
Abstract
The development of wound-dressing materials with superior therapeutic effects, controlled bioactive agent release, and optimal mechanical properties is crucial in healthcare. This study introduces innovative hydrogel films designed for the sustained release of the local anesthetic drug Procaine (PC), triggered by pH changes. These films are composed of MIL-101(Fe) particles and pectin polymers. MIL-101(Fe) was chosen for its high surface area, stability in aqueous environments, and biocompatibility, ensuring low toxicity to normal cells. MIL-101(Fe)-embedded-pectin hydrogels were synthesized and characterized using Fourier-transformed infrared (FTIR) spectroscopy, thermal gravimetric analysis (TGA), scanning electron microscopy (SEM), X-ray diffraction (XRD), inductively coupled plasma (ICP) spectrometry, particle size analysis, and goniometry. Rheological analysis assessed the hydrogels’ viscoelastic behavior, and UV-spectrophotometry was utilized for drug loading and release studies. The hydrogels exhibited shear-thinning properties, enhancing shape adaptability and recovery, crucial for wound-dressing applications. Controlled drug release was achieved by maintaining the PC solution’s pH between 8.2 and 9.8 during the drug-loading step. The hydrogel film’s impact on wound healing was evaluated through an in vitro wound healing assay, and cytotoxicity was assessed using a WST-1 cell proliferation assay with human dermal fibroblast cells. Results demonstrated that pectin composites enhance cell viability and support fibroblast cell migration without adverse effects, indicating their potential for effective wound healing applications. This study highlights the potential of MIL-101(Fe)-embedded-pectin hydrogels in advancing wound care technology.
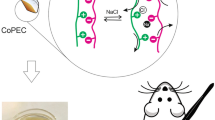
Graphical Abstract
MIL-101(Fe)-embedded pectin film as wound dressing

Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
1 Introduction
Nowadays, natural polymers and hydrogels are well-known materials used in wound-dressing applications [1,2,3,4,5,6,7]. Several commercial products are on the shelves to give patients a comfortable and safe life [8]. On the other hand, the motivation to produce a material with structural integrity, wound-healing properties, and active molecule diffusion with controlled release capabilities is still in progress.
Controlled release maintains optimal therapeutic concentration at the wound site, ensuring consistent healing and avoiding the peaks and troughs associated with conventional dosing. This method reduces wastage and delivers the drug directly to the target site in the precise amounts, leading to improved efficacy and reduced side effects. In addition, controlled drug release also allows for less frequent dosing, improving patient compliance. Furthermore, slow release or sustained exposure maintains therapeutic levels of drugs over an extended period [9,10,11,12].
Pectin is a natural polymer to prepare a wound dressing [13,14,15]. It is an environmentally friendly raw material mainly derived from by-products of the fruit juice and cider industry. The interaction of the carboxyl groups of the low methoxy pectin backbone with Ca+2 ions induces the formation of the ‘’egg box’’ structure, which provides superior properties to transport active molecules [14,15,16]. Pectin and its derivatives do not harm non-toxic, and pectin-based biomaterials exhibit noteworthy properties such as anti-tumor, anti-inflammatory, and antioxidant effects [17].
Pectin hydrogels are particularly well-suited for pH-sensitive drug release due to their high content of hydroxyl (OH) and carboxyl (COOH) groups, which can interact with the surrounding environment. These functional groups enable the hydrogel to respond to pH changes, altering its swelling behavior and drug release profile. In acidic conditions, the carboxyl groups become protonated, causing the hydrogel to shrink and release the drug more slowly. Conversely, in alkaline conditions, the carboxyl groups deprotonate, leading to increased swelling and faster drug release [18]. These hydrogels can be engineered to release small molecules and proteins [15, 19] present bioactive ligands to cells, and degrade at a tunable rate [20]. By adjusting the crosslinker and pectin polymer concentration, pectin hydrogels can be tailored as wound dressings. Thus, controlled release systems using pectin hydrogels can be designed to deliver different therapeutic agents at specific phases of wound healing. On the other hand, some properties of pectin hydrogels need to be improved, such as mechanical strength and drug release performance. Nano/microparticles are the most popular candidates for supporting structural integrity and improving drug delivery [15, 21].
