
ABSTRACT
The mitochondria-mediated caspase activation pathway is a major apoptotic pathway characterized by mitochondrial outer membrane permeabilization (MOMP) and subsequent release of cytochrome c into the cytoplasm to activate caspases. MOMP is regulated by the Bcl-2 family of proteins. This pathway plays important roles not only in normal development, maintenance of tissue homeostasis and the regulation of immune system, but also in human diseases such as immune disorders, neurodegeneration and cancer. In the past decades the molecular basis of this pathway and the regulatory mechanism have been comprehensively studied, yet a great deal of new evidence indicates that cytochrome c release from mitochondria does not always lead to irreversible cell death, and that caspase activation can also have non-death functions. Thus, many unsolved questions and new challenges are still remaining. Furthermore, the dysfunction of this pathway involved in cancer development is obvious, and targeting the pathway as a therapeutic strategy has been extensively explored, but the efficacy of the targeted therapies is still under development. In this review we will discuss the mitochondria-mediated apoptosis pathway and its physiological roles and therapeutic implications.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
INTRODUCTION
The term “apoptosis” was originally coined to describe a specific type of cell death characterized by specific cellular morphological changes, including membrane blebbing, cell shrinkage, nuclear fragmentation, chromatin condensation, and chromosomal DNA fragmentation. (Kerr, 2002; Kerr et al., 1972; Taylor et al., 2008). The nature of apoptosis as a process of “programmed” cell death was established at molecular level mainly by two waves of studies; the discovery of the oncogene product Bcl-2 as an inhibitor of apoptosis, by Korsmeyer, Cory, and others (Bakhshi et al., 1985; Hockenbery et al., 1990; Vaux et al., 1988), and the C. elegans genetic studies by Horvitz and colleagues leading to the identification of a pathway controlling development-associated death of a group of cells in the organism (Ellis and Horvitz, 1986; Horvitz, 1999; Horvitz et al., 1994). The prominent role of mitochondria in apoptosis was subsequently unveiled by Xiaodong Wang and colleagues through their discovery of the cytochrome c-mediated caspase activation pathway (Li et al., 1997; Liu et al., 1996; Zou et al., 1997).
THE BCL-2 FAMILY PROTEINS IN MITOCHONDRIAL APOPTOSIS
The first regulatory step for mitochondrial apoptosis is mediated by Bcl-2 family proteins. Bcl-2, also known as B-cell lymphoma 2, was the first member identified as an apoptosis inhibitory protein overexpressed in human follicular B-cell lymphomas due to t(14;18) chromosomal translocation (Bakhshi et al., 1985; Tsujimoto et al., 1985). Subsequently, three major mammalian groups of Bcl-2 family proteins have been identified. The original pro-survival group includes Bcl2, Bcl-xL, Mcl-1, etc.; an opposite functional group also called pro-apoptotic BH123 protein group includes Bax and Bak; and the third group called apoptosis initiator group is made of BH3 domain-only proteins including Bad, Bid, Bim, Puma, and Noxa (Fig. 1). Without apoptotic stress, Bcl-2 and Bcl-xL (pro-survival) form heterodimers with Bax and Bak (pro-apoptotic) to maintain the outer mitochondrial membrane (OMM) integrity and block mitochondrial apoptosis. In the presence of apoptotic stimuli, the expression of pro-apoptotic proteins Bax and/or BH3-only proteins (apoptosis initiator) increased, following which they bind to pro-survival Bcl-2 proteins to release Bax/Bak from inhibition. Free Bax and Bak form oligomers, leading to cytochrome c release from the intermembrane space of mitochondria to the cytoplasm, likely by forming a channel in OMM. The released cytochrome c activates the caspase cascade to induce apoptosis (Hardwick and Soane, 2013) (Fig. 2).

Schemes showing three groups of Bcl2- family proteins. BH: Bcl-homology domain; TM: transmembrane domains

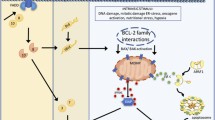
An overview of the mitochondria-mediated caspase activation pathway. Upon apoptotic stimuli such as DNA damage, growth factor deprivation, etc. BAX/BAK form oligomeric complexes to mediate cytochrome c release from the mitochondria to the cytosol. The released cytochrome c forms the apoptosome with Apaf-1 and subsequently activates the initiator caspase, caspase-9, which cleaves and activates effector caspases, caspase-3 and caspase-7, leading to ultimate apoptotic cell death. Other proapoptotic proteins including Smac, Omi, and ARTS also function to repress IAPs to enhance apoptosis. WD40: WD40 repeat domain; CARD: a caspase recruitment domain
To understand the roles of Bcl-2 family protein in vivo, many mouse models have been developed. Loss of Bcl-2 in mouse results in numerous defects, including growth retardation, short life span, polycystic kidney, apoptosis-induced atrophy in thymus and spleen (Kamada et al., 1995). Bcl-2 null mice also show defects in subpopulation of neurons during neonatal period (Michaelidis et al., 1996). Additionally, mice lacking Bcl-xL show early embryonic lethality due to the excess apoptosis of immature neurons in brain, spinal cord, and erythroid cells in the liver, indicating the role of Bcl-xL during neuron and erythrocyte maturation (Motoyama et al., 1999; Motoyama et al., 1995). The data strongly support the inhibitory roles of Bcl-2 and Bcl-xL in apoptosis, though the function may be tissue and developmental stage specific. On the contrary, the Bax/Bak knockout mice fail to promote MOMP and are resistant to various apoptotic stimuli, demonstrating the essential role of BAK and BAX in mitochondria-mediated apoptosis (Lindsten et al., 2000; Wei et al., 2001). Deletion of any single BH3-only gene in mice, on the other hand, does not result in obvious developmental defects (Ren et al., 2010; Villunger et al., 2011), although Bid deletion inhibits Fas-induced apoptosis in certain cell types (Yin et al., 1999). Intriguingly, mice with Bid, Bim, and Puma triple knockout showed embryonic lethality, and a subset of the viable triple null mice displayed similar developmental defects to those of Bax-/-Bak-/- mice with persistent interdigital webs of skin on their feet and imperforate vaginas, indicating these three BH3-only proteins in combination are essential for Bak/Bax activation (Ren et al., 2010; Villunger et al., 2011).
THE APOPTOSOME FORMATION AND CASPASE CASCADE AFTER CYTOCHROME C RELEASE
The second regulatory step of mitochondrial apoptosis is the formation of apoptosome. After MOMP is triggered, mitochondrial proteins such as cytochrome c can be released to the cytoplasm. The released cytochrome c binds to apoptotic protease activating factor-1 (Apaf-1), and activates nucleotide exchange activity of Apaf-1. The ADP/dADP-associated, inactive Apaf-1 becomes active, ATP/dATP-bound Apaf-1, and forms the apoptosome, a wheel-shaped homo-heptameric Apaf-1 complex. Interestingly, although the hydrolysis of dATP by Apaf-1 was initially thought to be essential for apoptosome function (Zou et al., 1997; Zou et al., 1999), more precise analysis demonstrate that dATP-binding but not hydrolysis is required for apoptosome function (Jiang and Wang, 2000). C-terminal WD40 repeats of Apaf-1 have auto-inhibitory activity, and either cytochrome c binding or deletion of these repeats can activate Apaf-1 (Hu et al., 1998; Riedl et al., 2005). Also it is important to have exogenous dATP/ATP present when cytochrome c binds to Apaf-1 to avoid the formation of non-functional aggregates (Kim et al., 2005). When activated Apaf-1 forms apoptosome, it binds and cleaves initiator procaspase-9, and converts it to an active form (Fig. 2).
Although the proteolytic processing of a caspase is usually necessary and sufficient for its activation (Thornberry and Lazebnik, 1998), cleaved caspase-9 needs to be associated with the apoptosome complex to be active (Jiang and Wang, 2000; Rodriguez and Lazebnik, 1999). In addition, even when all the possible cleavage sites of caspase-9 are mutated, the uncleaved caspase-9 can still be activated if it is associated with the functional apoptosome (Acehan et al., 2002; Jiang and Wang, 2000), indicating that proteolytic cleavage of caspase-9 is not required for its activation. Therefore, the holoenzyme formed by the apoptosome complex and caspase-9 is critical to activate downstream effector caspases, such as caspase-3, and caspase-7. On the other hand, although caspase-9 cleavage is not required for its activity, the cleavage significantly enhances the enzymatic activity of apoptosome-associated caspase-9 (Zou et al., 2003). Further, caspase-9 can undergo an autocatalysis process which does not change its own enzymatic activity, but is important for its regulation by inhibitors of apoptosis proteins (IAPs) (Twiddy and Cain, 2007), as we will discuss later.
