
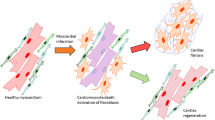
Abstract
Fibrotic remodeling is an adverse consequence of immune response-driven phenotypic modulation of cardiac cells following myocardial infarction (MI). MicroRNA-146b (miR-146b) is an active regulator of immunomodulation, but its function in the cardiac inflammatory cascade and its clinical implication in fibrotic remodeling following MI remain largely unknown. Herein, miR-146b-5p was found to be upregulated in the infarcted myocardium of mice and the serum of myocardial ischemia patients. Gain- and loss-of-function experiments demonstrated that miR-146b-5p was a hypoxia-induced regulator that governed the pro-fibrotic phenotype transition of cardiac cells. Overexpression of miR-146b-5p activated fibroblast proliferation, migration, and fibroblast-to-myofibroblast transition, impaired endothelial cell function and stress survival, and disturbed macrophage paracrine signaling. Interestingly, the opposite effects were observed when miR-146b-5p expression was inhibited. Luciferase assays and rescue studies demonstrated that the miR-146b-5p target genes mediating the above phenotypic modulations included interleukin 1 receptor associated kinase 1 (IRAK1) and carcinoembryonic antigen related cell adhesion molecule 1 (CEACAM1). Local delivery of a miR-146b-5p antagomir significantly reduced fibrosis and cell death, and upregulated capillary and reparative macrophages in the infarcted myocardium to restore cardiac remodeling and function in both mouse and porcine MI models. Local inhibition of miR-146b-5p may represent a novel therapeutic approach to treat cardiac fibrotic remodeling and dysfunction following MI.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Myocardial infarction is the leading cause of mortality and morbidity worldwide (Nagpal et al., 2016), indicating an urgent need for studies investigating the underlying mechanisms of MI to identify innovative therapeutic strategies.
Pathological cardiac remodeling is characterized by complex multicellular alterations, such as cardiomyocyte death, immune cell activation, and excessive deposition of the extracellular matrix, that exacerbate cardiac dysfunction, and often progresses to heart failure (Sutton and Sharpe, 2000; Prabhu and Frangogiannis, 2016a, b; Shiraishi et al., 2016). Cells are well known to sense their surrounding physical and signaling environments and respond accordingly by altering their functions and fates (Zhu et al., 2014a, 4b). Pioneering studies have demonstrated that adverse remodeling following MI is caused by the phenotypic modulation of cardiac cells, with the inappropriate and untimely activation and resolution of inflammation being a crucial driving factor (Epelman et al., 2015; Westman et al., 2016; Meyer et al., 2017; Huang and Frangogiannis, 2018). Thus, therapeutic modulation of immunoregulatory factors and reparative phenotypes of resident cells may be a promising approach for preventing post-infarction remodeling (Prabhu and Frangogiannis, 2016a, b).
MicroRNAs are functional, single-stranded, short noncoding RNAs that regulate a large array of biological processes through the degradation or inhibition of target messenger RNAs (mRNAs). The miR-146 family was the first family of microRNAs reported to be involved in mammalian immunomodulation (Taganov et al., 2006; Baltimore et al., 2008). However, the functions of these microRNAs in the cardiac inflammatory cascade and adverse remodeling remain unclear.
To address this, we generated two animal models (mouse and pig) to demonstrate the benefits of inhibiting miR-146b-5p post-MI. The results indicated that hypoxia induced miR-146b-5p expression through the NF-κB signaling pathway. miR-146b-5p gain-of-function was found to activate fibroblast proliferation, migration, and fibroblast to myofibroblast transition (FMT), impair endothelial cell function and stress survival, and disrupt macrophage paracrine signaling. The opposite effects were observed upon miR-146b-5p loss-of-function. IRAK1 and CEACAM1 were found to be the targets of miR-146b-5p in the above phenotypic modulations. The local delivery of a miR-146b-5p antagomir significantly reduced fibrosis and cell death, and upregulated capillary and reparative macrophages in infarcted myocardium to restore cardiac remodeling and function in both mice and porcine MI models.
Results
miR-146b-5p was upregulated in patients with chronic total occlusion (CTO) and in a murine MI model
CTO consists of the complete obstruction of a coronary artery, resulting in severe myocardial ischemia. Using quantitative real-time polymerase chain reaction (qPCR), we found that miR-146b-5p expression was significantly increased in the plasma of CTO patients compared to no-ischemia volunteers (n = 8 per group, P = 0.02; Fig. 1A). Clinical parameters and medical histories were available for all patients (Table S1). To elucidate the connection between plasma miR-146b-5p expression and myocardial ischemia, a mouse MI model was constructed, and qPCR was performed. Morphological observation and expression of the fibrotic genes COL1A1 (Col1) and ACTA2 (αSMA) were used to validate myocardial ischemia (Fig. 1B–D). We found that miR-146b-5p expression increased continuously in the infarct zone (reaching approximately 5-fold above the baseline), but not in the remote myocardium, over 14 days following the induction of MI (Fig. 1E).

miR-146b-5p is upregulated after myocardial infarction. (A) Levels of miR-146b-5p in the plasma of patients with chronic total occlusion (CTO) (n = 8 per group). (B) Representative images of infarcted mouse heart. Myocardium samples from different anatomic locations were harvested for the following analysis. (C and D) Expression levels of fibrotic genes Col1 (COL1A1) and αSMA (ACTA2) in myocardium samples (3 hearts, 5 samples per heart). (E) Expression levels of miR-146b-5p in mouse myocardium from different locations and time points, as measured by qPCR (n = 5 per group). Data are expressed as the mean ± SD. *P < 0.05 vs. control; **P < 0.01 vs. control. Data in panel (A), (C), and (D) were analyzed using Student’s unpaired t-test. Data in panel (E) was analyzed using one-way ANOVA followed by Tukey’s post-hoc analysis. Remo, remote zone; Infar, infarction zone.
Hypoxia upregulated miR-146b-5p in fibroblasts, endothelial cells, and macrophages may through NF-κB signaling
Given that miR-146b-5p is upregulated in the ischemic myocardium, we investigated which cell types expressed miR-146b-5p under ischemia and the mechanism by which it is induced.
We utilized a classic in vitro hypoxia model to expose the major resident cell types of the mouse heart, including cardiomyocytes, fibroblasts, endothelial cells, and macrophages, to ischemic conditions. After treatment with 1% oxygen for 24 h, miR-146b-5p expression was significantly induced in fibroblasts, endothelial cells, and macrophages, with the fibroblasts and macrophages showing the largest increases in expression (2.7- and 3.5-fold, P = 0.03 for both) (Fig. 2A). Hypoxia treatment failed to induce miR-146b-5p expression in cardiomyocytes (P = 0.18). Additionally, hypoxic fibroblasts exhibited considerably higher expression of miR-146b-5p compared to the normoxic control (Fig. S1A), which is consistent with our ischemic myocardium data (Fig. 1E).

Hypoxia-induced miR-146b-5p expression acts as a phenotypic modulator in fibroblasts, endothelial cells, and macrophages. (A) The expression profile of miR-146b-5p in primary cultured cardiac cells under hypoxia was studied using qPCR (n = 3 per group, 3 replicates). Multiple phenotypic studies were performed after 24 h of treatment with the miR-146b-5p mimic (miR-146b) or control (Ctrl) (B–H). (B) Representative images for EdU staining and EdU+ cell quantification of cardiac fibroblasts (n = 7 samples per group, 5 random fields per sample). Bar = 200 μm. (C and D), Representative migration images and quantification of data from Transwell and scratch assays in mouse fibroblasts (NIH3T3, n = 5 per group, 5 random fields per sample). Bar = 200 μm. (E) Representative images for immunostaining and quantification of expression of the fibroblast-to-myofibroblast transition genes Col1 (COL1A1) and αSMA (ACTA2) in cardiac fibroblasts (n = 3 per group, 7 fields per sample); bar = 5 μm. (F) Western blot analysis of the FMT genes in cardiac fibroblasts after treatment (n = 5). (G) Tube-formation assays of cardiac microvascular endothelial cells (CMVECs), with representative images on the left and quantification data on the right (n = 3 samples per group, 5 random fields per samples, 3 replicates). Bar = 200 μm. (H) ELISA of macrophage-secreted inflammatory factors (n = 5 per group). Data are expressed as the mean ± SD for at least three independent assays, unless otherwise noted. *P < 0.05; **P < 0.01; ns, not significant. Data were analyzed using Student’s t-test. CM, cardiomyocyte; FB, fibroblast; EC, endothelial cell; Mø, macrophage; IL, interleukin; TNF, tumor necrosis factor; MCP, monocyte chemotactic protein.