In recent years, using metal–organic frameworks (MOFs) for biomedical applications as drug delivery systems has been challenging due to their considerable pore size, high specific surface area, and long-term stability in water and air environments [22,23,24]. Generally, the purpose of developing MOFs as a drug delivery system is to design carriers that show little toxicity in the body, considering the biocompatibility of metal and bridge ligands [25, 26]. Horcajada et al. showed that MIL-100(Fe) and MIL-101(Fe) were stable in biologic solutions for a long time without any deterioration in drug delivery system performance [25]. Then, MIL-101(Fe) was developed as a biocompatible alternative and became a more convenient drug carrier [27]. Gordon et al. have used flexible microporous MIL-53(Fe) as a matrix for adsorption and in vitro delivery of acetaminophen, progesterone, and stavudine [28]. Wang et al. reported a MOF-based tumor-targeting doxorubicin delivery system improved by a one-pot and organic solvent-free “green” post-synthetic surface modification procedure, starting from the nanoscale MIL-101-N3(Fe) [29]. This system achieved effective drug cell inhibition with early drug release and reduced side effects. Haydar et al. synthesized and used Ca-MOF and Fe-MILs (53, 100, and 101) for flurbiprofen delivery [25]. Moreover, non-toxic iron-based MOFs such as MIL-53, MIL-88A, MIL-89, MIL-100, and MIL-101, were investigated as delivery systems for infection diseases [25], anticancer and antiviral agents against cancer and AIDS [30] and local tuberculosis therapy [31]. MIL-101(Fe) was also modified with polyamines for use as a carrier for the controlled release of naproxen, a nonsteroidal anti-inflammatory drug [32].
MOF and MOF-based polymer nanocomposites are not widely used in wound-healing systems and tissue engineering. So far, only a few studies have been found to investigate the potential of MOFs for tissue engineering and wound-healing applications [27,28,29,30]. A MOF-based hybrid nanocatalyst was investigated as a benign and self-activated cascade reagent for wound healing [27]. Polyacrylonitrile/Fe(III) metal–organic framework fibrous nanocomposites were fabricated for tissue engineering applications [33, 34]. For antibacterial, antithrombotic, and wound-healing applications, the researchers synthesized cobalt and nickel-based MOFs as an alternative way of polymers, zeolites, and functionalized silica nanoparticles to deliver nitric oxide, which is an important biologic signaling molecule [33, 35].
As a fundamental contribution to the literature, this study focuses on preparing MIL-101(Fe) embedded-pectin matrixes for a new dressing and investigating their drug release and wound-dressing performances. We combined the positive effects of pectin and MOFs in formulating a new drug-loaded wound dressing. Our motivation is also to ensure the structural integrity of hydrogels using MIL-101(Fe) particles. Procaine (PC), which is a local anesthetic molecule, was used as a model drug. The preparation of the pectin-MIL-101(Fe) composites and the effect of the MIL-101(Fe) particles on the performances of the drug release and wound-dressing properties of the hydrogels were reported for the first time to the best of our knowledge.
2 Materials and methods
2.1 Materials
Amidated low-methoxyl citrus pectin with a degree of esterification of 31% was provided by Herbstreith & Fox company (Germany). Glycerol (99 atom % 13C purity) and CaCl2.2H2O (> 99% purity) obtained from Sigma-Aldrich were used as plasticizers and crosslinkers. PC (≥ 97%) supplied by Pharmaris Sanowas was chosen as a model drug. Iron (III) chloride hexahydrate (FeCl3.6H2O, 98% Sigma-Aldrich), N,N-dimethylformamide (EMPARTA®) 99.5% Merck, and terephthalic acid 98% (Alfa Aesar) were used for the synthesis of MIL-101 (Fe). All chemicals were used as received without any further purification.