The importance of these key components in mitochondrial apoptotic pathway has been validated by mouse model studies. Cytochrome c with a K72A mutation is defective in interaction with Apaf-1, but retains its respiration-associated function (Yu et al., 2001). A knock-in mouse with cytochrome c K72A mutation shows strong resistance to DNA damage-induced apoptosis (Hao et al., 2005). Apaf-1 or caspase-9 knockout mice have the similar developmental defects as caspase-3 null mice with central nervous system and lymphocyte homeostasis defects caused by apoptotic deficiency (Cecconi et al., 1998; Hakem et al., 1998; Kuida et al., 1998; Kuida et al., 1996; Woo et al., 1998; Yoshida et al., 1998). Thus, the essential roles of cytochrome c, Apaf-1, caspases in this apoptotic pathway have been confirmed in vivo.
THE INHIBITORS OF APOPTOSIS (IAPS)
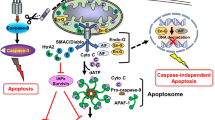
Whereas cytochrome c release from mitochondria leads to caspase activation and triggers apoptosis, the process is also tightly controlled by other endogenous regulators. The inhibitors of apoptosis (IAPs) family of proteins have BIR (baculovirus IAP repeats) domains. The BIR domain was originally discovered in baculovirus proteins (Crook et al., 1993) that can bind to caspases to inhibit their activity (Deveraux et al., 1997; Roy et al., 1997; LaCasse et al., 1998). IAP family proteins in mammals include X-chromosome linked IAP (XIAP), cellular IAP1 and 2 (cIAP1 and cIAP2), neuronal apoptosis inhibitory protein (NAIP), BRUCE (also called Apollon), Survivin, and ML-IAP (Deveraux and Reed, 1999; Dubrez et al., 2013; Harlin et al., 2001; LaCasse et al., 1998; Vucic et al., 2000). Similar to insect IAPs, mammalian IAPs can bind to caspase-3, 7, and 9 to inhibit apoptosis (Chai et al., 2001; Huang et al., 2001) (Fig. 3). Intriguingly, different IAP proteins may interact with a variety of pro-apoptotic proteins in tissue specific manner to inhibit apoptosis induced by diverse signals.

The structure of IAP family proteins. The IAP family protein has at least one baculovirus inhibitor of apoptosis protein repeat (BIR) domain. Several IAPs also contain a RING-zinc finger domain at the carboxy terminus with autoubiquitination and degradation activity. c-IAP1 and c-IAP2 have a caspase recruitment domain (CARD) between the BIR domains and the RING domain. BRUCE contains an ubiquitin-conjugation domain (UBC)
The relevance of IAP family proteins in vivo has been demonstrated by many mouse model studies. Survivin is essential in suppressing apoptosis during mouse development, Survivin null mice are lethal at early embryonic stage (Uren et al., 2000). Tissue specific deletion of Survivin in thymocytes causes mitotic defects and cell death (Okada et al., 2004), clearly indicating that the pro-survival role of Survivin in vivo. Similarly, Bruce/Apollon deletion in mouse causes activation of caspases and apoptosis in the placenta and yolk sac, leading to embryonic lethality. Bruce/Apollon-deficient MEFs are also sensitive to apoptosis (Hao et al., 2004; Ren et al., 2005). However, some IAP family proteins show functional redundancy with other IAP family members in vivo. Mice with XIAP deletion are normal and have no detectable defect in apoptosis with a compensating up-regulation of c-IAP1 and c-IAP2 (Harlin et al., 2001), while mice with deletion of cIAP1 in combination with cIAP2 or XIAP show embryonic lethality due to cardiovascular defects (Moulin et al., 2012). Although these in vivo studies have demonstrated important roles of IAP proteins in development, whether they exert these functions by directly inhibiting caspase activity, particularly, mitochondria-mediated caspase activation, is not defined.
IAP ANTAGONISTS AND THE INTERACTION WITH IAPS
Intriguingly, there is another family of proteins that functions to antagonize the anti-apoptotic activity of IAP proteins. This group of proteins was originally discovered in Drosophila by genetic screens. Pro-apoptotic genes Reaper, Hid, and Grim (RHG genes) were identified as suppressors of Drosophila IAP1 (dIAP1) (Chen et al., 1996; Goyal et al., 2000; Grether et al., 1995; Vucic et al., 1997; Vucic et al., 1998; Wang et al., 1999; White et al., 1996). The RHG proteins can compete with caspases to interact with the BIR domain of dIAP1 (Goyal et al., 2000). There are no obvious RHG homologous sequences in mammals. The mammalian RHG counterpart proteins were independently purified based on the apoptotic activity from two groups. Smac (second mitochondrial activator of caspases) was identified as a mitochondria-derived caspase activator in addition to cytochrome c (Du et al., 2000), and DIABLO was found by XIAP affinity purification (Verhagen et al., 2000). Interestingly, Smac and DIABLO turned out to be the same mitochondrial protein. The N-terminal AVPI motif of Smac/DIABLO specifically interacts with a groove region of the BIR3 domain of XIAP (Liu et al., 2000; Wu et al., 2000), which is sufficient to antagonize the inhibitory activity of BIR3 domain towards caspase-9 (Chai et al., 2000). Subsequently, other IAP antagonists were also identified from mitochondria in mammalian cells. For example, Omi/HtrA2 binds to XIAP, thereby antagonizing caspase-XIAP interaction. Interestingly, Omi/HtrA2 also degrades IAPs through its serine protease activity (Hegde et al., 2002; Martins et al., 2002; Suzuki et al., 2001; van Loo et al., 2002; Verhagen et al., 2002; Yang et al., 2003). ARTS/Sept4 is a septin-like IAP antagonist,which has a unique mechanism to regulate IAPs (Gottfried et al., 2004; Larisch et al., 2000). Unlike Smac and Omi localizing in mitochondria, ARTS is localized on the surface of the mitochondrial outer membrane, allowing it to interact with IAPs independent of MOMP (Edison et al., 2012).
Smac also suppresses the inhibitory activity of XIAP toward caspase-3 by cooperatively interacting with the BIR3 and BIR2 domains of XIAP. Thus, although the multiple BIR-domains of XIAP confer its concurrent inhibitory function to caspase-9 and caspase-3, it also makes the protein highly susceptible to inhibition by Smac (Gao et al., 2007). In addition to the BIR domains, most IAP proteins also have a RING domain with E3 ubiquitin ligase activity, which can cause ubiquitin-mediated degradation of active caspases and SMAC/Diablo (MacFarlane et al., 2002), indicating the RING domain of IAPs is also important for their anti-apoptotic function. Conversely, the serine protease activity of Omi/HtrA2 can also inactivate cIAPs and XIAP by proteolytic cleavage (Yang et al., 2003). Smac/Diablo can promote auto-ubiquination and degradation of cIAPs (Yang and Du, 2004). Thus IAPs and their antagonists have multiple ways in vivo to tightly regulate the mitochondrial apoptosis pathway (Figs. 2 and 3).
The function of these IAP antagonists may be redundant or tissue-specific in vivo as indicated by mouse models. Smac-deficient mice were viable and normal. Cultured Smac-null cells show normal response to all apoptotic signals, suggesting other IAP antagonist molecules can compensate the loss of Smac (Okada et al., 2002). HtrA2/Omi may work in a tissue specific manner or possess apoptosis-independent functions, since mice lacking HtrA2/Omi only show a neurodegenerative disorder similar to a Parkinson phenotype due to the loss of neurons in the striatum (Martins et al., 2004). Arts/Sept4-null mice show increased numbers of hematopoietic stem and progenitor cells, elevated XIAP protein, increased resistance to cell death, and accelerated tumor development in an Eμ-Myc background. These phenotypes are partially rescued by the inactivation of XIAP (Garcia-Fernandez et al., 2010). Thus, the apoptotic role of ARTS/Sept4 is specific to certain cell lineages and involved in cancer development.
CROSSTALK OF THE MITOCHONDRIAL PATHWAY WITH THE DEATH RECEPTOR-MEDIATED APOPTOSIS AND NECROSIS
In addition to the mitochondrial pathway, mammalian cells possess the death receptor-mediated apoptotic pathway that is triggered by the tumor necrosis factors (TNF family). The TNF family factors include Fas ligand, TNF-alpha, Apo3L, Apo2L, and TRAIL (TNF-related apoptosis inducing ligand) that can activate their corresponding receptors FasR, TNFR1, DR3, and DR4/DR5 (Ashkenazi et al., 2008; Tait and Green, 2010a). Upon receptor activation, the adaptor molecules such as FAS-associated death domain protein (FADD) are recruited to associate and activate caspase-8 or caspase-10, which leads to the cleavage and activation of caspase-3 and caspase-7. There is crosstalk between mitochondrial and death receptor pathways. Caspase-8/10 can activate mitochondrial apoptosis initiator protein BID, thus forming an amplification loop to enhance the mitochondrial pathway (Li et al., 1998; Luo et al., 1998). Conversely, Bcl-2 overexpression can completely block apoptosis induced by TNF ligands in various cell types known as Type-II cells (Jiang and Wang, 2004; Scaffidi et al., 1998), suggesting the mitochondrial amplification loop is required for sufficient activation of effector caspases by the death receptor pathway. This is further supported by the observation that Smac and Omi are released to antagonize IAPs by caspase-8-activated BID (Jost et al., 2009; Sun et al., 2002). Additionally, Smac/DIABLO overexpression can sensitize cells to TRAIL and overcome TRAIL resistance in malignant glioma xenografts model (Fulda et al., 2002). Small molecules mimicking Smac can sensitize various cell types to both TRAIL- and TNFα-induced apoptosis (Li et al., 2004). Further, in some Type-II cells, cellular apoptosis susceptibility protein (CAS) can be upregulated by death receptor ligands to stimulate of Apaf-1 (Kim et al., 2008). Therefore, the death receptor pathway can enhance the mitochondria-mediated pathway through multiple mechanisms.