Transforming growth factor-β (TGF-β) signaling is well known as one of the primary mechanisms mediating cardiac remodeling following ischemia/hypoxia. Therefore, we investigated whether miR-146b-5p expression was activated by TGF-β signaling. As shown in Fig. S1B, treatment with 10 ng/mL TGF-β for 24 h greatly increased the expression of the fibrotic marker αSMA (about 2-fold, P = 0.01) in mouse fibroblasts; however, this treatment failed to induce miR-146b-5p expression (P = 0.66). Additionally, a luciferase reporter-based promoter activity assay indicated that the NF-κB promoter was activated during the 24-h hypoxic period (Fig. S1C). Using JSH-23, an NF-κB transcriptional inhibitor (30 μmol/L, 24-h treatment), we demonstrated that the inhibition of NF-κB transcriptional activity significantly reduced the expression of miR-146b-5p in fibroblasts (P < 0.01, Fig. S1D), endothelial cells and cardiac macrophages (both P < 0.01, Fig. S1E) under hypoxic conditions, suggesting that hypoxia-induced miR-146b-5p expression is at least partially mediated through NF-κB signaling. Collectively, these findings demonstrate that miR-146b-5p is characteristically induced in fibroblasts, endothelial cells, and macrophages by hypoxia. NF-κB signaling rather than TGF-β signaling involve in hypoxia-induced upregulation of miR-146b-5p in fibroblast.
miR-146b-5p drove the pro-fibrotic transition of fibroblasts, impaired endothelial cell function and stress survival, and disrupted macrophage paracrine signaling
Pro-fibrotic myofibroblasts are derived from the FMT and are the main source of collagen and other ECM proteins during pathological fibrotic remodeling. To determine if miR-146b-5p regulates FMT, mouse fibroblasts were treated with miR-146b-5p mimic, miR-146b-5p inhibitor, and controls. The results of the gain-of-function experiments are shown in Fig. 2, and those of the loss-of-function experiments are provided in Fig. 3. We first performed a dose-response study in fibroblasts based on fibrotic gene expression (COL1A1) and found treatment with 20 nmol mimic to be a desirable concentration in regards to both economy and efficiency (Fig. S1F). Next, the pro-fibrotic phenotype was assessed in terms of proliferation, migration, and expression of FMT markers, as shown in Figs. 2B–F and 3A–E. Overexpression of miR-146b-5p markedly enhanced fibroblast proliferation by 1.6-fold (P = 0.01, Fig. 2B), as determined by EdU staining, while miR-146b-5p inhibition significantly reduced cell proliferation (P = 0.03, Fig. 3A). The Transwell assay indicated that the miR-146b-5p mimic increased fibroblast migration by 2-fold (P = 0.0001, Fig. 2C). Inhibition of miR-146b-5p significantly attenuated cell migration by up to 40% (P = 0.003, Fig. 3B). Similar trends were observed in the scratch assay (Figs. 2D and 3C). Furthermore, as shown in Fig. 2E, immunofluorescence staining revealed that the miR-146b-5p mimic increased the expression of the FMT markers COL1A1 (Col1) and ACTA2 (αSMA) by 1.8-fold and 1.1-fold (P = 0.02 and P = 0.84), respectively. Additionally, western blotting showed a 1.9-fold and 1.6-fold increase in Col1 and αSMA expression, respectively, after mimic treatment (Fig. 2F). As expected, the miR-146b-5p inhibitor significantly reduced Col1 and αSMA protein expression by 62% and 29%, respectively (P = 0.02 and P = 0.01; Fig. 3D and 3E).

Inhibition of miR-146b-5p consolidates the reparative phenotypes of fibroblasts, endothelial cells, and macrophages. Multiple phenotypic studies were performed after 24 h of treatment with miR-146b-5p inhibitor (Anti-146b) or control (Ctrl). (A) Representative images for EdU staining and EdU+ cell quantification of cardiac fibroblasts (n = 7 samples per group, 5 random fields per sample). (B and C), Representative migration images and quantification, according to Transwell and scratch assays of mouse fibroblasts (NIH3T3, n = 5 per group, 5 random fields per sample). (D and E) Representative western blot images and quantification of expression of the fibroblast-to-myofibroblast transition genes Col1 (COL1A1) and αSMA (ACTA2) in cardiac fibroblasts (n = 5 per group). (F) Tube-formation assays of cardiac microvascular endothelial cells (CMVECs), with representative images on the left and quantification data on the right (n = 3 samples per group, 5 random fields per samples, 3 replicates). (G) ELISA of macrophage-secreted inflammatory factors (n = 5 per group). Data are expressed as the mean ± SD for at least three independent assays unless otherwise noted. *P < 0.05 vs. control; **P < 0.01 vs. control; ns, not significant. Data were analyzed using Student’s t-test. IL, interleukin; TNF, tumor necrosis factor; MCP, monocyte chemotactic protein.
Primary cardiac microvascular endothelial cells (CMVECs) were cultured and revealed that both overexpression and inhibition of miR-146b-5p regulated tube formation in CMVECs (Figs. 2G and 3F) and survival under starvation (Fig. S2). Upregulation of miR-146b-5p enhanced malnutrition-induced death in CMVECs and inhibited tube formation in Matrigel. Loop count, tube length, and branch count were significantly decreased after treatment with the miR-146b-5p mimic (Fig. 2G), whereas these were partially increased by inhibition of miR-146b-5p (P = 0.002 for loops and P = 0.0002 for branch counts; Fig. 3F).
Cardiac macrophages can regulate inflammation, angiogenesis, and tissue repair after MI by secreting factors and undergoing polarization (Nahrendorf and Swirski 2013; Swirski and Nahrendorf, 2013; Ma et al., 2018; Dick et al., 2019). In the present study, using resident macrophages isolated from MI hearts revealed that miR-146b-5p overexpression significantly decreased secretion of interleukin-1β (IL-1β), IL-6, tumor necrosis factor-α (TNF-α), and monocyte chemotactic protein 1 (MCP-1) (Fig. 2H; P = 0.002, 0.0001, 0.0002 and 0.02, respectively). However, miR-146b-5p inhibition strongly increased the amount of IL-1β, IL-6, and MCP-1 released into the culture supernatant (Fig. 3G). A growing body of evidence has demonstrated that early inflammatory signals set the stage for cardiac repair in the infarcted heart (Frangogiannis, 2012, 2014), suggesting that inflammatory secretion regulated by miR-146b-5p may influence endogenous repair following MI.
We also investigated the effect of miR-146b-5p inhibition on cardiomyocyte viability and found that miR-146b-5p inhibition did not have a significant effect on cardiomyocyte viability in vitro for up to 72 h (Fig. S3).
