:3075–3079
Tolson KP, Gemelli T, Gautron L, Elmquist JK, Zinn AR, Kublaoui BM (2010) Postnatal Sim1 deficiency causes hyperphagic obesity and reduced Mc4r and oxytocin expression. J Neurosci 30:3803–3812
Tomiyama AJ (2018) Stress and obesity. Annu Rev Psychol 70:703–718
Tramunt B, Smati S, Grandgeorge N, Lenfant F, Arnal JF, Montagner A, Gourdy P (2020) Sex differences in metabolic regulation and diabetes susceptibility. Diabetologia 63:453–461
Tsai SF, Wu HT, Chen PC, Chen YW, Yu M, Tzeng SF, Wu PH, Chen PS, Kuo YM (2018) Stress aggravates high-fat-diet-induced insulin resistance via a mechanism that involves the amygdala and is associated with changes in neuroplasticity. Neuroendocrinology 107:147–157
Turcot V, Lu Y, Highland HM, Schurmann C, Justice AE, Fine RS, Bradfield JP, Esko T, Giri A, Graff M et al (2018) Protein-altering variants associated with body mass index implicate pathways that control energy intake and expenditure in obesity. Nat Genet 50:26–41
Turer EE, San Miguel M, Wang KW, McAlpine W, Ou F, Li X, Tang M, Zang Z, Wang J, Hayse B et al (2018) A viable hypomorphic Arnt2 mutation causes hyperphagic obesity, diabetes and hepatic steatosis. Dis Model Mech 11(12):dmm035451
Turnbaugh PJ, Ley RE, Mahowald MA, Magrini V, Mardis ER, Gordon JI (2006) An obesity-associated gut microbiome with increased capacity for energy harvest. Nature 444:1027–1031
Tye KM (2018) Neural circuit motifs in valence processing. Neuron 100:436–452
Ulrich-Lai YM, Herman JP (2009) Neural regulation of endocrine and autonomic stress responses. Nat Rev Neurosci 10:397–409
Valdearcos M, Douglass JD, Robblee MM, Dorfman MD, Stifler DR, Bennett ML, Gerritse I, Fasnacht R, Barres BA, Thaler JP et al (2017) Microglial inflammatory signaling orchestrates the hypothalamic immune response to dietary excess and mediates obesity susceptibility. Cell Metab 26:185-197.e183
van Veen JE, Kammel LG, Bunda PC, Shum M, Reid MS, Park JW, Zhang Z, Massa MG, Arneson D, Hrncir HJB (2019) Single cell profiling of the VMH reveals a sexually dimorphic regulatory node of energy expenditure. https://doi.org/10.1101/549725
Van Wagoner NJ, Benveniste EN (1999) Interleukin-6 expression and regulation in astrocytes. J Neuroimmunol 100:124–139
Venzala E, Garcia-Garcia AL, Elizalde N, Tordera RM (2013) Social vs. environmental stress models of depression from a behavioural and neurochemical approach. Eur Neuropsychopharmacol 23:697–708
Vercruysse P, Vieau D, Blum D, Petersen A, Dupuis L (2018) Hypothalamic alterations in neurodegenerative diseases and their relation to abnormal energy metabolism. Front Mol Neurosci 11:2
Veyrat-Durebex C, Quirion R, Ferland G, Dumont Y, Gaudreau P (2013) Aging and long-term caloric restriction regulate neuropeptide Y receptor subtype densities in the rat brain. Neuropeptides 47:163–169
Vicente-Gutierrez C, Bonora N, Bobo-Jimenez V, Jimenez-Blasco D, Lopez-Fabuel I, Fernandez E, Josephine C, Bonvento G, Enriquez JA, Almeida A et al (2019) Astrocytic mitochondrial ROS modulate brain metabolism and mouse behaviour. Nat Metab 1:201–211
Villeda SA, Luo J, Mosher KI, Zou B, Britschgi M, Bieri G, Stan TM, Fainberg N, Ding Z, Eggel A et al (2011) The ageing systemic milieu negatively regulates neurogenesis and cognitive function. Nature 477:90–94
Wahl S, Drong A, Lehne B, Loh M, Scott WR, Kunze S, Tsai PC, Ried JS, Zhang W, Yang Y et al (2017) Epigenome-wide association study of body mass index, and the adverse outcomes of adiposity. Nature 541:81–86
Wallen WJ, Belanger MP, Wittnich C (2001) Sex hormones and the selective estrogen receptor modulator tamoxifen modulate weekly body weights and food intakes in adolescent and adult rats. J Nutr 131:2351–2357
Walters RG, Jacquemont S, Valsesia A, de Smith AJ, Martinet D, Andersson J, Falchi M, Chen F, Andrieux J, Lobbens S et al (2010) A new highly penetrant form of obesity due to deletions on chromosome 16p11.2. Nature 463:671–675
Wang C, He Y, Xu P, Yang Y, Saito K, Xia Y, Yan X, Hinton A Jr, Yan C, Ding H et al (2018) TAp63 contributes to sexual dimorphism in POMC neuron functions and energy homeostasis. Nat Commun 9:1544