2.2 Synthesis of MIL-101(Fe) nanoparticles
Equimolar amounts of terephthalic acid (0.346 mmol, 0.0575 g) and FeCl3.6H2O (0.346 mmol, 0.0935 g) were dissolved in 15 mL of dimethylformamide (DMF). The solution was placed in an XP1500 microwave vessel and sealed. The reaction mixture was then rapidly heated to 150 °C and held at this temperature for 10 min. After cooling to room temperature, the particles were isolated by centrifuging and were washed with DMF and ethanol. The obtained orange powder was dried and activated overnight at 100 °C, which enables the removal of the trapped or confined solvent in the voids and pores [21].
2.3 Drug loading to MIL-101(Fe) nanoparticles
First, MIL-101 (Fe) particles were dried at 150 °C overnight. Then, 10 mL buffer solution at pH 5.8, 8.2, 9.1, or 9.8 containing 30 mg PC was added to the 30 or 50 mg MIL-101 (Fe) particles. The mixture was first treated on an orbital shaker at 5 °C and 100 rpm for 5 h, then stirred in the ultrasonic bath for 5 min. The drug concentration of the solution was periodically determined using a Lambda 1050 UV/Vis spectrophotometer at 289.9 nm wavelength. The drug-loading content (DLC) of nanoparticles was calculated according to Eq. (1) [36].
2.4 Preparation of drug-loaded hydrogel films
Hydrogel films were prepared using an ionotropic gelation method [15, 37], adding drug-loaded MIL-101(Fe) particles to the pectin matrix before crosslinking. CaCl2 was used as a crosslinker, and glycerol was used as a plasticizer to prepare a pectin hydrogel [15]. First, 600 mg of pectin powder was dissolved in ultra-de-ionized water as 2% w w−1. Then, 300 mg of glycerol solution (5% w w−1) was kindly added to the pectin solution, and this mixture was stirred at 100 rpm for 2 h to obtain a homogenous solution. Drug-loaded MIL-101(Fe) solution was added to the pectin–glycerol mixture, and the obtained mixture was stirred at 100 rpm for 24 h in the dark medium. The mixture was poured onto 10 mL of CaCl2 solution (0.7% w w−1) in a petri plate, and the resulting crosslinked hydrogel was then dried at 25 °C on an orbital shaker. The hydrogels were designated using the abbreviation xM-PCy, where x indicates the amount of nanoparticles per gram hydrogel, and y indicates the pH of the drug solution. To demonstrate the contribution of MIL-101(Fe) particles on the drug release performance of the composites, a series of drug-loaded hydrogels were synthesized without MIL-101 (Fe) particles (PCy-C).
Additionally, two comparison samples were prepared from pectin and MIL-101(Fe) particles without the drug. For this purpose, 30 or 50 mg MIL-101(Fe) particles were dispersed in a 10 mL mixture prepared by mixing 5 mL of ethanol and 5 mL of pure ultra-pure bi-distilled water (30 and 50 M, respectively) and sonicated in an ultrasonic water bath for 10 min. Then, MIL-101(Fe) dispersion was added to the pectin–glycerol solution and stirred at 100 rpm for 24 h in the dark medium. The same procedure was followed to develop a crosslinked hydrogel film with CaCl2 solution. The code of all samples is shown in Table 1. A pectin hydrogel was also formulated without including any drug or MIL-101(Fe) particles, serving as a comparative sample following the identical procedure outlined above.
2.5 Drug release from MIL-101(Fe) particles and hydrogel films
For the drug release from particles, the drug-loaded MOFs were filtered, washed and dried at room temperature. To investigate the drug release profiles, defined amounts of the drug-loaded particles were suspended in a pH 6.4 Tris buffer solution at room temperature.
In vitro, drug release profiles of the hydrogel films were determined in Tris buffer solution at pH 6.4. A drug-loaded dry disk with 1.2 cm diameter was placed in vials containing buffer solution and was mixed at 25 ± 1 °C on an orbital shaker.
A UV spectrophotometer determined the drug amount in the solution at 289.9 nm wavelength. For this purpose, an 850 µL sample was periodically taken from the solution, measured, and immediately poured back from the cuvette into the vial. The drug concentration was determined using a calibration curve. The measurements were made at least in triplicate, and an average was calculated.