While the functional interplay between the mitochondrial pathway and death receptor-mediated apoptosis is well established, recent evidence suggests that the mitochondrial pathway also communicates with death receptor-induced programmed necrosis (also called necroptosis). The typical morphologies of necrosis are the formation of intracellular vacuoles, organelle swelling, and plasma membrane rupture (Chan, 2012). Although necrosis is originally thought to be passive, it has been unambiguously demonstrated that at least at certain contexts, necrosis can be programmed. For example, death receptor-mediated necrosis requires a kinase cascade, including receptor interacting protein (RIP) kinases RIP1 and RIP3, and the effector protein MLKL (Cho et al., 2009; He et al., 2009; Sun et al., 2012; Zhang et al., 2009). Death receptor-mediated necrosis plays an important role during development and maintenance of adaptive immune response (Han et al., 2011; Li et al., 2012; Linkermann and Green, 2014), and there is intimate crosstalk between this pathway and death receptor-mediated apoptosis, an alternative outcome of the death receptor signaling. For example, caspase-8 activity inhibits RIP3-dependent necrosis (Oberst et al., 2011) and RIP3 in turn suppresses death receptor-mediated apoptosis (Newton et al., 2014). Intriguingly, the mitochondrial apoptotic pathway also shares some regulatory components with necrosis. For example, necrosis caused by hepatic and cerebral ischemia/reperfusion is reduced by inhibition of Bax, and the effect is even stronger than that caused by inhibition of initial apoptotic signal, suggesting Bax plays an important role to promote necrotic cell death under this context (Ben-Ari et al., 2007; Hetz et al., 2005). In addition, Bmf, a pro-apoptotic Bcl-2 protein, is another example of mitochondrial pathway regulator that has been implicated in TNFα-induced necrosis (Hitomi et al., 2008).
CELL FATE DETERMINATION AND NON-CANONICAL FUNCTIONS OF THE MITOCHONDRIAL PATHWAY
It was originally believed that once MOMP is triggered, cells are doomed to die even when downstream caspase activation is completely inhibited (Cheng et al., 2001; Goldstein et al., 2005; Goldstein et al., 2000). However, new evidence shows that cells can survive with partial MOMP and induction of modest cytochrome c release. As mentioned previously, cells have developed multiple mechanisms to regulate caspase activation downstream of cytochrome c release, which strongly suggests that apoptosis can still be avoided even after cytochrome c release. For example, Apaf-1 or caspase-9 knockout mice show the resistance to cell death in the developing neuronal cells (Cecconi et al., 1998; Hakem et al., 1998; Kuida et al., 1998; Yoshida et al., 1998). As both caspase-9 and Apaf-1 function downstream of cytochrome c release, these studies demonstrate that deficiencies downstream cytochrome c release can also block cell death, and thus cytochrome c release is not always the “point of no-return” of mitochondrial apoptosis. Also, when cleaved BID induces modest cytochrome c release, if downstream caspase activation is inhibited, the same cells can fully recover and proliferate (Tait et al. 2010b).
Additionally, cytochrome c release may have non-apoptotic functions. For example, cytochrome c-mediated caspase activation in hippocampal neurons does not lead to apoptosis, yet it is required for brain development and function (Li et al., 2010), indicating that cytochrome c release has non canonical functions at least in neurons. Furthermore, caspase activation is also involved in many biological processes, including sperm and red blood cell differentiation (Kuranaga and Miura, 2007; Lamkanfi et al., 2007; Zermati et al., 2001), and axonal pruning (Nikolaev et al., 2009). Interestingly, caspase-3 deficient mice have increased B cells with enhanced proliferation and hyperproliferation under mitogen treatment (Woo et al., 2003), indicating that caspase-3 can also be involved in cell cycle arrest. In these caspase-dependent events, caspase activity does not result in cell death, but is involved in cellular component clearance and loss of cell mass. The mechanisms underlying how cells determine if cytochrome c-mediated caspase activation should lead to apoptotic cell death or a specific non-death biological function remain unclear.
Besides non-canonical function of caspases, other members of the mitochondrial pathway are also involved in non-death processes in cells. Some of the Bcl-2 family proteins regulate calcium homeostasis, glucose metabolism, and mitochondrial dynamics (Chen et al., 2004; Danial et al., 2010; Danial et al., 2003; Popgeorgiev et al., 2011; Rolland and Conradt, 2010). Apaf-1 is involved in DNA damage induced cell cycle arrest independent of caspase activation (Zermati et al., 2007). The members of the IAP family, Survivin, is involved in kinetochore function (Skoufias et al., 2000; Speliotes et al., 2000), while cIAP1 and cIAP2 are critical regulators of the NF-κB signaling (Beug et al., 2012). Human NAIP regulates the host response to L. pneumophila infection and inhibits apoptosis or promotes pyroptosis in response to specific cellular signals (Katagiri et al., 2012). As proteins of mitochondrial pathway are important for many developmental and cellular events independent of cell death, it is important to determine whether the phenotypes caused by alteration of these proteins are related to mitochondria-mediated apoptosis or their non-canonical functions.
THE ROLE OF MITOCHONDRIAL APOPTOSIS PATHWAY IN CANCER AND CANCER TREATMENT
As we discussed in previous sections, apoptosis is essential for multiple physiological processes. Because aberrant apoptotic cell death is one of the hallmarks of tumorigenesis and tumor progression, cancer cells develop various mechanisms to deregulate the mitochondrial pathway, which leads to apoptotic resistance and survival advantage.
Many components of the mitochondrial apoptosis pathway are deregulated in cancer cells. The elevated expression of pro-survival Bcl-2 gene has been identified in many different cancers, including melanoma, breast, prostate, chronic lymphocytic leukemia, and lung cancer. The high expression of Bcl-2 imparts therapeutic resistance of these cancer cells. Tremendous effort has been spent on developing drugs to target the Bcl-2 pro-survival family members. The first clinical trial agent that targets Bcl-2 is oblimersen sodium (a Bcl-2 antisense oligonucleotide compound). This oligonucleotide specifically binds to human bcl-2 mRNA, resulting in its degradation (Herbst and Frankel, 2004) (Fig. 4).

The therapeutic agents developed to target the mitochondrial apoptotic pathway. Oblimersen sodium is a Bcl-2 antisense oligonucleotide compound. BH3 mimetic compounds include ABT- 737, ABT-263, and JY-1-106. Nutlin and MI-219 block Mdm2 and p53 interaction to activate p53 transcription activity to induce the expression of Puma and Bax. Smac mimetics and the antisense oligonucleotide AEG35156 are inhibitors of XIAP. 4-Pyridineethanol (PETCM), gambonic acid, and the gambonic acid derivative MX-206 can activate caspases-3
Another strategy to target the Bcl-2 family proteins (Bcl-2, Bcl-w, Bcl-xL, MCL-1) is to develop potent BH3 mimetic compounds. These BH3 mimetic compounds bind the hydrophobic groove of anti-apoptotic Bcl-2 proteins in place of BH3-only proteins, allowing Bax and other pro-apoptotic proteins to induce MOMP and apoptotic death. ABT-737 and the orally form ABT-263 developed by Abbott are successful examples. ABT-263 induces tumor regression in the xenograft models of small cell lung cancer and acute lymphoblastic leukemia (Ackler et al., 2008; Tse et al., 2008). More recently, another BH3 mimetic compound JY-1-106 is demonstrated to induce apoptosis in lung cancer, colon cancer, and mesothelioma (Cao et al., 2013).