Collectively, these results demonstrate that miR-146b-5p regulates the pro-fibrotic phenotype and FMT of fibroblasts, tube formation, starvation survival of CMVECs, and inflammatory factor secretion by macrophages, indicating an important regulatory role for miR-146b-5p in the phenotypic modulation of cardiac cells following MI.
miR-146b-5p may modulate cardiac cell phenotypes by targeting IRAK1 and CEACAM1
In the present study, miR-146b-5p was found to directly bind to the 3′UTRs of IRAK1 and CEACAM1 based on luciferase reporter assays (Fig. S4A and S4B). IRAK1 is a major downstream effector of IL-1β signaling. CEACAM1 is a cell-cell adhesion molecule involved in differentiation, angiogenesis, apoptosis, and the innate and adaptive immune responses (Horst, 2006; Huang et al., 2015). Previously, little was known regarding the pathophysiological roles of IRAK1 and CEACAM1 in the heart. Interestingly, we found that miR-146b-5p exhibits preferential targeting in cardiac cells. In protein level, miR-146b-5p was more likely to inhibit IRAK1 expression in fibroblasts and macrophages, whereas it tended to inhibit CEACAM1 expression in endothelial cells (Fig. S4C–E). The IRAK1 overexpression was able to significantly rescue miR-146b-induced phenotypic changes in fibroblasts and macrophages (Fig. S4F and S4G). Additionally, treatment with the miR-146b-5p mimic reduced endothelial cell survival by 66.4% under starvation, and decreased tube length, loop count, and branch count in tube formation assays (23.1%, 48.7%, and 49.3%, respectively). Lentivirus-mediated upregulation of CEACAM1 significantly rescued miR-146b-induced endothelial cell survival (Fig. S4H) and tube formation (P = 0.01, Fig. S4I). Taken together, these results demonstrate that miR-146b-5p stimulates the pro-fibrotic transition of cardiac cells, possibly through IRAK1 and CEACAM1.
miR-146b-5p inhibition reduced cardiac fibrosis and apoptosis, promoted angiogenesis, and increased reparative macrophages in an MI mouse model
To assess whether miR-146b-5p is a viable therapeutic target for fibrotic remodeling following ischemic injury, an in vivo inhibition strategy was devised. Mice were treated with the miR-146b-5p antagomir immediately after MI surgery (Fig. 4A). Compared to the antagomir negative control group (Anti-Ctrl), the miR-146b-5p inhibition group (Anti-146b) exhibited a marked decrease in heart weight/tibia length (HW/TL, Fig. 4B), and the left ventricular (LV) wall thickness and infarction area were significantly restored (n = 8, P = 0.0001; Fig. 4C). Specifically, the Anti-Ctrl group had a higher HW/TL ratio (9.93% ± 0.56%) than the Anti-146b group (8.82% ± 0.45%). Moreover, the infarction area was 22.60% ± 3.56% and 13.12% ± 3.31% in the Anti-Ctrl and Anti-146b groups, respectively (P = 0.001). Echocardiography showed that miR-146b-5p inhibition restored the left ventricular ejection fraction (LVEF), fractional shortening (FS), and left ventricular internal diastolic diameter (LVIDd) (Fig. 4D). Significant differences in LVEF were observed at day 14 post-MI and remained stable to day 28 post-MI. The LVEF at 28 days post-MI were 20.43% ± 4.9% and 34.39% ± 3.8% in the Anti-Ctrl group and Anti-146b group, respectively (P = 0.01). Compared to the Anti-Ctrl group, the FS in the Anti-146b group was significantly higher at 28 days post-MI (P = 0.02). Despite only one significant difference being found regarding the LVIDd between these two groups at day 21, the average LVIDd in the Anti-Ctrl group remained higher than in the Anti-146b group (Fig. 4D).

Therapeutic modulation of miR-146b-5p restores cardiac function in the MI mouse. (A) Workflow for animal experiments to determine the effect of miR-146b-5p inhibition on infarcted hearts in vivo. The miR-146b-5p antagomir (Anti-146b) or control (Anti-Ctrl) was locally administered after MI surgery. The cardiac structure and function were studied for up to 4 weeks. (B) The ratio of heart weight to tibia length (n = 8 per group). (C) Representative images for hematoxylin and eosin (HE) and Masson’s staining and quantification of infarcted area (n = 8 samples per group, 10 random fields per sample). (D) Echocardiographic analyses of cardiac functions at 7, 14, 21, and 28 days post-MI and antagomir treatment (n = 7 per group). (E and F) Representative images for TUNEL staining and quantification of TUNEL+ apoptotic cells. Blue signals indicate cell nuclei. Brown signals represent TUNEL+ apoptotic cells (n = 7 per group, 10 random fields per sample); bar = 50 μm. (G and H) Representative images for CD31 staining and quantification of CD31+ vessels. Blue signals indicate cell nuclei. Brown signals indicate CD31+ vessels (n = 7 per group, 10 random fields per sample); bar = 50 μm. (I and J) Representative images for CD206 staining and quantification of CD206+ reparative macrophages. Blue signals indicate cell nuclei. Brown signals indicate CD206+ macrophages (n = 7 per group, 10 random fields per sample); bar = 100 μm. Data are expressed as the mean ± SD. *P < 0.05 vs. Anti-Ctrl group; **P < 0.01. Data in panel (D) were analyzed using two-way ANOVA followed by Tukey’s post-hoc analysis. Other data were analyzed using one-way ANOVA followed by Tukey’s post-hoc analysis. HW, heart weight; TL, tibia length; LVEF, left ventricular ejection fraction; FS, fractional shortening; LVIDd, left ventricular internal diastolic diameter; Anti-146b, antagomir miR-146b-5p; Anti-Ctrl, antagomir control.
TUNEL staining revealed that miR-146b-5p inhibition significantly reduced the number of apoptotic cells following MI from 21% ± 5.73% to 6% ± 3.94% (P = 0.0002; Fig. 4E and 4F). Immunohistochemical staining demonstrated that Anti-146b treatment significantly increased the number of CD31+ microvessels in the ischemic myocardium (Fig. 4G and 4H). In addition, we found CD206+ reparative macrophages to be increased by 9.38-fold following antagomir treatment compared to the Anti-Ctrl (Fig. 4I and 4J).
Therapeutic silencing of miR-146b-5p improved cardiac fibrosis and ventricular strain in a porcine MI model
We next studied the therapeutic potential of miR-146b-5p in a porcine model. A small-scale MI was induced by left circumflex artery (LCX) ligation surgery in minipigs, as developed and validated by Shen et al. (1999). A homologous antagomir was used for the porcine study. After ligation, 200 nmol antagomir in 1 mL PBS was immediately injected into the ischemic myocardium and adjacent border zone. The porcine cardiac structure and functions were evaluated by histological assays and echocardiography (Figs. 5, 6; Tables 1 and 2). Echocardiography indicated a marked improvement of cardiac function with miR-146b-5p silencing four weeks after MI. The LVEF in the antagomir-treated pigs (Anti-146b) was restored to 48.89 ± 13.45, while the LVEF in the antagomir control pigs (Anti-Ctrl) decreased to 37.26 ± 5.47 (n = 8, P = 0.039; Fig. 5A). Detailed echocardiographic results were showed in Table 1. Only one experimental animal in the antagomir control group died during the study (at day 21) (Fig. 5B). Morphologic observation and Masson staining of cardiac sections showed that antagomir treatment (Anti-146b) significantly reduced the infracted area from 7.13% ± 2.33% to 3.59% ± 1.02% (% of entire cross-section area; P = 0.02 vs. Anti-Ctrl) (Fig. 5C–E). Immunohistochemical staining demonstrated that miR-146b-5p antagomir treatment significantly decreased the expression of collagen type 1 and increased the number of CD31+ microvessels in the porcine MI heart, from 70.50 ± 12.80 to 26.86 ± 6.35 (P = 0.0001) and 24.92 ± 4.59 to 64.67 ± 7.74 (P = 0.0001), respectively, when compared with the Anti-Ctrl group (Fig. 5F and 5G). TUNEL staining demonstrated that miR-146b-5p inhibition markedly reduced the number of TUNEL+ apoptotic cells following MI from 70.57 ± 16.43 to 44.71 ± 19.39 TUNEL+ cells per view (P = 0.02; Fig. S5).