2.6 Characterization methods
The samples’ Fourier transform infrared (FTIR) spectra were analyzed in the 500–4000 cm−1 range using the attenuated total reflectance (ATR) technique at room temperature with a Perkin-Elmer Spectrum One FTIR Spectrometer.
Thermogravimetric analysis (TGA) was performed on a Perkin-Elmer Diamond TG/DTA thermogravimetric analyzer under nitrogen flow (100 mL min−1) with a heating rate of 10 °C min−1.
The films were prepared for measurement by cracking in liquid nitrogen, and their morphologic properties were determined using a Jeol (JSM-6390) brand scanning electron microscope (SEM).
The atomic and molecular structure of MIL-101 (Fe) particles was investigated using the X-ray diffraction method (XRD). XRD analysis of the hydrogel films was also performed.
Particle size analysis of MIL-101 (Fe) particles was performed with Malvern Zetasizer Nano S. Wet chemical analysis of the metal cations was carried out using Inductively Coupled Plasma (ICP-OES Optima 2100 DV).
The static contact angle of the water on the surface of the hydrogel films was measured at room temperature with the contact angle measurement device KSV CAM200.
Dynamic rheological measurements of the hydrogels were determined using Anton Paar (Graz, Austria) Physica Rheometer MCR301 with a 25 mm plate-plate geometry at room temperature. Viscotherm VT2 controls the temperature of the sample. All measurements recorded the gap between the probe and samples at 2.7 ± 0.3 mm. First, a dynamic strain sweep measurement was performed at 10 rad s−1 to determine the linear viscoelastic range (LVE). Then, the storage modulus (G′) and the loss modulus (G″) were measured. At the same time, the angular frequency (ω) was varied from 0.1 to 100 rad s−1. Complex viscosity (η*) and damping factor (tan δ) were calculated from G′ and G″ using Eqs. (2) and (3), respectively.
2.7 In vitro characterization of hydrogel films for evaluation as a wound-dressing material
The pectin film containing 30 mg MOF without the drug (30 M-C) was evaluated for in vitro characterization of the hydrogel films. Various standard properties were tested, such as flexibility, average mass per unit area, thickness, fluid handling capacity, dehydration rate, dispersion property, and pH shift capacity [38].
The average mass per unit area (g m−2) and the thickness of hydrogel films (mm) were determined according to BS EN 12127: 1998 [39] and BS EN ISO 9073-2: 1997 standards [40], respectively. The dry-hydrogel films were cut into 5 × 5 cm squares, weighed, and the weight was divided into areas for mass determination. The thickness was measured with an electronic caliper.
Shore A hardness of the hydrogels was determined by a durometer, according to ASTM D2240 [41]. The Erichsen Model 266S cylindrical mandrel bending tester measured the samples’ flexibility with 14 stainless steel mandrels of 2–32 mm diameter [42]. The diameter of the largest cylinder that did not cause cracking in the film was reported.
The fluid handling capacity of the samples was determined by the test methods of BS EN 13726-1: 2002 [43]. First, the films were cut to size 5 × 5 cm, weighed (wD1), and placed in a petri dish. 2.298 g of NaCl and 0.368 g of CaCl2.2H2O were added to 1 L of de-ionized water to prepare test solution A. Test solution A was added to the petri dish at a ratio of 1:40 (w v−1) and heated to 37 ± 1 °C in the incubator for 30 min. The hydrogel films were then removed from the incubator and weighed. The excess solution on the film was suspended from a corner using a tweezer to allow it to drip for 30 s. The film was re-weighed to calculate the mass of the solution absorbed by the sample (WW). Equation (4) was used for calculations.
Before determining the in vitro dehydration rate, the samples were cut to size 5 × 5 cm and dried in an incubator at 37 ± 1 °C for 24 h. Then, they were immersed in de-ionized water for 30 min at 37 ± 1 °C and taken for re-weighing (wW). It was dried for 24 h at 37 ± 1 °C and weighed (wD2). The dehydration rate was calculated according to Eq. (5).