On the other hand, the pro-apoptotic Bax and BH3-only proteins Puma, Noxa are the transcriptional targets of p53 tumor suppressor. Since it is well known that one of the mechanisms for p53 to suppress tumorigenesis is mediated by its apoptosis function, activation of p53 pathway can be an appealing therapeutic strategy to treat cancer. The most common mechanism to inactivate p53 function in human tumors is missense mutations; several compounds have been developed to restore activity of mutant p53. A synthetic 22-mer peptide corresponding to the carboxy-terminal amino acid residues 361–382 of p53 was the first compound identified to restore mutant p53 activity in tumor cells thereby inducing apoptosis (Selivanova et al., 1997). PRIMA-1 has been shown to have a similar function (Bykov et al., 2002). More recently, a compound (NSC319726) from the thiosemicarbazone family was shown to specifically restore the activity of p53R175H mutation (Yu et al., 2012). However, all these compounds still need to be tested in patients for efficacy. Additionally, other mechanisms, such as overexpression of p53 negative regulators Mdm2 and Mdm4, have been proven to be alternative ways to inactivate wild type p53 function in human tumors (Oliner et al., 1992; Toledo and Wahl, 2007; Wade et al., 2010). In tumors with wild type p53, activation of p53 to induce apoptosis can be achieved by blocking Mdm2 or Mdm4 binding to p53 (Martins et al., 2006; Shchors et al., 2013; Ventura et al., 2007; Wang et al., 2011; Xue et al., 2007). Several chemicals, such as Nutlin and MI-219, have been developed to block the interaction between Mdm2 and p53 (Shangary et al., 2008; Vassilev et al., 2004). Chemicals targeting Mdm4 are still under development.
Increased expression of pro-apoptotic proteins, such as Apaf-1 and Smac are associated with longer survival in cancer patients (Endo et al., 2009; Huang et al., 2010; McIlwain et al., 2013; Provencio et al., 2010; Strater et al., 2010; Zlobec et al., 2007). Conversely, over-expression of IAP proteins are frequently detected in various human cancers and associated with poor prognosis (Barrett et al., 2011; Fulda and Vucic, 2012; Mizutani et al., 2007; Tamm et al., 2000). Thus, blocking IAP proteins in human tumors may improve patient survival. Smac mimetics induce apoptosis through their ability to suppress IAPs by direct inhibition and/or proteasomal degradation of some members of the IAP family. These compounds can target cancer cells with IAPs overexpression, and some of these compounds are currently in clinical trials (Chen and Huerta, 2009; Fulda and Vucic, 2012; Lu et al., 2008). Also an antisense oligonucleotide against XIAP (AEG35156) has been developed to treat patients with pancreatic, breast, non-small cell lung cancer, AML, and lymphoma (Mahadevan et al., 2013; Schimmer et al., 2009).
Additionally, decreased expression of caspase-3 is frequently observed in cancer cells and is associated with chemoresistance. Conversely, activation of caspase-3 often increases cancer cell sensitivity to apoptosis (Devarajan et al., 2002; Guicciardi and Gores, 2013). 4-pyridineethanol (PETCM), gambonic acid, and the gambonic acid derivative MX-206 were identified by high-throughput screens for caspases 3 activation in vitro. Some of these molecules have been reported to induce apoptosis in cancer cell lines (Jiang et al. 2003; Zhang et al. 2004; Fischer and Schulze-Osthoff 2005).
More recently, many studies showed that combination therapies can achieve better therapeutic effect. When ABT-737 is administrated together with paclitaxel, it can enhance the cytotoxic effect of paclitaxel (Lieber et al., 2011). Although the alkylating agent temozolomide (TMZ) is commonly used in treating melanoma, it has low response rate by itself. Combining ABT-737 with TMZ can induce strong apoptosis in multiple human melanoma cell lines and in a mouse xenograft model at much lower concentrations (Reuland et al., 2011). To activate apoptosis in tumors, SMAC mimetic compounds (SMCs) have disappointing effects as single agents in tumors with low expression of death-inducing proteins. However, Smac mimetic BV6, which antagonizes XIAP, cIAP1, and cIAP2, together with the demethylating agent 5-azacytidine or 5-aza-2’-deoxycytidine can induce cell death more efficiently in otherwise resistant AML cells (Steinhart et al., 2013). In conclusion, many drugs are under development to target different components of the mitochondrial apoptotic pathway to treat cancer patients (Fig. 4). Further investigation is needed to improve the efficacy of these leading compounds in humans.
PERSPECTIVE
Tremendous progresses have been made for our understanding of the molecular mechanisms and biological function of mitochondrial apoptotic pathway, leading to potential therapeutic development to target the components of the pathway. Recent work also led to the discovery of novel functional interactions between the mitochondrial pathway and other death pathways, including programmed necrosis. In addition, it becomes clear that the function of the mitochondrial pathway is context-dependent and cell death is not necessarily always its “intended” biological outcome. Therefore, it is important to decode the context-specific regulatory mechanisms of the pathway, and to dissect the function of the pathway in a spatial and temporal specific manner in vivo. Further investigation is needed in order to achieve a more complete understanding of the mechanisms and biology of the mitochondria-mediated caspase activation pathway, and for eventual therapeutic application targeting this important pathway.
Abbreviations
- Apaf-1:
-
apoptotic protease activating factor-1
- BIR:
-
baculovirus IAP repeats
- CAS:
-
cellular apoptosis susceptibility protein
- FADD:
-
FAS-associated death domain protein
- IAPs:
-
inhibitors of apoptosis proteins
- MOMP:
-
mitochondrial outer membrane permeabilization
- NAIP:
-
neuronal apoptosis inhibitory protein
- OMM:
-
outer mitochondrial membrane
- RIP:
-
receptor interacting protein
- XIAP:
-
X-chromosome linked IAP
References
Acehan D, Jiang X, Morgan DG, Heuser JE, Wang X, Akey CW (2002) Three-dimensional structure of the apoptosome: implications for assembly, procaspase-9 binding, and activation. Mol Cell 9:423–432
Ackler S, Xiao Y, Mitten MJ, Foster K, Oleksijew A, Refici M, Schlessinger S, Wang B, Chemburkar SR, Bauch J et al (2008) ABT-263 and rapamycin act cooperatively to kill lymphoma cells in vitro and in vivo. Mol Cancer Ther 7:3265–3274
Ashkenazi A, Holland P, Eckhardt SG (2008) Ligand-based targeting of apoptosis in cancer: the potential of recombinant human apoptosis ligand 2/Tumor necrosis factor-related apoptosis-inducing ligand (rhApo2L/TRAIL). J Clin Oncol 26:3621–3630
Bakhshi A, Jensen JP, Goldman P, Wright JJ, McBride OW, Epstein AL, Korsmeyer SJ (1985) Cloning the chromosomal breakpoint of t(14;18) human lymphomas: clustering around JH on chromosome 14 and near a transcriptional unit on 18. Cell 41:899–906
Barrett RM, Colnaghi R, Wheatley SP (2011) Threonine 48 in the BIR domain of survivin is critical to its mitotic and anti-apoptotic activities and can be phosphorylated by CK2 in vitro. Cell Cycle 10:538–548
Ben-Ari Z, Pappo O, Cheporko Y, Yasovich N, Offen D, Shainberg A, Leshem D, Sulkes J, Vidne BA, Hochhauser E (2007) Bax ablation protects against hepatic ischemia/reperfusion injury in transgenic mice. Liver Transplant 13:1181–1188
Beug ST, Cheung HH, Lacasse EC, Korneluk RG (2012) Modulation of immune signalling by inhibitors of apoptosis. Trends Immunol 33(11):535–545