Therapeutic modulation of miR-146b-5p relieves cardiac fibrosis and remodeling in MI pig. (A) The LVEF of the experimental pigs was analyzed using echocardiography at different time points (sham group: n = 4, other groups: n = 8 each). (B) The survival analysis of the experimental groups. The survival curve was plotted using the Kaplan-Meier method. Pigs were sacrificed 4 weeks after MI. (C) Representative slices of the porcine myocardium. Infarction areas are indicated by the blue dashed line. Bar = 1 cm. (D) Infarction area analysis. Five whole heart sections per group were randomly selected, and the percentage of infarction area to total area in cross section was calculated. (E–G) The representative images for histological and immunohistochemical staining are shown (10 random fields per group, bar = 100 μm), indicating fibrosis, as per Masson’s staining (E); expression of collagen type 1 (Col1, F); and CD31+ capillaries (G). Cell apoptosis was analyzed by TUNEL staining (Fig. S5). Data are expressed as the mean ± SD. *P < 0.05; **P < 0.01. Data were analyzed using one-way ANOVA followed by Tukey’s post-hoc analysis. 1H, 1 h; 2W, 2 weeks; 4W, 4 weeks; Anti-146b, antagomir miR-146b-5p; Anti-Ctrl, antagomir control
Speckle tracking echocardiography (STE) analysis is a novel way to evaluate ventricular wall motion in large animals (Torres et al., 2018). Our study indicated that miR-146b-5p inhibition significantly improved the cardiac strain and ventricular wall motion of the ischemic myocardium four weeks after MI (Fig. 6). Figure 6A–C shows the cardiac strain classification, infarcted area, and segment definition of experimental hearts. In Fig. 6D, each line represents the strain changes of relevant segments during all phases of the cardiac cycle. We found that radial, rotation, and part circumferential strains were significantly improved in the Anti-146b group compared to the MI and Anti-Ctrl groups. No significant improvement was observed regarding the longitudinal strain among the groups. Figure 6E–G shows a bulls’ eye diagram with each segment in the cross section of the myocardium labelled according to its peak strain (%) at four weeks post-MI, in three directions: radial (E), circumferential (F), and longitudinal (G). Details regarding peak myocardial strains (%) of the ischemic area are shown in Table 2. Taken together, these data demonstrate that silencing miR-146b-5p significantly alleviates cardiac fibrosis and cell death to enhance angiogenesis and cardiac function in a porcine MI model.


Therapeutic modulation of miR-146b-5p improves myocardial strains in MI pig. (A) The cardiac strains in different directions. (B and C) The definition of myocardial segmentation and the representative echocardiographic images of pig heart obtained 4 weeks post-MI (at end systole). (C) For speckle tracking, the left ventricle is divided into six anatomical zones in both the cross-sectional and longitudinal direction. (D) Representative segmental strain curves of the left ventricular (LV) mid-wall over one cardiac cycle at 4 weeks post-MI in the longitudinal, radial, circumferential, and rotation directions. (E–G) Bull’s eye diagrams of the absolute value of peak myocardial segmental strain in the radial (E), circumferential (F), and longitudinal (G) directions at 4 weeks post MI. BS, basal septum; MS, mid septum; AS, apical septal; AL, apical lateral; ML, mid lateral; BL, basal lateral; Ant-sept, anterior septum; Ant, anterior; Lat, lateral; Post, posterior; Inf, inferior; Sept, septum; Anti-146b, antagomir miR-146b-5p; Anti-Ctrl, antagomir control
Discussion
In adult mammals, the resident heart cells have been demonstrated to be key cellular effectors in the inflammatory response during cardiac injury and repair (Westman et al., 2016; Fu et al., 2018). In the present study, we found that miR-146b-5p expression was upregulated in fibroblasts, endothelial cells, and macrophages under hypoxic conditions. Cardiac fibrosis is predominantly orchestrated by myofibroblasts derived from FMT (Prabhu and Frangogiannis, 2016). The TGFβ-Smad signaling pathway is the core signaling pathway governing FMT. Nagpal et al. demonstrated that inhibition of FMT by targeting TGF-β-induced miR-125b expression successfully prevented fibrotic remodeling after MI (Nagpal et al., 2016). In the present study, miR-146b-5p was found to be a novel facilitator of FMT; it was activated by hypoxia rather than by the classic pro-fibrotic TGF-β signaling.
CMVECs play a vital role in the regulation of fibrotic remodeling (Liu et al., 2019). Previous work found that miR-146 represses endothelial cell activity by targeting endothelial nitric oxide synthase in response to stimulation by the pro-inflammatory cytokine IL-1β (Cheng et al., 2013). We revealed that miR-146b-5p impaired CMVEC tube formation in Matrigel and reduced cell viability under starvation in vitro. By using an antagomir, we found that ischemic myocardium with inhibited miR-146b-5p expression showed a marked increase in the number of CD31+ microvessels (about 6-fold compared to control). Due to the local upregulation of IL-1β following MI, miR-146b-5p inhibition may preserve cardiac endothelial cell activity and function, and our data provides direct evidence for this in vivo.
Cardiac macrophages participate in homeostatic maintenance and tissue repair by producing reparative inflammatory mediators or undergoing polarization (Shiraishi et al., 2016; Ferraro et al., 2019). Some pioneering studies have reported that miR-146b regulates macrophage paracrine signaling and activation under various inflammatory environments (He et al., 2016; Peng et al., 2016; Deng et al., 2019; Zhang et al., 2019); however, little is known about the effects of miR-146b on cardiac macrophages. In the present study, the four most abundant cytokines in wounds (IL-1β, IL-6, TNF-α, and MCP-1) (Mescher, 2017) were found to be significantly reduced in the presence of a miR-146b-5p mimic, whereas many were significantly increased by treatment with an miR-146b-5p inhibitor. Figure 4I and 4J show the first direct in vivo evidence of miR-146b-5p inhibition increasing the number of reparative CD206+ macrophages, which may also aid in the repair of the ischemic myocardium.
The human miR-146 family is composed of two genes: MIR146A and MIR146B. These two miRNAs are located on different chromosomes and have differential regulatory functions (Paterson and Kriegel, 2017). Most published literature has focused on the effect of the miR-146 family in cancer or the immune system, and there are only a few reports of its functions in cardiovascular disease (Gao et al., 2015; Wang et al., 2015; Cheng et al., 2017; Desjarlais et al., 2019). Specifically, Di et al. found that miR-146b was upregulated in the ischemia/reperfusion(I/R) myocardium. miR-146b overexpression reduced the infarct size in the I/R model and attenuated I/R-induced H9C2 apoptosis in vitro (Di et al., 2017). Li et al. found that miR-146b protected H9C2 cells against hypoxia (Li et al., 2015). These contrasting results may be explained by our use of primary neonatal cardiomyocytes rather than a cell line, and our use of a permanent MI model rather than an I/R model. Furthermore, our data demonstrated that miR-146b-5p expression was not significantly changed in primary cardiomyocytes after 24 h under hypoxic conditions, suggesting that the upregulation of miR-146b detected in the I/R myocardium may be derived from non-cardiomyocyte cells, such as fibroblasts. Additionally, the mature miR-146b includes miR-146b-3p and -5p; each strand has a unique binding site and function. We investigated miR-146b-5p in the present study. These differences in experimental settings may have led to different results. Here, we found that miR-146b-5p regulates the reparative phenotype of cardiac cells and demonstrated the feasibility of alleviating cardiac remodeling and dysfunction by inhibiting miR-146b-5p in both small (mice) and large (pig) mammals. To the best of our knowledge, this is the first study investigating the therapeutic potential of miR-146b-5p on cardiac remodeling and dysfunction in a pig MI model.
In recent years, pioneering studies have demonstrated that the silencing of specific microRNAs, including miR-199a (Gabisonia et al., 2019), miR-15 (Hullinger et al., 2012), miR-21 (Thum et al., 2008), miR-92a (Hinkel et al., 2013), and miR-125b (Nagpal et al., 2016) reduces cardiac fibrosis and dysfunction in vivo. In the present study, we found that miR-146b-5p expression was regionally increased in the ischemic myocardium; we then injected a miR-146b-5p antagomir into the infarcted and peri-infarcted myocardium. Our in vivo study of antagomir efficacy identified a single dosage that significantly inactivated the effects of miR-146 on target genes for up to three days (Fig. S6), suggesting a short-term inhibition of miR-146b-5p was achieved in the present study. Given that miR-146b is an anti-inflammatory factor, its long-term inhibition may prevent the timely resolution of inflammation following MI, which has been demonstrated to be detrimental. In other words, the dosage and intervention time must account for the biphasic nature of cardiac inflammation following MI. In murine models, reparative inflammatory signals were triggered within the first four days after MI. We thus postulate that the effective window for miR-146b-5p intervention should not exceed the inflammatory phase immediately after MI.