The dispersion characteristics of the samples were examined according to BS EN 13726-2: 2001 [44]. The sample was cut to 5 × 5 cm and placed in a 250 mL conical flask, to which 50 mL of test A solution was added. The vial was gently rotated for 60 s without causing a vortex, and the integrity of the hydrogel was visually examined. The results were expressed as to whether there was distribution by the standards. All measurements were performed at least three times.
To investigate the pH change of the external solution in the presence of the hydrogel film, first, the hydrogel was immersed in de-ionized water at a ratio of 1:100 (w v−1). It was held at room temperature for 3 and 24 h, and then the pH of the de-ionized water was measured using a WTW Portable pH meter [34]. Measurements were performed at least three times, and the mean was reported.
2.8 In vitro cell culture and wound-healing assay
The effects of the samples on cell viability were assessed by WST-1 cell proliferation assay (Roche, Mannheim, Germany) according to the manufacturer’s instructions. In PCS-201-012, human dermal fibroblast cells (ATCC) were plated at 96 well plates at a density of 5 × 103 cells / well and were cultured at 37 °C in a humidified atmosphere containing 5% CO2 for 24 h to bind the cells. Then, the medium was removed and filled with 350 µL of fresh medium. 2 mg of samples were placed in each well. After 24 h of incubation, the medium and samples were removed and replaced with 100 µL of fresh culture medium and 10 µL of WST-1 reagent. The absorbance of formazan crystals was measured at 440 nm using a microplate reader (Molecular Devices, CA, US-111A). The absorbance values of the treated groups were presented as the percentage of absorbance compared to the control group. Three copies of the samples were used in each experiment, and three independent experiments were performed. The results were analyzed with a one-way analysis of variance (ANOVA) followed by the Bonferroni post hoc test using SPSS version 13.0 (SPSS, Chicago, IL, USA).
The effect of hydrogels on wound healing was evaluated by in vitro wound-healing test. PCS-201-012 human dermal fibroblast cells (American Tissue Culture Collection, VA, USA) were cultivated in DMEM (PAN Biotech GmbH, Aidenbach, Germany) supplemented with 10% fetal bovine serum (Biochrom, Berlin, Germany) and penicillin/streptomycin (PAN Biotech GmbH, Aidenbach, Germany).
The cells were seeded in 24-well tissue culture plates. When cells were combined, a monolayer was plated with a micropipette tip. Subsequently, 23 mg hydrogel fragments were placed in the wells in 1500 µL medium and incubated for 48 h. Free cell area images were taken before and after incubation with a microscope (Carl Zeiss, Jena, Germany).
2.9 Statistical analysis
Data analysis is conducted using Minitab16 (Minitab Inc., State College, Pennsylvania, PA, USA). For rheological, drug loading, and release data, a two-way ANOVA is performed with pH and MOF concentration as the independent variables. All ANOVA analyses are followed by Tukey’s test for pairwise and simultaneous comparisons of the independent variables, maintaining a 95% confidence level.
3 Results and discussion
In this study, we chose MIL-101(Fe) particles as a drug carrier system, PC as a model drug, and pectin as a polymer matrix to prepare a wound dressing. We selected MIL-101(Fe), pectin, and PC for specific reasons. MIL-101(Fe) particles have been reported to be biocompatible [27], while pectin functions as a biopolymer and demonstrate biocompatibility [13]. PC is a recognized safe molecule known for its ability to alleviate pain in the vicinity of the 3wound and expedite the healing process [45]. MIL-101(Fe) particles were first synthesized to design a new drug carrier system, and PC molecules were loaded into the particles. Then, the drug-loaded MIL-101(Fe) particles were dispersed into the pectin matrix to prepare a wound dressing. A schematic representation of MIL-101(Fe)-embedded hydrogel films are shown in Fig. 1. Hydrogels’ drug release and wound-dressing performance were investigated after determining their structural and physicochemical characterizations.