Bykov VJ, Issaeva N, Shilov A, Hultcrantz M, Pugacheva E, Chumakov P, Bergman J, Wiman KG, Selivanova G (2002) Restoration of the tumor suppressor function to mutant p53 by a low-molecular-weight compound. Nat Med 8:282–288
Cao X, Yap JL, Newell-Rogers MK, Peddaboina C, Jiang W, Papaconstantinou HT, Jupitor D, Rai A, Jung KY, Tubin RP et al (2013) The novel BH3 alpha-helix mimetic JY-1-106 induces apoptosis in a subset of cancer cells (lung cancer, colon cancer and mesothelioma) by disrupting Bcl-xL and Mcl-1 protein-protein interactions with Bak. Mol Cancer 12:42
Cecconi F, Alvarez-Bolado G, Meyer BI, Roth KA, Gruss P (1998) Apaf1 (CED-4 homolog) regulates programmed cell death in mammalian development. Cell 94:727–737
Chai J, Du C, Wu JW, Kyin S, Wang X, Shi Y (2000) Structural and biochemical basis of apoptotic activation by Smac/DIABLO. Nature 406:855–862
Chai J, Shiozaki E, Srinivasula SM, Wu Q, Datta P, Alnemri ES, Shi Y, Dataa P (2001) Structural basis of caspase-7 inhibition by XIAP. Cell 104:769–780
Chan FK (2012) Fueling the flames: mammalian programmed necrosis in inflammatory diseases. Cold Spring Harb Perspect Biol 4(11). doi:10.1101/cshperspect.a008805
Chen DJ, Huerta S (2009) Smac mimetics as new cancer therapeutics. Anticancer Drugs 20:646–658
Chen P, Nordstrom W, Gish B, Abrams JM (1996) Grim, a novel cell death gene in Drosophila. Genes Dev 10:1773–1782
Chen R, Valencia I, Zhong F, McColl KS, Roderick HL, Bootman MD, Berridge MJ, Conway SJ, Holmes AB, Mignery GA et al (2004) Bcl-2 functionally interacts with inositol 1,4,5-trisphosphate receptors to regulate calcium release from the ER in response to inositol 1,4,5-trisphosphate. J cell biol 166:193–203
Cheng EH, Wei MC, Weiler S, Flavell RA, Mak TW, Lindsten T, Korsmeyer SJ (2001) BCL-2, BCL-X(L) sequester BH3 domain-only molecules preventing BAX- and BAK-mediated mitochondrial apoptosis. Mol Cell 8:705–711
Cho YS, Challa S, Moquin D, Genga R, Ray TD, Guildford M, Chan FK (2009) Phosphorylation-driven assembly of the RIP1-RIP3 complex regulates programmed necrosis and virus-induced inflammation. Cell 137:1112–1123
Crook NE, Clem RJ, Miller LK (1993) An apoptosis-inhibiting baculovirus gene with a zinc finger-like motif. J Virol 67:2168–2174
Danial NN, Gramm CF, Scorrano L, Zhang CY, Krauss S, Ranger AM, Datta SR, Greenberg ME, Licklider LJ, Lowell BB et al (2003) BAD and glucokinase reside in a mitochondrial complex that integrates glycolysis and apoptosis. Nature 424:952–956
Danial NN, Gimenez-Cassina A, Tondera D (2010) Homeostatic functions of BCL-2 proteins beyond apoptosis. Adv Exp Med Biol 687:1–32
Devarajan E, Sahin AA, Chen JS, Krishnamurthy RR, Aggarwal N, Brun AM, Sapino A, Zhang F, Sharma D, Yang XH et al (2002) Down-regulation of caspase 3 in breast cancer: a possible mechanism for chemoresistance. Oncogene 21:8843–8851
Deveraux QL, Takahashi R, Salvesen GS, Reed JC (1997) X-linked IAP is a direct inhibitor of cell-death proteases. Nature 388:300–304
Deveraux QL, Reed JC (1999) IAP family proteins–suppressors of apoptosis. Genes Dev 13:239–252
Du C, Fang M, Li Y, Li L, Wang X (2000) Smac, a mitochondrial protein that promotes cytochrome c-dependent caspase activation by eliminating IAP inhibition. Cell 102:33–42
Dubrez L, Berthelet J, Glorian V (2013) IAP proteins as targets for drug development in oncology. Onco Targets Ther 9:1285–1304
Edison N, Zuri D, Maniv I, Bornstein B, Lev T, Gottfried Y, Kemeny S, Garcia-Fernandez M, Kagan J, Larisch S (2012) The IAP-antagonist ARTS initiates caspase activation upstream of cytochrome C and SMAC/Diablo. Cell Death Differ 19:356–368
Ellis HM, Horvitz HR (1986) Genetic control of programmed cell death in the nematode C. elegans. Cell 44:817–829
Endo K, Kohnoe S, Watanabe A, Tashiro H, Sakata H, Morita M, Kakeji Y, Maehara Y (2009) Clinical significance of Smac/DIABLO expression in colorectal cancer. Oncol Rep 21:351–355
Fischer U, Schulze-Osthoff K (2005) New approaches and therapeutics targeting apoptosis in disease. Pharmacol Rev 57:187–215
Fulda S, Vucic D (2012) Targeting IAP proteins for therapeutic intervention in cancer. Nat Rev Drug Discov 11:109–124
Fulda S, Wick W, Weller M, Debatin KM (2002) Smac agonists sensitize for Apo2L/TRAIL- or anticancer drug-induced apoptosis and induce regression of malignant glioma in vivo. Nat Med 8:808–815
Gao Z, Tian Y, Wang J, Yin Q, Wu H, Li YM, Jiang X (2007) A dimeric Smac/diablo peptide directly relieves caspase-3 inhibition by XIAP. Dynamic and cooperative regulation of XIAP by Smac/Diablo. J Biol Chem 282:30718–30727
Garcia-Fernandez M, Kissel H, Brown S, Gorenc T, Schile AJ, Rafii S, Larisch S, Steller H (2010) Sept4/ARTS is required for stem cell apoptosis and tumor suppression. Genes Dev 24:2282–2293
Goldstein JC, Waterhouse NJ, Juin P, Evan GI, Green DR (2000) The coordinate release of cytochrome c during apoptosis is rapid, complete and kinetically invariant. Nat Cell Biol 2:156–162
Goldstein JC, Munoz-Pinedo C, Ricci JE, Adams SR, Kelekar A, Schuler M, Tsien RY, Green DR (2005) Cytochrome c is released in a single step during apoptosis. Cell Death Differ 12:453–462
Gottfried Y, Rotem A, Lotan R, Steller H, Larisch S (2004) The mitochondrial ARTS protein promotes apoptosis through targeting XIAP. EMBO J 23:1627–1635
Goyal L, McCall K, Agapite J, Hartwieg E, Steller H (2000) Induction of apoptosis by Drosophila reaper, hid and grim through inhibition of IAP function. EMBO J 19:589–597
Grether ME, Abrams JM, Agapite J, White K, Steller H (1995) The head involution defective gene of Drosophila melanogaster functions in programmed cell death. Genes Dev 9:1694–1708
Guicciardi ME, Gores GJ (2013) Unshackling caspase-7 for cancer therapy. J Clin Investig 123:3706–3708
Hakem R, Hakem A, Duncan GS, Henderson JT, Woo M, Soengas MS, Elia A, de la Pompa JL, Kagi D, Khoo W et al (1998) Differential requirement for caspase 9 in apoptotic pathways in vivo. Cell 94:339–352
Han J, Zhong CQ, Zhang DW (2011) Programmed necrosis: backup to and competitor with apoptosis in the immune system. Nat Immunol 12:1143–1149
Hao Y, Sekine K, Kawabata A, Nakamura H, Ishioka T, Ohata H, Katayama R, Hashimoto C, Zhang X, Noda T et al (2004) Apollon ubiquitinates SMAC and caspase-9, and has an essential cytoprotection function. Nat Cell Biol 6:849–860
Hao Z, Duncan GS, Chang CC, Elia A, Fang M, Wakeham A, Okada H, Calzascia T, Jang Y, You-Ten A et al (2005) Specific ablation of the apoptotic functions of cytochrome C reveals a differential requirement for cytochrome C and Apaf-1 in apoptosis. Cell 121:579–591
Hardwick JM, Soane L (2013) Multiple functions of BCL-2 family proteins. Cold Spring Harb Perspect Biol 5(2). doi:10.1101/cshperspect.a008722
Harlin H, Reffey SB, Duckett CS, Lindsten T, Thompson CB (2001) Characterization of XIAP-deficient mice. Mol Cell Biol 21:3604–3608
He S, Wang L, Miao L, Wang T, Du F, Zhao L, Wang X (2009) Receptor interacting protein kinase-3 determines cellular necrotic response to TNF-alpha. Cell 137:1100–1111
Hegde R, Srinivasula SM, Zhang Z, Wassell R, Mukattash R, Cilenti L, DuBois G, Lazebnik Y, Zervos AS, Fernandes-Alnemri T et al (2002) Identification of Omi/HtrA2 as a mitochondrial apoptotic serine protease that disrupts inhibitor of apoptosis protein-caspase interaction. J Biol Chem 277:432–438
Herbst RS, Frankel SR (2004) Oblimersen sodium (Genasense bcl-2 antisense oligonucleotide): a rational therapeutic to enhance apoptosis in therapy of lung cancer. Clin Cancer Res 10:4245s–4248s
Hetz C, Vitte PA, Bombrun A, Rostovtseva TK, Montessuit S, Hiver A, Schwarz MK, Church DJ, Korsmeyer SJ, Martinou JC et al (2005) Bax channel inhibitors prevent mitochondrion-mediated apoptosis and protect neurons in a model of global brain ischemia. J Biol Chem 280:42960–42970