Although promising, there are remaining hurdles that must be overcome to achieve this goal in clinical settings. One major concern is the pleiotropic nature of microRNAs (Mendell and Olson, 2012). Although local treatment (Hinkel et al., 2013; Yang et al., 2018a, b) can partially prevent the risk of undesired side effects induced by systemic anti-microRNA treatment, current anti-microRNA therapy is not yet cell-specific and results in a general systemic inhibition of microRNA expression. Although the above-mentioned technical hurdles have not yet been addressed, this study provides a basis for improving our current understanding of immunomodulatory microRNAs and fibrotic remodeling post-MI in large mammals. Our data indicate that targeting immunoregulatory microRNAs may represent a novel therapeutic strategy for complementary use with current ischemic heart disease medications.
Materials and methods
Human samples
Human blood samples were collected from Shanghai East Hospital and Zhongshan Hospital with informed consent and were approved by the hospital ethics committee in accordance with the Helsinki Declaration. Arterial blood (5 mL) was drawn from patients diagnosed with CTO through a catheter before percutaneous recanalization. Equivalent blood samples were obtained from health volunteers by arteriopuncture. Clinical parameters and medical histories were available for all patients (Table S1).
Animal studies
All experimental procedures were approved by the local Institutional Animal Care and Use Committee and performed in accordance with the related guidelines.
Myocardial infarction surgery and antagomir treatments
Male C57BL/6 mice aged 10–12 weeks were purchased from Shanghai SLAC Laboratories. Surgery was carried out as previously described (Hinkel et al., 2013; Yang et al., 2018a, b). Briefly, mice were anesthetized with isoflurane (2%) and ventilated using a rodent ventilator (Harvard Apparatus Inc.) throughout the surgical procedure. The descending coronary artery was ligated with a silk suture (Nakada et al., 2017). The chest was then closed in layers. Sham-operated animals underwent the same procedure without ligation. For in vivo miRNA treatment, a total of 20 nmol miR-146b-5p antagomir (miR30003475-4-5, Ribobio, China) or negative control antagomir (miR3N0000001-4-20, Ribobio, China) was dissolved in 40 µL of PBS and injected into the infarct and peri-infarct zone using a syringe (Nakada et al., 2017). Echocardiography was performed before and after left anterior descending coronary artery ligation using a Supersonic Aixplorer Vevo 2100 system.
Female Bama minipigs (6 months, 20 kg) were randomly assigned to the following groups: sham group (Sham, n = 4); MI + saline group (MI, n = 8); MI + miR-146b-5p antagomir group (Anti-146b, n = 8); and MI + antagomir negative control group (Anti-Ctrl, n = 8). The surgical procedure to induce myocardial ischemia has been described previously (Shen et al., 1999; Purcell et al., 2014). Briefly, the animals were anesthetized using 2% isoflurane, intubated, and ventilated using an animal anesthesia ventilator (Matrx model 3000; USA). The left circumflex artery (LCX) was ligated with a 4-0 silk suture. Commercially available miR-146b-5p antagomir or negative control antagomir (200 nmol in 1 mL PBS; miR30003475-4-5 and miR3N0000001-4-20, Ribobio, China) was injected intramyocardially into the infarct and peri-infarct zone using a syringe (Krutzfeldt et al., 2005). The chest was then closed in layers and the animals were allowed to recover. Standard post-operative care was administered until the animals fed normally. One hour after surgery, an echocardiograph was performed to assess cardiac function. Postoperative animals with LVEF ranging from 30% to 60% were enrolled and followed up for four weeks. Three animals died of lethal arrhythmia during surgery.
Cardiac function analyses
Mouse cardiac function was assessed according to a previously published protocol (Yuan et al., 2018). For minipigs, cardiac function was measured using a TOSHIBA Artida/SSH-880CV. Briefly, minipigs were anesthetized and placed in the supine position. The end-diastolic and end-systolic volumes (LVEDV and LVESV) and LVEF were calculated using the Simpson method. Wall thickness (LVPWd and LVPWs) and left ventricle diameter (LVIDd and LVIDs) were measured at early and end-diastole in the oblique right parasternal long-axis, four-chamber view. STE analysis was performed as previously described (Torres et al., 2018). Continuous strain measurements of different ventricle segments over the whole cardiac cycle were obtained.
Histology and immunostaining
Heart samples were harvested, fixed with paraformaldehyde, and embedded in paraffin. Hematoxylin and eosin (HE) and Masson’s trichrome staining were performed. To quantify the fibrosis area, ten fields of view were randomly selected from eight cardiac sections and the percentage of Masson’s trichrome positive-stained area relative to total myocardial area was calculated. TUNEL staining was performed using a TUNEL apoptosis detection kit (40306ES50, Yeasen, China), according to the manufacturer’s instructions. Immunohistochemical staining was performed according to the standard protocol and using the following antibodies: Collagen type 1 (Col I, 1:1,000; ab34710, Abcam), αSMA (1:500; #19245, CST), CD31 (1:1,000; ab182981, Abcam), CD206 (1 µg/mL; ab64693, Abcam), goat anti-rabbit IgG H&L (HRP) (1:10,000; ab205718, Abcam), and goat anti-mouse IgG H&L (HRP) (1:10,000; ab205719, Abcam). For immunofluorescence, cells were fixed in cold methanol for 5 min, then permeabilized and blocked in phosphate buffered solution (0.5% Triton X-100) with 1% bovine serum albumin for 1 h. Immunostaining was performed using the following antibodies: Collagen type 1 (ab34710, Abcam), αSMA (#19245, CST), and the corresponding secondary antibodies.
Cell culture and treatments
The primary culture of mouse neonatal cardiomyocytes and cardiac fibroblasts was performed as we have previously published (Liu et al., 2019). In brief, neonatal cardiomyocytes were isolated from 1-day-old C57BL/6 mice by enzyme digestion (0.06% trypsin and 0.025% collagenase). Single cell suspensions were plated on 100-mm culture dishes in high-glucose Dulbecco’s modified Eagle’s medium (DMEM) with 14% fetal bovine serum (FBS) for 2 h. Non-attached cardiomyocytes were transferred to another dish to culture and 100 μmol/L bromodeoxyuridine was added to block fibroblast proliferation. The attached cardiac fibroblast fraction was cultured in DMEM with 10% FBS and 1% penicillin/streptomycin. Cells were cultured in serum-free medium for 4 h before treatment. NIH3T3 fibroblasts were obtained from the ATCC and cultured following the classic protocol (Peres et al., 2018).
CMVECs were isolated as previously described (Liu et al., 2019). Briefly, minced mouse myocardium tissue was digested using 1 mg/mL warm collagenase type 1 (C5849, Sigma), 5 units/mL dispase (345235, Collaborative), and 60 units/mL DNase 1 (10104159001, Roche) for 45 min. After filtration through a 70-μm cell strainer, single cell pellets were incubated with anti-CD31 magnetic microbeads (antibody: 553370, BD; microbeads: 11035, Invitrogen) for 20 min following the manufacturer’s instructions. After magnetic separation, CMVECs were cultured in endothelial cell growth medium (CC-3162, Lonza).
Mouse cardiac macrophages were isolated as previously described (Jia et al., 2019), with modifications. Specifically, two days after MI surgery, the myocardium of the left ventricular anterior wall was minced and enzymatically digested with 1.5 mg/mL collagenase type 2, 0.25 mg/mL elastase, and 0.5 mg/mL DNase 1 for 1 h at 37 °C. The cell suspension was passed through a 70-μm cell strainer and macrophages were isolated by positive selection using CD11b (130-049-601, Miltenyi Biotec) and F4/80 microbeads (130-110-443, Miltenyi Biotec) with a magnetic cell sorting separator (Miltenyi Biotec) according to the manufacturer’s protocol.