Schematic representation of MIL-101(Fe)-embedded hydrogel films
3.1 Synthesis and characterization of MIL-101(Fe) particles
X-ray diffraction analysis carried out the structural characterization of synthesized MIL-101(Fe) powder. The XRD pattern of crystalline MIL-101(Fe) is indicated in Fig. 2a. The sample exhibited two sharp peaks at about 2θ = 10°, which indicates high crystallinity. However, a displacement of peaks is evident compared to the literature [46], which might be due to the use of different analysis conditions. The conversion efficiency of the reaction is about 20%.

a XRD pattern, and b TGA thermogram of MIL-101(Fe)
Figure 2b shows the TGA thermogram of the MIL-101(Fe) crystals, which consists of three steps. The first weight loss (≈ 5 wt%) observed before 150 °C related to the non-bounded solvent molecules within the structure. Then, the bounded solvent species were released up to approximately 400 °C. The third weight loss, associated with the decomposition of the organic ligand, occurred between 400 and 500 °C. The result is in good agreement with the literature [46].
Figure 3 shows the SEM images of the MIL-101(Fe) crystals. It was observed that the MOF samples retained their typical octahedral crystal shape even after the drug loading.

SEM images of; a MIL-101(Fe), and b pectin/pectin composite
Particle size analysis is of critical importance in characterizing MOFs. In this study, we have two significant limitations for MIL-101(Fe) particle sizes; first, MIL-101(Fe) should be in nanosized due to their application in drug delivery system, and second, to control nanosized MIL-101(Fe)’ dispersion in pectin medium and to avoid agglomeration of nanoparticles. The particle size distribution analysis of MIL-101(Fe) reveals a highly monodisperse sample, with the mean particle size centered around 820 nm (Fig. 4). The narrow and sharp peak observed in the distribution indicates low polydispersity, reflecting the uniformity in particle size. The minimal spread of particle sizes around the mean suggests a small standard deviation, corroborating the consistency and homogeneity of the particles. These characteristics are indicative of a well-defined and reproducible particle size distribution within the sample [47].

Particle size distribution of MIL-101(Fe)
3.2 Characterization of hydrogels
The Fourier transform infrared (FTIR) spectra of the hydrogels, pure pectin, MIL-101(Fe) particles, and PC are shown in Fig. 5. characteristic peak for pure pectin is at around 1742 cm−1 coming from esterified carboxyl groups [44]. The peaks at around 1600 cm−1 for –COO− groups and at 1420 cm−1 for C–OH stretching of the carboxylic group are the other characteristic peaks of low-methoxyl pectin [15, 48, 49]. The characteristic peaks of MIL-101(Fe) are observed at 1392 and 1599 cm‐1, corresponding to O–C–O bonds. InAdditionally, the bands relative to C=C and C–H bonds are observed at 1506 and 749 cm‐1, respectively. The stretching and bending vibrational bands in the 600–1600 cm‐1 range correspond to the benzene ring [45]. Due to the C–N stretching vibrations of the aromatic amine in the PC structure, the peaks appeared in the 1360–1250 cm−1 region [15, 50]. We could not observe the specific MIL-101(Fe) and PC peaks in all drug-loaded hydrogels because they overlapped with the pectin peaks.

FTIR spectra of the samples in the range of a 500–4000 cm−1 and b 600–1800 cm−1
SEM images of the MIL-101(Fe) particles, pristine pectin hydrogel and the pectin composites with MIL-101(Fe) are shown in Fig. 3. In the image of the neat pectin hydrogel, ellipse-shaped slits show the egg-box structure of crosslinked-pectin chains. Adding MIL-101(Fe) particles to the pectin matrix (30 M) formed a more porous structure, an advantage for fluid deposition and drug release. As expected, the size of the ellipse-shaped slits was larger in one dimension after adding drug-loaded MIL-101(Fe) particles into the pectin matrix (30 M-PC5.8). It can be concluded from these data that drug-loaded MIL-101(Fe) was successfully dispersed in the pectin matrix.
3.3 In vitro drug loading and release behavior
3.3.1 In vitro drug loading and release from MIL-101(Fe) particles
The study investigated the loading efficiency of PC (PC) into MIL-101(Fe) under varying pH conditions and MOF-to-drug ratios. The results were employed the ANOVA method complemented by Tukey’s test to evaluate results.