Hitomi J, Christofferson DE, Ng A, Yao J, Degterev A, Xavier RJ, Yuan J (2008) Identification of a molecular signaling network that regulates a cellular necrotic cell death pathway. Cell 135:1311–1323
Hockenbery D, Nunez G, Milliman C, Schreiber RD, Korsmeyer SJ (1990) Bcl-2 is an inner mitochondrial membrane protein that blocks programmed cell death. Nature 348:334–336
Horvitz HR (1999) Genetic control of programmed cell death in the nematode Caenorhabditis elegans. Cancer Res 59:1701s–1706s
Horvitz HR, Shaham S, Hengartner MO (1994) The genetics of programmed cell death in the nematode Caenorhabditis elegans. Cold Spring Harb Symp Quant Biol 59:377–385
Hu Y, Ding L, Spencer DM, Nunez G (1998) WD-40 repeat region regulates Apaf-1 self-association and procaspase-9 activation. J Biol Chem 273:33489–33494
Huang Y, Park YC, Rich RL, Segal D, Myszka DG, Wu H (2001) Structural basis of caspase inhibition by XIAP: differential roles of the linker versus the BIR domain. Cell 104:781–790
Huang H, Zhang XF, Zhou HJ, Xue YH, Dong QZ, Ye QH, Qin LX (2010) Expression and prognostic significance of osteopontin and caspase-3 in hepatocellular carcinoma patients after curative resection. Cancer Sci 101:1314–1319
Jiang X, Wang X (2000) Cytochrome c promotes caspase-9 activation by inducing nucleotide binding to Apaf-1. J Biol Chem 275:31199–31203
Jiang X, Kim HE, Shu H, Zhao Y, Zhang H, Kofron J, Donnelly J, Burns D, Ng SC, Rosenberg S, Wang X et al (2003) Distinctive roles of PHAP proteins and prothymosin-alpha in a death regulatory pathway. Science 299:223–226
Jiang X, Wang X (2004) Cytochrome C-mediated apoptosis. Annu Rev Biochem 73:87–106
Jost PJ, Grabow S, Gray D, McKenzie MD, Nachbur U, Huang DC, Bouillet P, Thomas HE, Borner C, Silke J et al (2009) XIAP discriminates between type I and type II FAS-induced apoptosis. Nature 460:1035–1039
Kamada S, Shimono A, Shinto Y, Tsujimura T, Takahashi T, Noda T, Kitamura Y, Kondoh H, Tsujimoto Y (1995) bcl-2 deficiency in mice leads to pleiotropic abnormalities: accelerated lymphoid cell death in thymus and spleen, polycystic kidney, hair hypopigmentation, and distorted small intestine. Cancer Res 55:354–359
Katagiri N, Shobuike T, Chang B, Kukita A, Miyamoto H (2012) The human apoptosis inhibitor NAIP induces pyroptosis in macrophages infected with Legionella pneumophila. Microbes Infect 14:1123–1132
Kerr JF (2002) History of the events leading to the formulation of the apoptosis concept. Toxicology 181–182:471–474
Kerr JF, Wyllie AH, Currie AR (1972) Apoptosis: a basic biological phenomenon with wide-ranging implications in tissue kinetics. Br J Cancer 26:239–257
Kim HE, Du F, Fang M, Wang X (2005) Formation of apoptosome is initiated by cytochrome c-induced dATP hydrolysis and subsequent nucleotide exchange on Apaf-1. Proc Natl Acad Sci USA 102:17545–17550
Kim HE, Jiang X, Du F, Wang X (2008) PHAPI, CAS, and Hsp70 promote apoptosome formation by preventing Apaf-1 aggregation and enhancing nucleotide exchange on Apaf-1. Mol Cell 30:239–247
Kuida K, Zheng TS, Na S, Kuan C, Yang D, Karasuyama H, Rakic P, Flavell RA (1996) Decreased apoptosis in the brain and premature lethality in CPP32-deficient mice. Nature 384:368–372
Kuida K, Haydar TF, Kuan CY, Gu Y, Taya C, Karasuyama H, Su MS, Rakic P, Flavell RA (1998) Reduced apoptosis and cytochrome c-mediated caspase activation in mice lacking caspase 9. Cell 94:325–337
Kuranaga E, Miura M (2007) Nonapoptotic functions of caspases: caspases as regulatory molecules for immunity and cell-fate determination. Trends Cell Biol 17:135–144
LaCasse EC, Baird S, Korneluk RG, MacKenzie AE (1998) The inhibitors of apoptosis (IAPs) and their emerging role in cancer. Oncogene 17:3247–3259
Lamkanfi M, Festjens N, Declercq W, Vanden Berghe T, Vandenabeele P (2007) Caspases in cell survival, proliferation and differentiation. Cell Death Differ 14:44–55
Larisch S, Yi Y, Lotan R, Kerner H, Eimerl S, Tony Parks W, Gottfried Y, Birkey Reffey S, de Caestecker MP, Danielpour D et al (2000) A novel mitochondrial septin-like protein, ARTS, mediates apoptosis dependent on its P-loop motif. Nat Cell Biol 2:915–921
Li P, Nijhawan D, Budihardjo I, Srinivasula SM, Ahmad M, Alnemri ES, Wang X (1997) Cytochrome c and dATP-dependent formation of Apaf-1/caspase-9 complex initiates an apoptotic protease cascade. Cell 91:479–489
Li H, Zhu H, Xu CJ, Yuan J (1998) Cleavage of BID by caspase 8 mediates the mitochondrial damage in the Fas pathway of apoptosis. Cell 94:491–501
Li L, Thomas RM, Suzuki H, De Brabander JK, Wang X, Harran PG (2004) A small molecule Smac mimic potentiates TRAIL- and TNFalpha-mediated cell death. Science 305:1471–1474
Li Z, Jo J, Jia JM, Lo SC, Whitcomb DJ, Jiao S, Cho K, Sheng M (2010) Caspase-3 activation via mitochondria is required for long-term depression and AMPA receptor internalization. Cell 141:859–871
Li J, McQuade T, Siemer AB, Napetschnig J, Moriwaki K, Hsiao YS, Damko E, Moquin D, Walz T, McDermott A et al (2012) The RIP1/RIP3 necrosome forms a functional amyloid signaling complex required for programmed necrosis. Cell 150:339–350
Lieber J, Eicher C, Wenz J, Kirchner B, Warmann SW, Fuchs J, Armeanu-Ebinger S (2011) The BH3 mimetic ABT-737 increases treatment efficiency of paclitaxel against hepatoblastoma. BMC Cancer 11:362
Lindsten T, Ross AJ, King A, Zong WX, Rathmell JC, Shiels HA, Ulrich E, Waymire KG, Mahar P, Frauwirth K et al (2000) The combined functions of proapoptotic Bcl-2 family members bak and bax are essential for normal development of multiple tissues. Mol Cell 6:1389–1399
Linkermann A, Green DR (2014) Necroptosis. N Engl J Med 370:455–465
Liu X, Kim CN, Yang J, Jemmerson R, Wang X (1996) Induction of apoptotic program in cell-free extracts: requirement for dATP and cytochrome c. Cell 86:147–157
Liu Z, Sun C, Olejniczak ET, Meadows RP, Betz SF, Oost T, Herrmann J, Wu JC, Fesik SW (2000) Structural basis for binding of Smac/DIABLO to the XIAP BIR3 domain. Nature 408:1004–1008
Lu J, Bai L, Sun H, Nikolovska-Coleska Z, McEachern D, Qiu S, Miller RS, Yi H, Shangary S, Sun Y et al (2008) SM-164: a novel, bivalent Smac mimetic that induces apoptosis and tumor regression by concurrent removal of the blockade of cIAP-1/2 and XIAP. Cancer Res 68:9384–9393
Luo X, Budihardjo I, Zou H, Slaughter C, Wang X (1998) Bid, a Bcl2 interacting protein, mediates cytochrome c release from mitochondria in response to activation of cell surface death receptors. Cell 94:481–490
MacFarlane M, Merrison W, Bratton SB, Cohen GM (2002) Proteasome-mediated degradation of Smac during apoptosis: XIAP promotes Smac ubiquitination in vitro. J Biol Chem 277:36611–36616
Mahadevan D, Chalasani P, Rensvold D, Kurtin S, Pretzinger C, Jolivet J, Ramanathan RK, Von Hoff DD, Weiss GJ (2013) Phase I trial of AEG35156 an antisense oligonucleotide to XIAP plus gemcitabine in patients with metastatic pancreatic ductal adenocarcinoma. Am J Clin Oncol 36:239–243
Martins LM, Iaccarino I, Tenev T, Gschmeissner S, Totty NF, Lemoine NR, Savopoulos J, Gray CW, Creasy CL, Dingwall C et al (2002) The serine protease Omi/HtrA2 regulates apoptosis by binding XIAP through a reaper-like motif. J Biol Chem 277:439–444
Martins LM, Morrison A, Klupsch K, Fedele V, Moisoi N, Teismann P, Abuin A, Grau E, Geppert M, Livi GP et al (2004) Neuroprotective role of the Reaper-related serine protease HtrA2/Omi revealed by targeted deletion in mice. Mol Cell Biol 24:9848–9862
Martins CP, Brown-Swigart L, Evan GI (2006) Modeling the therapeutic efficacy of p53 restoration in tumors. Cell 127:1323–1334