Wang C, Xu Y (2019) Mechanisms for Sex Differences in Energy Homeostasis. J Mol Endocrinol 62:R129-r143
Wang Z, Li V, Chan GC, Phan T, Nudelman AS, Xia Z, Storm DR (2009) Adult type 3 adenylyl cyclase-deficient mice are obese. PLoS ONE 4:e6979
Webb EA, AlMutair A, Kelberman D, Bacchelli C, Chanudet E, Lescai F, Andoniadou CL, Banyan A, Alsawaid A, Alrifai MT et al (2013) ARNT2 mutation causes hypopituitarism, post-natal microcephaly, visual and renal anomalies. Brain 136:3096–3105
Wolden-Hanson T, Marck BT, Matsumoto AM (2004) Blunted hypothalamic neuropeptide gene expression in response to fasting, but preservation of feeding responses to AgRP in aging male Brown Norway rats. Am J Physiol Regul Integr Comp Physiol 287:R138–R146
Woodward CJ, Emery PW (1989) Energy balance in rats given chronic hormone treatment. 2. Effects of corticosterone. Br J Nutr 61:445–452
Wu MV, Manoli DS, Fraser EJ, Coats JK, Tollkuhn J, Honda S, Harada N, Shah NM (2009) Estrogen masculinizes neural pathways and sex-specific behaviors. Cell 139:61–72
Wu Z, Kim ER, Sun H, Xu Y, Mangieri LR, Li DP, Pan HL, Xu Y, Arenkiel BR, Tong Q (2015) GABAergic projections from lateral hypothalamus to paraventricular hypothalamic nucleus promote feeding. J Neurosci 35:3312–3318
Xiao R, Liu JF, Xu XZS (2015) Thermosensation and longevity. J Comp Physiol A 201:857–867
Xiao YZ, Yang M, Xiao Y, Guo Q, Huang Y, Li CJ, Cai DS, Luo XH (2020) Reducing hypothalamic stem cell senescence protects against aging-associated physiological decline. Cell Metab 31:534
Xu AW, Ste-Marie L, Kaelin CB, Barsh GS (2007) Inactivation of signal transducer and activator of transcription 3 in proopiomelanocortin (Pomc) neurons causes decreased pomc expression, mild obesity, and defects in compensatory refeeding. Endocrinology 148:72–80
Xu B, Goulding EH, Zang K, Cepoi D, Cone RD, Jones KR, Tecott LH, Reichardt LF (2003) Brain-derived neurotrophic factor regulates energy balance downstream of melanocortin-4 receptor. Nat Neurosci 6:736–742
Xu P, Cao X, He Y, Zhu L, Yang Y, Saito K, Wang C, Yan X, Hinton AO Jr, Zou F et al (2015) Estrogen receptor-alpha in medial amygdala neurons regulates body weight. J Clin Investig 125:2861–2876
Xu P, Grueter BA, Britt JK, McDaniel L, Huntington PJ, Hodge R, Tran S, Mason BL, Lee C, Vong L et al (2013) Double deletion of melanocortin 4 receptors and SAPAP3 corrects compulsive behavior and obesity in mice. Proc Natl Acad Sci USA 110:10759–10764
Xu P, He Y, Cao X, Valencia-Torres L, Yan X, Saito K, Wang C, Yang Y, Hinton A Jr, Zhu L et al (2017) Activation of serotonin 2C receptors in dopamine neurons inhibits binge-like eating in mice. Biol Psychiatry 81:737–747
Xu Y, Jones JE, Kohno D, Williams KW, Lee CE, Choi MJ, Anderson JG, Heisler LK, Zigman JM, Lowell BB et al (2008) 5-HT2CRs expressed by pro-opiomelanocortin neurons regulate energy homeostasis. Neuron 60:582–589
Xu Y, Lu Y, Cassidy RM, Mangieri LR, Zhu C, Huang X, Jiang Z, Justice NJ, Xu Y, Arenkiel BR et al (2019) Identification of a neurocircuit underlying regulation of feeding by stress-related emotional responses. Nat Commun 10:3446
Xu Y, Nedungadi TP, Zhu L, Sobhani N, Irani BG, Davis KE, Zhang X, Zou F, Gent LM, Hahner LD et al (2011) Distinct hypothalamic neurons mediate estrogenic effects on energy homeostasis and reproduction. Cell Metab 14:453–465
Yan J, Zhang H, Yin Y, Li J, Tang Y, Purkayastha S, Li L, Cai D (2014) Obesity- and aging-induced excess of central transforming growth factor-beta potentiates diabetic development via an RNA stress response. Nat Med 20:1001–1008
Yang SB, Tien AC, Boddupalli G, Xu AW, Jan YN, Jan LY (2012) Rapamycin ameliorates age-dependent obesity associated with increased mTOR signaling in hypothalamic POMC neurons. Neuron 75:425–436
Yang Y, Smith DL Jr, Keating KD, Allison DB, Nagy TR (2014) Variations in body weight, food intake and body composition after long-term high-fat diet feeding in C57BL/6J mice. Obesity (Silver Spring) 22:2147–2155
Yang Y, van der Klaauw AA, Zhu L, Cacciottolo TM, He Y, Stadler LKJ, Wang C, Xu P, Saito K, Hinton A Jr et al (2019) Steroid receptor coactivator-1 modulates the function of Pomc neurons and energy homeostasis. Nat Commun 10:1718