At pH 9.1, with MOF:PC ratios of 1:0.6, 1:1, and 1:1.5, the cumulative drug-loading percentages were 55, 56, and 58%, respectively, indicating that higher drug ratios slightly increased the loading capacity (p value = 0.000) (Fig. 6a). This suggests that as more PC is available, MIL-101(Fe) can accommodate more drug molecules, likely due to enhanced interactions between PC and the MOF in an alkaline environment. Conversely, at pH 8.2 with a 1:1 ratio, the loading efficiency dropped to 33%, suggesting that the lower pH might reduce electrostatic attraction or alter the protonation state of PC, hindering its encapsulation within the MOF’s pores (p value = 0.000). Since the PC molecule is at the deprotonated state at pH 9.8, at this pH level with a 1:1 ratio, the loading efficiency was 32%, which suggests that despite the alkaline nature of the environment, other factors such as steric hindrance or rigidity of the deprotonated amine groups may influence the interaction between PC and MIL-101(Fe), resulting in a lower loading efficiency compared to pH 9.1 [51]. For practical understanding, the actual weights of PC loaded per 1 mg of MOF were calculated as follows: 0.33 mg at a 1:1 ratio and pH 8.2, 0.55 mg at a 1:0.6 ratio and pH 9.1, 0.56 mg at a 1:1 ratio and pH 9.1, 0.58 mg at a 1:1.5 ratio and pH 9.1, and 0.57 mg at a 1:1 ratio and pH 9.8. These results demonstrate that increasing the MOF-to-drug ratio from 1:0.6 to 1:1.5 at pH 9.1 enhances the drug loading, although the increase is incremental. This trend indicates a saturation point beyond which the efficiency gain diminishes, suggesting that the MOF’s capacity for drug loading is approaching its limit. These findings highlight the significant impact of pH (p value = 0.000) and drug ratio on the loading efficiency, underscoring the importance of optimizing these parameters for enhanced drug delivery applications (p value = 0.000).

Cumulative drug loading and release; a drug loading to MIL-101(Fe) particles, b drug release from MIL-101(Fe) particles, c drug release from pectin hydrogels with MIL-101(Fe) particles, d drug release from pectin hydrogels without MIL-101(Fe) particles
The release profile of PC from MIL-101(Fe) at pH 6.4, demonstrates a distinct biphasic release pattern (Fig. 6b). In the initial phase, there is a rapid release of approximately 50% of the drug PC within the first hour, attributed to the desorption of drug molecules loosely bound to the MOF surface or located near pore entrances. Following this burst, the release rate slows down, reaching around 80% cumulative release by the 10-h mark and stabilizing close to 85% by 24 h. This sustained release phase is governed by the gradual diffusion of PC from the inner pores of the MIL-101(Fe) framework, controlled by the drug-MOF interactions and the pore structure.
3.3.2 In vitro drug release from pectin-MIL-101(Fe)
PC release performance of the hydrogels with different MIL-101(Fe) concentration (0, 30 and 50 mg/g hydrogel) and varying drug-loading pH (5.8, 8.2, 9.1 and 9.8) (Table 1) is displayed in Fig. 6c and d [14, 15]. According to the obtained data, Tukey’s pairwise comparison reveals that while the inclusion of MIL-101(Fe) particles in the hydrogel significantly influences the release amount (p value < 0.000), there was no significant difference in the release profiles of hydrogels based on the drug-loading medium pH levels (p value > 0.5).