McIlwain DR, Berger T, Mak TW (2013) Caspase functions in cell death and disease. Cold Spring Harb Perspect Biol 5:a008656
Michaelidis TM, Sendtner M, Cooper JD, Airaksinen MS, Holtmann B, Meyer M, Thoenen H (1996) Inactivation of bcl-2 results in progressive degeneration of motoneurons, sympathetic and sensory neurons during early postnatal development. Neuron 17:75–89
Mizutani Y, Nakanishi H, Li YN, Matsubara H, Yamamoto K, Sato N, Shiraishi T, Nakamura T, Mikami K, Okihara K et al (2007) Overexpression of XIAP expression in renal cell carcinoma predicts a worse prognosis. Int J Oncol 30:919–925
Motoyama N, Wang F, Roth KA, Sawa H, Nakayama K, Negishi I, Senju S, Zhang Q, Fujii S et al (1995) Massive cell death of immature hematopoietic cells and neurons in Bcl-x-deficient mice. Science 267:1506–1510
Motoyama N, Kimura T, Takahashi T, Watanabe T, Nakano T (1999) bcl-x prevents apoptotic cell death of both primitive and definitive erythrocytes at the end of maturation. J Exp Med 189:1691–1698
Moulin M, Anderton H, Voss AK, Thomas T, Wong WW, Bankovacki A, Feltham R, Chau D, Cook WD, Silke J et al (2012) IAPs limit activation of RIP kinases by TNF receptor 1 during development. EMBO J 31:1679–1691
Newton K, Dugger DL, Wickliffe KE, Kapoor N, de Almagro MC, Vucic D, Komuves L, Ferrando RE, French DM, Webster J et al (2014) Activity of protein kinase RIPK3 determines whether cells die by necroptosis or apoptosis. Science 343:1357–1360
Nikolaev A, McLaughlin T, O’Leary DD, Tessier-Lavigne M (2009) APP binds DR6 to trigger axon pruning and neuron death via distinct caspases. Nature 457:981–989
Oberst A, Dillon CP, Weinlich R, McCormick LL, Fitzgerald P, Pop C, Hakem R, Salvesen GS, Green DR (2011) Catalytic activity of the caspase-8-FLIP(L) complex inhibits RIPK3-dependent necrosis. Nature 471:363–367
Okada H, Suh WK, Jin J, Woo M, Du C, Elia A, Duncan GS, Wakeham A, Itie A, Lowe SW et al (2002) Generation and characterization of Smac/DIABLO-deficient mice. Mol Cell Biol 22:3509–3517
Okada H, Bakal C, Shahinian A, Elia A, Wakeham A, Suh WK, Duncan GS, Ciofani M, Rottapel R, Zuniga-Pflucker JC et al (2004) Survivin loss in thymocytes triggers p53-mediated growth arrest and p53-independent cell death. J Exp Med 199:399–410
Oliner JD, Kinzler KW, Meltzer PS, George DL, Vogelstein B (1992) Amplification of a gene encoding a p53-associated protein in human sarcomas.[see comment]. Nature 358:80–83
Popgeorgiev N, Bonneau B, Ferri KF, Prudent J, Thibaut J, Gillet G (2011) The apoptotic regulator Nrz controls cytoskeletal dynamics via the regulation of Ca2+ trafficking in the zebrafish blastula. Dev Cell 20:663–676
Provencio M, Martin P, Garcia V, Candia A, Sanchez AC, Bellas C (2010) Caspase 3a: new prognostic marker for diffuse large B-cell lymphoma in the rituximab era. Leuk Lymphoma 51:2021–2030
Ren J, Shi M, Liu R, Yang QH, Johnson T, Skarnes WC, Du C (2005) The Birc6 (Bruce) gene regulates p53 and the mitochondrial pathway of apoptosis and is essential for mouse embryonic development. Proc Natl Acad Sci USA 102:565–570
Ren D, Tu HC, Kim H, Wang GX, Bean GR, Takeuchi O, Jeffers JR, Zambetti GP, Hsieh JJ, Cheng EH (2010) BID, BIM, and PUMA are essential for activation of the BAX- and BAK-dependent cell death program. Science 330:1390–1393
Reuland SN, Goldstein NB, Partyka KA, Cooper DA, Fujita M, Norris DA, Shellman YG (2011) The combination of BH3-mimetic ABT-737 with the alkylating agent temozolomide induces strong synergistic killing of melanoma cells independent of p53. PLoS One 6:e24294
Riedl SJ, Li W, Chao Y, Schwarzenbacher R, Shi Y (2005) Structure of the apoptotic protease-activating factor 1 bound to ADP. Nature 434:926–933
Rodriguez J, Lazebnik Y (1999) Caspase-9 and APAF-1 form an active holoenzyme. Genes Dev 13:3179–3184
Rolland SG, Conradt B (2010) New role of the BCL2 family of proteins in the regulation of mitochondrial dynamics. Curr Opin Cell Biol 22:852–858
Roy N, Deveraux QL, Takahashi R, Salvesen GS, Reed JC (1997) The c-IAP-1 and c-IAP-2 proteins are direct inhibitors of specific caspases. Embo J 16:6914–6925
Scaffidi C, Fulda S, Srinivasan A, Friesen C, Li F, Tomaselli KJ, Debatin KM, Krammer PH, Peter ME (1998) Two CD95 (APO-1/Fas) signaling pathways. EMBO J 17:1675–1687
Schimmer AD, Estey EH, Borthakur G, Carter BZ, Schiller GJ, Tallman MS, Altman JK, Karp JE, Kassis J, Hedley DW et al (2009) Phase I/II trial of AEG35156 X-linked inhibitor of apoptosis protein antisense oligonucleotide combined with idarubicin and cytarabine in patients with relapsed or primary refractory acute myeloid leukemia. J Clin Oncol 27:4741–4746
Selivanova G, Iotsova V, Okan I, Fritsche M, Strom M, Groner B, Grafstrom RC, Wiman KG (1997) Restoration of the growth suppression function of mutant p53 by a synthetic peptide derived from the p53 C-terminal domain. Nat Med 3:632–638
Shangary S, Qin D, McEachern D, Liu M, Miller RS, Qiu S, Nikolovska-Coleska Z, Ding K, Wang G, Chen J et al (2008) Temporal activation of p53 by a specific MDM2 inhibitor is selectively toxic to tumors and leads to complete tumor growth inhibition. Proc Natl Acad Sci USA 105:3933–3938
Shchors K, Persson AI, Rostker F, Tihan T, Lyubynska N, Li N, Swigart LB, Berger MS, Hanahan D, Weiss WA et al (2013) Using a preclinical mouse model of high-grade astrocytoma to optimize p53 restoration therapy. Proc Natl Acad Sci USA 110:E1480–E1489
Skoufias DA, Mollinari C, Lacroix FB, Margolis RL (2000) Human survivin is a kinetochore-associated passenger protein. J Cell Biol 151:1575–1582
Speliotes EK, Uren A, Vaux D, Horvitz HR (2000) The survivin-like C. elegans BIR-1 protein acts with the Aurora-like kinase AIR-2 to affect chromosomes and the spindle midzone. Mol Cell 6:211–223
Steinhart L, Belz K, Fulda S (2013) Smac mimetic and demethylating agents synergistically trigger cell death in acute myeloid leukemia cells and overcome apoptosis resistance by inducing necroptosis. Cell Death Dis 4:e802
Strater J, Herter I, Merkel G, Hinz U, Weitz J, Moller P (2010) Expression and prognostic significance of APAF-1, caspase-8 and caspase-9 in stage II/III colon carcinoma: caspase-8 and caspase-9 is associated with poor prognosis. Int J Cancer 127:873–880
Sun XM, Bratton SB, Butterworth M, MacFarlane M, Cohen GM (2002) Bcl-2 and Bcl-xL inhibit CD95-mediated apoptosis by preventing mitochondrial release of Smac/DIABLO and subsequent inactivation of X-linked inhibitor-of-apoptosis protein. J Biol Chem 277:11345–11351
Sun L, Wang H, Wang Z, He S, Chen S, Liao D, Wang L, Yan J, Liu W, Lei X et al (2012) Mixed lineage kinase domain-like protein mediates necrosis signaling downstream of RIP3 kinase. Cell 148:213–227
Suzuki Y, Imai Y, Nakayama H, Takahashi K, Takio K, Takahashi R (2001) A serine protease, HtrA2, is released from the mitochondria and interacts with XIAP, inducing cell death. Mol Cell 8:613–621
Tait SW, Green DR (2010) Mitochondria and cell death: outer membrane permeabilization and beyond. Nat Rev Mol Cell Biol 11:621–632
Tait SW, Parsons MJ, Llambi F, Bouchier-Hayes L, Connell S, Munoz-Pinedo C, Green DR (2010) Resistance to caspase-independent cell death requires persistence of intact mitochondria. Dev Cell 18:802–813
Tamm I, Kornblau SM, Segall H, Krajewski S, Welsh K, Kitada S, Scudiero DA, Tudor G, Qui YH, Monks A et al (2000) Expression and prognostic significance of IAP-family genes in human cancers and myeloid leukemias. Clin Cancer Res 6:1796–1803
Taylor RC, Cullen SP, Martin SJ (2008) Apoptosis: controlled demolition at the cellular level. Nat Rev Mol Cell Biol 9:231–241