A commercially available miR-146b-5p mimic (miR10003475-1-5, Ribobio) and inhibitor (miR20003475-1-5, Ribobio) were used for gain- and loss-of-function experiments in vitro according to the manufacturer’s protocol. Dose-response curves were determined using cardiac fibroblasts at 10, 20, and 50 nmol. In vitro hypoxia experiments were conducted using Genbag (45534, BioMerieux). Cells were incubated under hypoxic conditions for 24 h.
Cell proliferation analyses
Cell proliferation analyses were performed using cell counting kit-8 (CCK-8) (CK04-11, Dojindo, Japan) and the Cell-Light EdU kit (C10310-3, Ribobio, China). Briefly, 100 μL of cell suspension containing 3,000 cells was added to a 96-well plate. Fibroblasts were transfected with miR-146b-5p mimic, miR-146b-5p inhibitor, or the control (Ctrl) using the riboFECT™ CP Transfection Kit (C10511-1, Ribobio) according to the manufacturer’s instructions and cultured for an additional 24 h. For CCK-8 analysis, 10 μL of the CCK-8 reagent was added to the medium and incubated for 3 h. The OD levels were measured using a spectrophotometric microplate reader at 450 nm. For EdU analysis, the reagent was added to the medium and incubated for 2 h to label EdU. Cell images were then acquired at five random fields per sample using a Leica fluorescence microscope. The data presented are the average of three independent assays performed in triplicate.
Cell migration analyses
Cell migration analyses were performed via a Transwell assay (8.0 μm; 3422, Corning, USA) and a scratch wound assay. Fibroblasts were transfected with the miR-146b-5p mimic, miR-146b-5p inhibitor, or control using the riboFECT™ CP Transfection Kit (C10511-1, Ribobio) according to the manufacturer’s instructions. Transfected cells were harvested and resuspended in serum-free DMEM. An aliquot (200,000 cells/200 µL) of cells in serum-free DMEM was dispensed into the Transwell inserts, and DMEM with 20% serum was placed in the lower chamber. After 8 h of culture, the non-migrated cells were carefully removed and the migrated cells in the bottom were stained with crystal violet solution. Images were captured at five random fields by microscopy. The migrated cells were counted by two volunteers blinded to the study.
For the scratch wound assay, fibroblasts were seeded into 6-well plates and transfected with the miR-146b-5p mimic, miR-146b-5p inhibitor, or control as described above. The transfected cells were subjected to serum starvation for 24 h after reaching confluence. The cells were scratched in a crisscross manner and rinsed with DMEM to remove any cellular debris prior to culturing in DMEM supplemented with 10% FBS. The wound area was documented at 0 h (A) and 6 h (B) by photomicrographic images and measured with ImageJ software by two experienced investigators who were blinded to the treatments. The wound width percentages were calculated as: (A−B)/A ×100%.
In vitro endothelial function assays: starvation, survival, and tube formation
In vitro endothelial function assays were performed as previously described (Kilic et al., 2005; Li et al., 2018). Briefly, CMVECs were transfected with the miR-146b-5p mimic, miR-146b-5p inhibitor, or control. Cells were cultured in normal endothelial growth medium until reaching confluence. The full medium was then replaced with starvation medium (DMEM containing 2% FBS without other supplements). The starvation medium was renewed every 3 days. After 3 days of culture, cell viability was assessed morphologically using phase-contrast microscopy. The criteria for viability were the state of confluence and the adherence of endothelial cells.
In the tube formation assay, 2 × 104 mouse CMVECs were seeded onto 96-well plates pre-coated with Matrigel and incubated for 6 h. Tube formation was quantified by counting the number of loops and branch points, then calculating the total tube length. Five random fields were captured per group and three independent experiments were performed and analyzed.
Cytokine measurement
Cardiac macrophages were cultured in the absence of cytokines and were transfected with the miR-146b-5p mimic, miR-146b-5p inhibitor, or control, as described previously (Yang et al., 2018a, b). The culture supernatant was collected two times—immediately after transfection was completed, and 6 hours’ culture following transfection completion. The levels of interleukin-1β (IL-1β), IL-6, tumor necrosis factor-α (TNF-α), and monocyte chemotactic protein 1(MCP-1) in the culture supernatant were measured using commercially available ELISA kits, according to the manufacturer’s instructions. All samples were assayed in triplicate.
qPCR
qPCR was performed as previously described (Yang et al., 2018a, b), with minor modifications. In brief, total RNA was collected using Trizol reagent (Invitrogen). Long RNA was reverse transcribed using the PrimeScriptTM RT reagent kit with gDNA Eraser (RR047A, TaKaRa). For miRNA, a miRNA cDNA synthesis kit (A11193-051, Invitrogen) was used for polyadenylation and reverse transcription. qPCR was performed using TB Green® Advantage® qPCR Premix (639676, Clontech) on a QuantStudioTM 6 Flex Real-time PCR System according to the manufacturer’s protocol. The miRNA primers were purchased from Ribobio (Guangzhou, China), hsa-miR-146b-5p primers (MQPS0000653-1-200), mmu-miR-146b-5p primers (MQPS0002464-1-200), and U6 primers (MQPS0000002-1-200) were adopted. Additional primers used for qPCR are listed in Table S2. The relative mRNA or miRNA expression levels were normalized to GAPDH or U6 snRNA, respectively, using the delta-delta Ct method. Three biological replicates per group were used for qPCR.
Western blot
Cells were lysed in RIPA lysis buffer supplemented with protease inhibitor pellets (Amersham Biosciences, USA). Insoluble debris were removed by centrifugation (25,000 ×g) for 30 min at 4 °C. Cell lysates were boiled with loading buffer for 10 min. Equal amounts of protein (20 μg) were separated by SDS-PAGE and assessed by Western blot for the indicated protein(s) using the antibodies described above. Experiments were repeated three times and GAPDH was used as the loading control.
Luciferase reporter construct and assay
The luciferase reporter assay was performed using the Dual-Luciferase® reporter assay kit (Promega, USA) and Promega GloMax 96 according to the manufacturer’s protocol. The pGM-NF-kappa B-Luc plasmid was purchased from Genomeditech (GM-021001, China). To assemble the 3′UTR reporter, the binding sites of miR-146b-5p on mouse IRAK1 and CEACAM1 mRNA 3′UTRs were amplified and cloned into a pGL3-promoter vector (Promega) to construct the wild-type (wt) and mutant (mut) luciferase reporters. Fibroblasts were co-transfected with individual reporter vectors (Luc-CEACAM1-wt, Luc-CEACAM1-mut, Luc-IRAK1-wt, Luc-IRAK1-mut; 0.2 μg/2.5 × 104 cells) and control or miR-146b-5p mimics (20 nmol/L) using the riboFECT™ CP Reagent (C10511-1, Ribobio), according to the manufacturer’s instructions. Luciferase and Renilla activities were measured 48 h after transfection using a standard protocol. The relative luciferase unit was defined as the ratio of luciferase versus Renilla activity compared to that of the control (set to 1).
Statistical analysis
Data are presented as the mean ± standard deviation (SD) for at least three independent assays unless otherwise noted. Student’s t-test was used to compare the differences between two groups. One-way ANOVA with Tukey’s post-hoc test was used for comparisons between multiple groups. The survival curve was plotted using the Kaplan-Meier method. GraphPad Prism 8 was used for the statistical analyses. Differences with P-values < 0.05 were considered statistically significant.
Author contributions
Concepts, H.Z. and J.G.; resources, H.Z. and Z.L; experiments and data analysis, Y.L., H.C., H.L., and Y.D.; manuscript preparation, Y.L., H.L. and H.Z.; revision and editing, all authors.