Yaswen L, Diehl N, Brennan MB, Hochgeschwender U (1999) Obesity in the mouse model of pro-opiomelanocortin deficiency responds to peripheral melanocortin. Nat Med 5:1066–1070
Yau YH, Potenza MN (2013) Stress and eating behaviors. Minerva Endocrinol 38:255–267
Yeo GS, Connie Hung CC, Rochford J, Keogh J, Gray J, Sivaramakrishnan S, O’Rahilly S, Farooqi IS (2004) A de novo mutation affecting human TrkB associated with severe obesity and developmental delay. Nat Neurosci 7:1187–1189
Yeo GS, Farooqi IS, Aminian S, Halsall DJ, Stanhope RG, O’Rahilly S (1998) A frameshift mutation in MC4R associated with dominantly inherited human obesity. Nat Genet 20:111–112
Yi CX, van der Vliet J, Dai J, Yin G, Ru L, Buijs RM (2006) Ventromedial arcuate nucleus communicates peripheral metabolic information to the suprachiasmatic nucleus. Endocrinology 147:283–294
Yi CX, Walter M, Gao Y, Pitra S, Legutko B, Kälin S, Layritz C, García-Cáceres C, Bielohuby M, Bidlingmaier M et al (2017) TNFα drives mitochondrial stress in POMC neurons in obesity. Nat Commun 8:15143
Yu IC, Lin HY, Liu NC, Sparks JD, Yeh S, Fang LY, Chen L, Chang C (2013) Neuronal androgen receptor regulates insulin sensitivity via suppression of hypothalamic NF-kappaB-mediated PTP1B expression. Diabetes 62:411–423
Zhang G, Li J, Purkayastha S, Tang Y, Zhang H, Yin Y, Li B, Liu G, Cai D (2013) Hypothalamic programming of systemic ageing involving IKK-β, NF-κB and GnRH. Nature 497:211–216
Zhang Y, Kim MS, Jia B, Yan J, Zuniga-Hertz JP, Han C, Cai D (2017a) Hypothalamic stem cells control ageing speed partly through exosomal miRNAs. Nature 548:52–57
Zhang Y, Reichel JM, Han C, Zuniga-Hertz JP, Cai D (2017b) Astrocytic process plasticity and IKKβ/NF-κB in central control of blood glucose, blood pressure, and body weight. Cell Metab 25:1091-1102.e1094
Zhu C, Xu Y, Jiang Z, Tian JB, Cassidy RM, Cai ZL, Shu G, Xu Y, Xue M, Arenkiel BR et al (2020) Disrupted hypothalamic CRH neuron responsiveness contributes to diet-induced obesity. EMBO Rep 21:e49210
Zhu L, Yang Y, Xu P, Zou F, Yan X, Liao L, Xu J, O’Malley BW, Xu Y (2013) Steroid receptor coactivator-1 mediates estrogenic actions to prevent body weight gain in female mice. Endocrinology 154:150–158
Acknowledgements
We apologize for not being able to cite many additional important contributions from peers due to space limitation. This study was funded by the National Key R&D Program of China (2019YFA0801900,2018YFA0800300), the National Natural Science Foundation of China (91749104 and 31971074), the Science and Technology Innovation Action Plan of Shanghai Science and Technology Committee (18140901300), the Open Research Fund of the National Key Laboratory of Genetic Engineering (SKLGE1803), the Shanghai Pujiang Talent Project (18PJ1400700) to T.L., and Dr. Tong is the holder of Cullen Chair in Molecular Medicine at McGovern Medical School of UTHealth.
Author information
Authors and Affiliations
Contributions
All authors were involved in the development of this review. TL drafted the section of “GENETIC BASIS FOR HUMAN OBESITY”, “GUT MICROBIOTA AND BRAIN-REGULATED METABOLISM”; YX drafted the section of “SEX DIFFERENCES IN METABOLIC REGULATION”; C-XY drafted the section of “HYPOTHALAMIC GLIAL IMMUNE AND METABOLIC CHANGES IN OBESITY AND DIABETES”; QT drafted the section of “STRESS, FEEDING AND OBESITY DEVELOPMENT”; DC drafted the section of “AGING IN THE CENTRAL NERVOUS SYSTEM OF ORGANISMS”. TL, QT and DC drafted the rest part of the review and critically revised the overall review.
Corresponding authors
Ethics declarations
Conflict of interest
All the authors of this paper declare that they have no conflict of interest that are relevant to the content of this article.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Liu, T., Xu, Y., Yi, CX. et al. The hypothalamus for whole-body physiology: from metabolism to aging. Protein Cell 13, 394–421 (2022). https://doi.org/10.1007/s13238-021-00834-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13238-021-00834-x