Thornberry NA, Lazebnik Y (1998) Caspases: enemies within. Science 281:1312–1316
Toledo F, Wahl GM (2007) MDM2 and MDM4: p53 regulators as targets in anticancer therapy. Int J Biochem Cell Biol 39:1476–1482
Tse C, Shoemaker AR, Adickes J, Anderson MG, Chen J, Jin S, Johnson EF, Marsh KC, Mitten MJ, Nimmer P et al (2008) ABT-263: a potent and orally bioavailable Bcl-2 family inhibitor. Cancer Res 68:3421–3428
Tsujimoto Y, Cossman J, Jaffe E, Croce CM (1985) Involvement of the bcl-2 gene in human follicular lymphoma. Science 228:1440–1443
Twiddy D, Cain K (2007) Caspase-9 cleavage, do you need it? Biochem J 405:e1–e2
Uren AG, Wong L, Pakusch M, Fowler KJ, Burrows FJ, Vaux DL, Choo KH (2000) Survivin and the inner centromere protein INCENP show similar cell-cycle localization and gene knockout phenotype. Curr Biol 10:1319–1328
van Loo G, van Gurp M, Depuydt B, Srinivasula SM, Rodriguez I, Alnemri ES, Gevaert K, Vandekerckhove J, Declercq W, Vandenabeele P (2002) The serine protease Omi/HtrA2 is released from mitochondria during apoptosis. Omi interacts with caspase-inhibitor XIAP and induces enhanced caspase activity. Cell Death Differ 9:20–26
Vassilev LT, Vu BT, Graves B, Carvajal D, Podlaski F, Filipovic Z, Kong N, Kammlott U, Lukacs C, Klein C et al (2004) In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science 303:844–848
Vaux DL, Cory S, Adams JM (1988) Bcl-2 gene promotes haemopoietic cell survival and cooperates with c-myc to immortalize pre-B cells. Nature 335:440–442
Ventura A, Kirsch DG, McLaughlin ME, Tuveson DA, Grimm J, Lintault L, Newman J, Reczek EE, Weissleder R, Jacks T (2007) Restoration of p53 function leads to tumour regression in vivo. Nature 445:661–665
Verhagen AM, Ekert PG, Pakusch M, Silke J, Connolly LM, Reid GE, Moritz RL, Simpson RJ, Vaux DL (2000) Identification of DIABLO, a mammalian protein that promotes apoptosis by binding to and antagonizing IAP proteins. Cell 102:43–53
Verhagen AM, Silke J, Ekert PG, Pakusch M, Kaufmann H, Connolly LM, Day CL, Tikoo A, Burke R, Wrobel C et al (2002) HtrA2 promotes cell death through its serine protease activity and its ability to antagonize inhibitor of apoptosis proteins. J Biol Chem 277:445–454
Villunger A, Labi V, Bouillet P, Adams J, Strasser A (2011) Can the analysis of BH3-only protein knockout mice clarify the issue of ‘direct versus indirect’ activation of Bax and Bak? Cell Death Differ 18:1545–1546
Vucic D, Kaiser WJ, Harvey AJ, Miller LK (1997) Inhibition of reaper-induced apoptosis by interaction with inhibitor of apoptosis proteins (IAPs). Proc Natl Acad Sci USA 94:10183–10188
Vucic D, Kaiser WJ, Miller LK (1998) Inhibitor of apoptosis proteins physically interact with and block apoptosis induced by Drosophila proteins HID and GRIM. Mol Cell Biol 18:3300–3309
Vucic D, Stennicke HR, Pisabarro MT, Salvesen GS, Dixit VM (2000) ML-IAP, a novel inhibitor of apoptosis that is preferentially expressed in human melanomas. Curr Biol 10:1359–1366
Wade M, Wang YV, Wahl GM (2010) The p53 orchestra: Mdm2 and Mdmx set the tone. Trends Cell Biol 20:299–309
Wang SL, Hawkins CJ, Yoo SJ, Muller HA, Hay BA (1999) The Drosophila caspase inhibitor DIAP1 is essential for cell survival and is negatively regulated by HID. Cell 98:453–463
Wang Y, Suh YA, Fuller MY, Jackson JG, Xiong S, Terzian T, Quintas-Cardama A, Bankson JA, El-Naggar AK, Lozano G (2011) Restoring expression of wild-type p53 suppresses tumor growth but does not cause tumor regression in mice with a p53 missense mutation. J Clin Investig 121:893–904
Wei MC, Zong WX, Cheng EH, Lindsten T, Panoutsakopoulou V, Ross AJ, Roth KA, MacGregor GR, Thompson CB, Korsmeyer SJ (2001) Proapoptotic BAX and BAK: a requisite gateway to mitochondrial dysfunction and death. Science 292:727–730
White K, Tahaoglu E, Steller H (1996) Cell killing by the Drosophila gene reaper. Science 271:805–807
Woo M, Hakem R, Soengas MS, Duncan GS, Shahinian A, Kagi D, Hakem A, McCurrach M, Khoo W, Kaufman SA et al (1998) Essential contribution of caspase 3/CPP32 to apoptosis and its associated nuclear changes. Genes Dev 12:806–819
Woo M, Hakem R, Furlonger C, Hakem A, Duncan GS, Sasaki T, Bouchard D, Lu L, Wu GE, Paige CJ et al (2003) Caspase-3 regulates cell cycle in B cells: a consequence of substrate specificity. Nat Immunol 4:1016–1022
Wu G, Chai J, Suber TL, Wu JW, Du C, Wang X, Shi Y (2000) Structural basis of IAP recognition by Smac/DIABLO. Nature 408:1008–1012
Xue W, Zender L, Miething C, Dickins RA, Hernando E, Krizhanovsky V, Cordon-Cardo C, Lowe SW (2007) Senescence and tumour clearance is triggered by p53 restoration in murine liver carcinomas. Nature 445:656–660
Yang QH, Du C (2004) Smac/DIABLO selectively reduces the levels of c-IAP1 and c-IAP2 but not that of XIAP and livin in HeLa cells. J Biol Chem 279:16963–16970
Yang QH, Church-Hajduk R, Ren J, Newton ML, Du C (2003) Omi/HtrA2 catalytic cleavage of inhibitor of apoptosis (IAP) irreversibly inactivates IAPs and facilitates caspase activity in apoptosis. Genes Dev 17:1487–1496
Yin XM, Wang K, Gross A, Zhao Y, Zinkel S, Klocke B, Roth KA, Korsmeyer SJ (1999) Bid-deficient mice are resistant to Fas-induced hepatocellular apoptosis. Nature 400:886–891
Yoshida H, Kong YY, Yoshida R, Elia AJ, Hakem A, Hakem R, Penninger JM, Mak TW (1998) Apaf1 is required for mitochondrial pathways of apoptosis and brain development. Cell 94:739–750
Yu T, Wang X, Purring-Koch C, Wei Y, McLendon GL (2001) A mutational epitope for cytochrome C binding to the apoptosis protease activation factor-1. J Biol Chem 276:13034–13038
Yu X, Vazquez A, Levine AJ, Carpizo DR (2012) Allele-specific p53 mutant reactivation. Cancer Cell 21:614–625
Zermati Y, Garrido C, Amsellem S, Fishelson S, Bouscary D, Valensi F, Varet B, Solary E, Hermine O (2001) Caspase activation is required for terminal erythroid differentiation. J Exp Med 193:247–254
Zermati Y, Mouhamad S, Stergiou L, Besse B, Galluzzi L, Boehrer S, Pauleau AL, Rosselli F, D’Amelio M, Amendola R et al (2007) Nonapoptotic role for Apaf-1 in the DNA damage checkpoint. Mol Cell 28:624–637
Zhang HZ, Kasibhatla S, Wang Y, Herich J, Guastella J, Tseng B, Drewe J, Cai SX (2004) Discovery, characterization and SAR of gambogic acid as a potent apoptosis inducer by a HTS assay. Bioorg Med Chem 12:309–317
Zhang DW, Shao J, Lin J, Zhang N, Lu BJ, Lin SC, Dong MQ, Han J (2009) RIP3, an energy metabolism regulator that switches TNF-induced cell death from apoptosis to necrosis. Science 325:332–336
Zlobec I, Steele R, Terracciano L, Jass JR, Lugli A (2007) Selecting immunohistochemical cut-off scores for novel biomarkers of progression and survival in colorectal cancer. J Clin Pathol 60:1112–1116
Zou H, Henzel WJ, Liu X, Lutschg A, Wang X (1997) Apaf-1, a human protein homologous to C. elegans CED-4, participates in cytochrome c-dependent activation of caspase-3. Cell 90:405–413
Zou H, Li Y, Liu X, Wang X (1999) An APAF-1.cytochrome c multimeric complex is a functional apoptosome that activates procaspase-9. J Biol Chem 274:11549–11556
Zou H, Yang R, Hao J, Wang J, Sun C, Fesik SW, Wu JC, Tomaselli KJ, Armstrong RC (2003) Regulation of the Apaf-1/caspase-9 apoptosome by caspase-3 and XIAP. J Biol Chem 278:8091–8098
COMPLIANCE WITH ETHICS GUIDELINES
Shunbin Xiong, Tianyang Mu, Guowen Wang, and Xuejun Jiang declare that they have no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
About this article

Cite this article
Xiong, S., Mu, T., Wang, G. et al. Mitochondria-mediated apoptosis in mammals. Protein Cell 5, 737–749 (2014). https://doi.org/10.1007/s13238-014-0089-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13238-014-0089-1