References
Aurora AB, Porrello ER, Tan W, Mahmoud AI, Hill JA, Bassel-Duby R, Sadek HA, Olson EN (2014) Macrophages are required for neonatal heart regeneration. J Clin Invest 124:1382–1392
Baltimore D, Boldin MP, O’Connell RM, Rao DS, Taganov KD (2008) MicroRNAs: new regulators of immune cell development and function. Nat Immunol 9:839–845
Cheng HS, Sivachandran N, Lau A, Boudreau E, Zhao JL, Baltimore D, Delgado-Olguin P, Cybulsky MI, Fish JE (2013) MicroRNA-146 represses endothelial activation by inhibiting pro-inflammatory pathways. EMBO Mol Med 5:1017–1034
Cheng HS, Besla R, Li A, Chen Z, Shikatani EA, Nazari-Jahantigh M, Hammoutène A, Nguyen M, Geoffrion M, Cai L et al (2017) Paradoxical suppression of atherosclerosis in the absence of microRNA-146a. Circ Res 121:354–367
Deng F, He S, Cui S, Shi Y, Tan Y, Li Z, Huang C, Liu D, Zhi F, Peng L (2019) A molecular targeted immunotherapeutic strategy for ulcerative colitis via dual-targeting nanoparticles delivering miR-146b to intestinal macrophages. J Crohns Colitis 13:482–494
Desjarlais M, Dussault S, Rivard F, Harel S, Sanchez V, Hussain SNA, Rivard A (2019) Forced expression of microRNA-146b reduces TRAF6-dependent inflammation and improves ischemia-induced neovascularization in hypercholesterolemic conditions. Atherosclerosis 289:73–84
Di YF, Li DC, Shen YQ, Wang CL, Zhang DY, Shang AQ, Hu T (2017) MiR-146b protects cardiomyocytes injury in myocardial ischemia/reperfusion by targeting Smad4. Am J Transl Res 9:656–663
Dick SA, Macklin JA, Nejat S, Momen A, Clemente-Casares X, Althagafi MG, Chen J, Kantores C, Hosseinzadeh S, Aronoff L et al (2019) Self-renewing resident cardiac macrophages limit adverse remodeling following myocardial infarction. Nat Immunol 20:29–39
Epelman S, Liu PP, Mann DL (2015) Role of innate and adaptive immune mechanisms in cardiac injury and repair. Nat Rev Immunol 15:117–129
Ferraro B, Leoni G, Hinkel R, Ormanns S, Paulin N, Ortega-Gomez A, Viola JR, de Jong R, Bongiovanni D, Bozoglu T et al (2019) Pro-angiogenic macrophage phenotype to promote myocardial repair. J Am Coll Cardiol 73:2990–3002
Frangogiannis NG (2012) Regulation of the inflammatory response in cardiac repair. Circ Res 110:159–173
Frangogiannis NG (2014) The inflammatory response in myocardial injury, repair, and remodelling. Nat Rev Cardiol 11:255–265
Fu X, Khalil H, Kanisicak O, Boyer JG, Vagnozzi RJ, Maliken BD, Sargent MA, Prasad V, Valiente-Alandi I, Blaxall BC et al (2018) Specialized fibroblast differentiated states underlie scar formation in the infarcted mouse heart. J Clin Invest 128:2127–2143
Gabisonia K, Prosdocimo G, Aquaro GD, Carlucci L, Zentilin L, Secco I, Ali H, Braga L, Gorgodze N, Bernini F et al (2019) MicroRNA therapy stimulates uncontrolled cardiac repair after myocardial infarction in pigs. Nature 569:418–422
Gao M, Wang X, Zhang X, Ha T, Ma H, Liu L, Kalbfleisch JH, Gao X, Kao RL, Williams DL et al (2015) Attenuation of cardiac dysfunction in polymicrobial sepsis by microRNA-146a is mediated via targeting of IRAK1 and TRAF6 expression. J Immunol 195:672–682
He X, Tang R, Sun Y, Wang YG, Zhen KY, Zhang DM, Pan WQ (2016) MicroR-146 blocks the activation of M1 macrophage by targeting signal transducer and activator of transcription 1 in hepatic schistosomiasis. eBiomedicine 13:339–347
Hinkel R, Penzkofer D, Zühlke S, Fischer A, Husada W, Xu Q, Baloch E, van Rooij E, Zeiher AM, Kupatt C et al (2013) Inhibition of MicroRNA-92a protects against ischemia/reperfusion injury in a large-animal model. Circulation 128:1066–1075
Horst AK (2006) Carcinoembryonic antigen-related cell adhesion molecule 1 modulates vascular remodeling in vitro and in vivo. J Clin Invest 116:1596–1605
Huang S, Frangogiannis NG (2018) Anti-inflammatory therapies in myocardial infarction: failures, hopes and challenges. Br J Pharmacol 175:1377–1400
Huang Y, Zhu C, Kondo Y, Anderson AC, Gandhi A, Russell A, Dougan SK, Petersen B, Melum E, Pertel T et al (2015) CEACAM1 regulates TIM-3-mediated tolerance and exhaustion. Nature 517:386–390
Huang S, Li X, Zheng H, Si X, Li B, Wei G, Li C, Chen Y, Chen Y, Liao W et al (2019) Loss of super-enhancer-regulated circRNA Nfix induces cardiac regeneration after myocardial infarction in adult mice. Circulation 139:2857–2876
Hullinger TG, Montgomery RL, Seto AG, Dickinson BA, Semus HM, Lynch JM, Dalby CM, Robinson K, Stack C, Latimer PA et al (2012) Inhibition of miR-15 protects against cardiac ischemic injury. Circ Res 110:71–81
Jia D, Jiang H, Weng X, Wu J, Bai P, Yang W, Wang Z, Hu K, Sun A, Ge J (2019) Interleukin-35 promotes macrophage survival and improves wound healing after myocardial infarction in mice. Circ Res 124:1323–1336
Kilic N, Oliveira-Ferrer L, Wurmbach J, Loges S, Chalajour F, Vahid SN, Weil J, Fernando M, Ergun S (2005) Pro-angiogenic signaling by the endothelial presence of CEACAM1. J Biol Chem 280:2361–2369
Krutzfeldt J, Rajewsky N, Braich R, Rajeev KG, Tuschl T, Manoharan M, Stoffel M (2005) Silencing of microRNAs in vivo with ‘antagomirs’. Nature 438:685–689
Li JW, He SY, Feng ZZ, Zhao L, Jia WK, Liu P, Zhu Y, Jian Z, Xiao YB (2015) MicroRNA-146b inhibition augments hypoxia-induced cardiomyocyte apoptosis. Mol Med Rep 12:6903–6910
Li H, Liao Y, Gao L, Zhuang T, Huang Z, Zhu H, Ge J (2018) Coronary serum exosomes derived from patients with myocardial ischemia regulate angiogenesis through the miR-939-mediated nitric oxide signaling pathway. Theranostics 8:2079–2093
Liu J, Zhuang T, Pi J, Chen X, Zhang Q, Li Y, Wang H, Shen Y, Tomlinson B, Chan P et al (2019) Endothelial forkhead box transcription factor P1 regulates pathological cardiac remodeling through transforming growth factor-β1-endothelin-1 signal pathway. Circulation 140:665–680
Ma Y, Mouton AJ, Lindsey ML (2018) Cardiac macrophage biology in the steady-state heart, the aging heart, and following myocardial infarction. Transl Res 191:15–28
Mendell JT, Olson EN (2012) MicroRNAs in stress signaling and human disease. Cell 148:1172–1187
Mescher AL (2017) Macrophages and fibroblasts during inflammation and tissue repair in models of organ regeneration. Regeneration 4:39–53
Meyer IS, Jungmann A, Dieterich C, Zhang M, Lasitschka F, Werkmeister S, Haas J, Müller OJ, Boutros M, Nahrendorf M et al (2017) The cardiac microenvironment uses non-canonicalWNT signaling to activate monocytes after myocardial infarction. EMBO Mol Med 9:1279–1293
Nagpal V, Rai R, Place AT, Murphy SB, Verma SK, Ghosh AK, Vaughan DE (2016) MiR-125b is critical for fibroblast-to-myofibroblast transition and cardiac fibrosis. Circulation 133:291–301
Nahrendorf M, Swirski FK (2013) Monocyte and macrophage heterogeneity in the heart. Circ Res 112:1624–1633
Nakada Y, Canseco DC, Thet S, Abdisalaam S, Asaithamby A, Santos CX, Shah AM, Zhang H, Faber JE, Kinter MT et al (2017) Hypoxia induces heart regeneration in adult mice. Nature 541:222–227
Paterson MR, Kriegel AJ (2017) MiR-146a/b: a family with shared seeds and different roots. Physiol Genomics 49:243–252
Peng L, Zhang H, Hao Y, Xu F, Yang J, Zhang R, Lu G, Zheng Z, Cui M, Qi C et al (2016) Reprogramming macrophage orientation by microRNA 146b targeting transcription factor IRF5. eBiomedicine 14:83–96
Peres DL, Schuman ML, Aisicovich M, Toblli JE, Pirola CJ, Landa MS, Garcia SI (2018) Angiotensin II requires an intact cardiac thyrotropin-releasing hormone (TRH) system to induce cardiac hypertrophy in mouse. J Mol Cell Cardiol 124:1–11
Prabhu SD, Frangogiannis NG (2016a) The biological basis for cardiac repair after myocardial infarction. Circ Res 119:91–112
Prabhu SD, Frangogiannis NG (2016b) The biological basis for cardiac repair after myocardial infarction: from inflammation to fibrosis. Circ Res 119:91–112
Purcell BP, Lobb D, Charati MB, Dorsey SM, Wade RJ, Zellars KN, Doviak H, Pettaway S, Logdon CB, Shuman JA et al (2014) Injectable and bioresponsive hydrogels for on-demand matrix metalloproteinase inhibition. Nat Mater 13:653–661
Shen Y, Lynch JJ, Shannon RP, Wiedmann RT (1999) A novel heart failure model induced by sequential coronary artery occlusions and tachycardiac stress in awake pigs. Am J Physiol-Heart C 277:H388–H398
Shiraishi M, Shintani Y, Shintani Y, Ishida H, Saba R, Yamaguchi A, Adachi H, Yashiro K, Suzuki K (2016) Alternatively activated macrophages determine repair of the infarcted adult murine heart. J Clin Invest 126:2151–2166
Sutton MG, Sharpe N (2000) Left ventricular remodeling after myocardial infarction: pathophysiology and therapy. Circulation 101:2981–2988
Swirski FK, Nahrendorf M (2013) Macrophage-stem cell crosstalk after myocardial infarction*. J Am Coll Cardiol 62:1902–1904
Taganov KD, Boldin MP, Chang KJ, Baltimore D (2006) NF-kappaB-dependent induction of microRNA miR-146, an inhibitor targeted to signaling proteins of innate immune responses. Proc Natl Acad Sci USA 103:12481–12486
Thum T, Gross C, Fiedler J, Fischer T, Kissler S, Bussen M, Galuppo P, Just S, Rottbauer W, Frantz S et al (2008) MicroRNA-21 contributes to myocardial disease by stimulating MAP kinase signalling in fibroblasts. Nature 456:980–984
Torres WM, Jacobs J, Doviak H, Barlow SC, Zile MR, Shazly T, Spinale FG (2018) Regional and temporal changes in left ventricular strain and stiffness in a porcine model of myocardial infarction. Am J Physiol Heart Circ Physiol 315:H958–H967
Wang J, Wang Y, Han J, Li Y, Xie C, Xie L, Shi J, Zhang J, Yang B, Chen D et al (2015) Integrated analysis of microRNA and mRNA expression profiles in the left atrium of patients with nonvalvular paroxysmal atrial fibrillation: Role of miR-146b-5p in atrial fibrosis. Heart Rhythm 12:1018–1026
Westman PC, Lipinski MJ, Luger D, Waksman R, Bonow RO, Wu E, Epstein SE (2016) Inflammation as a driver of adverse left ventricular remodeling after acute myocardial infarction. J Am Coll Cardiol 67:2050–2060
Yang F, Chen Q, He S, Yang M, Maguire EM, An W, Afzal TA, Luong LA, Zhang L, Xiao Q (2018a) miR-22 Is a novel mediator of vascular smooth muscle cell phenotypic modulation and neointima formation. Circulation 137:1824–1841
Yang L, Wang B, Zhou Q, Wang Y, Liu X, Liu Z, Zhan Z (2018b) MicroRNA-21 prevents excessive inflammation and cardiac dysfunction after myocardial infarction through targeting KBTBD7. Cell Death Dis 9:769
Yuan J, Liu H, Gao W, Zhang L, Ye Y, Yuan L, Ding Z, Wu J, Kang L, Zhang X et al (2018) MicroRNA-378 suppresses myocardial fibrosis through a paracrine mechanism at the early stage of cardiac hypertrophy following mechanical stress. Theranostics 8:2565–2582
Zhang L, Fu Y, Wang H, Guan Y, Zhu W, Guo M, Zheng N, Wu Z (2019) Severe fever with thrombocytopenia syndrome virus-induced macrophage differentiation is regulated by miR-146. Front Immunol 10:1095
Zhu H, Sun A, Zhu H, Li Z, Huang Z, Zhang S, Ma X, Zou Y, Hu K, Ge J (2014a) Aldehyde dehydrogenase-2 is a host factor required for effective bone marrow mesenchymal stem cell therapy. Arterioscler Thromb Vasc Biol 34:894–901
Zhu H, Sun A, Zou Y, Ge J (2014b) Inducible metabolic adaptation promotes mesenchymal stem cell therapy for ischemia. Arterioscler Thromb Vasc Biol 34:870–876
ACKNOWLEDGMENTS
The authors are grateful for technical assistance from Dr. Tiantong Ou, Mrs. Dongling Zhu, Dr. Fang Lin, and Dr. Xiaoting Liang and helpful discussions with Prof. Yuzhen Zhang, Prof. Aijun Sun, and Prof. Kai Hu.
ABBREVIATIONS
AL, apical lateral; Anti-146b, miR-146b-5p inhibition group; Anti-Ctrl, antagomir negative control; Ant, anterior; Ant-sept, anterior septum; AS, apical septal; BS, basal septum; BL, basal lateral; CCK-8, cell counting kit-8; CEACAM1, carcinoembryonic antigen related cell adhesion molecule 1; CMVEC, primary cardiac microvascular endothelial cell; CTO, chronic total occlusion; DMEM, Dulbecco’s modified Eagle’s medium; EdU, 5-ethynyl-2’-deoxyuridine; EF, ejection fraction; FMT, fibroblast-to-myofibroblast transition; FS, fractional shortening; HE, hematoxylin and eosin; HW/TL, heart weight/tibia length; IF, immunofluorescence; IL-1β, interleukin-1β; IL-6, interleukin-6; Inf, inferior; I/R, ischemia/reperfusion; IRAK1, interleukin 1 receptor associated kinase 1; Lat, lateral; MCP-1, monocyte chemotactic protein 1; LCX, left circumflex artery; LV, left ventricle; LVEDV, left ventricular end-diastolic volume; LVESV, left ventricular end-systolic volume; LVIDd, left ventricular internal diastolic diameter; LVIDs, left ventricular internal systolic diameter; STE, speckle tracking echocardiography; LVPWd, left ventricular posterior wall diastolic thickness; LVPWs, left ventricular posterior wall systolic thickness; Luc, luciferase; MS, mid septum; ML, mid lateral; Post, posterior; Sept, septum; TNF-α, tumor necrosis factor-α; TUNEL, terminal deoxynucleotidyl transferase dUTP nick end labeling;
COMPLIANCE WITH ETHICS GUIDELINES
Yiteng Liao, Hao Li, Hao Cao, Yun Dong, Lei Gao, Zhongming Liu, Junbo Ge, and Hongming Zhu declare that they have no conflict of interest. All procedures followed were in accordance with the ethical standards of the responsible committee on human experimentation (institutional and national) and with the Helsinki Declaration of 1975, as revised in 2000 (5). Informed consent was obtained from all patients for being included in the study. All institutional and national guidelines for the care and use of laboratory animals were followed.
Author information
Authors and Affiliations
Corresponding authors
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Liao, Y., Li, H., Cao, H. et al. Therapeutic silencing miR-146b-5p improves cardiac remodeling in a porcine model of myocardial infarction by modulating the wound reparative phenotype. Protein Cell 12, 194–212 (2021). https://doi.org/10.1007/s13238-020-00750-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13238-020-00750-6