To highlight the contribution of MIL-101(Fe) particles on the drug release profile, all drug release data were compared with those of the comparison samples prepared without MIL-101(Fe) particles (Fig. 6d). The comparison samples (PC5.8-C, PC8.2-C, PC9.1-C, and PC9.8-C) showed the uncontrolled release of the drug. 60–80% of drug release was performed in 7 h. This is strong evidence for the contribution of MIL-101(Fe) to controlled drug release for wound-dressing applications (p value = 0.000). Two hydrogels, 30 M-PC9.1 and 50 M-PC9.1, synthesized to investigate particle amount on drug release behavior, gave similar drug release profiles by following different pathways (Fig. 6c). An increasing in the amount of MIL-101(Fe) particles from 30 mg per gram hydrogel (30 M-PC9.1) to 50 mg per gram hydrogel (50 M-PC9.1) caused an insignificant (p value = 0.922) slight increase in drug release (Fig. 6c). The reason for this behavior and its relationship with the hydrogel structure can be explained by the complex viscosity data discussed in the next section. Overall, the drug release amounts of the hydrogels after 25 h are shown in Table 1.
The positive contribution of the MIL-101(Fe) particles on the drug release performance of the hydrogels can be proven, except for the hydrogel prepared by loading the drug at pH 5.8.
In comparison, the release profile of PC-loaded MIL-101(Fe) dispersed into pectin reveals a markedly slower release. In the initial hour, less than 20% of PC is released, with a cumulative release of around 30% by the 6-h mark, nearly 60% by 10 h, and approximately 70% by 24 h. The presence of pectin creates an additional barrier, significantly slowing the diffusion of PC from the MOF. This barrier effect of pectin modulates the drug release kinetics, resulting in a more controlled and prolonged release profile compared to the MIL-101(Fe) alone.
The comparison highlights that while the PC-loaded MIL-101(Fe) particles provides an initial rapid release followed by sustained release, the incorporation of pectin significantly slows down the release rate, making it more gradual and controlled. This controlled release is particularly beneficial for applications requiring extended drug delivery, as it can maintain therapeutic drug levels over a longer period, potentially enhancing efficacy and reducing the frequency of administration [52,53,54]. The differences in release rate and cumulative percentages underscore the impact of the pectin barrier in modulating the release behavior of the drug. In the presence of MIL-101(Fe) particles, the drug is released for a more extended period, which ensures long-term use of the system due to the cage structure and of the MIL-101(Fe) [55, 56]. Furthermore, Fe ions in the MIL-101(Fe) structure might interact electrostatically with COO- groups on the pectin chains, resulting in an additional crosslinking. This interaction increases the crosslinking density of the pectin hydrogel, which in turn decreases the drug release profile from the pectin chains [15].
On the other hand, as illustrated in Fig. 6a, the hydrogel prepared with drug-loaded MIL-101(Fe) at pH 5.8 (30 M-PC5.8) is not suitable for controlled release of the drug because of the burst drug release and the large standard deviations. Drug release is between 10 and 40% for other films. 30 M-PC8.2, 30 M-PC9.1, 30 M-PC9.8, and 50 M-PC9.1 showed a more controlled drug release than that of 30 M-PC5.8. This can be attributed to the fact that under acidic conditions, the acid-sensitive MOF structure is corroded by H+, causing the shell to lose its protective function and leading to the release of the drug from the core[56, 57].
As mentioned above, we designed a group of experiments to ensure the stability of MIL101(Fe) in all drug solutions at various pH. MIL-101(Fe) particles were added into two various leaching solutions at pH 5.8 and 9.1, and they were shaken at 100 rpm for 24 h at 5 °C in the dark. The samples were coded as MIL-101(Fe)5.8 and MIL-101(Fe)9.1, respectively. After the separation of the solid and liquid phases by filtration, solid particles were dried. SEM analyzed their structures, and the results were compared to the neat MIL-101(Fe) results. SEM images shown in Fig. 3 indicate the changes in the MIL-101(Fe) structure. The regular 3D shape of MIL-101(Fe) particles shown in Fig. 3a was partly lost, and a distorted structure was obtained for MIL-101(Fe)5.8. On the other hand, MIL-101(Fe)9.1 particles retained their original structure.
To support SEM outcomes, the iron ion content of the leaching solutions was also analyzed using inductively coupled plasma (ICP). The ICP analysis determined a relatively small amount of Fe (3.8 ppm) for the pH 9.1 solution. In contrast, a higher amount of Fe (55.91 ppm) was observed in the pH 5.8 medium. These results also confirm a lower MIL-101 (Fe) structure degradation in the pH 9.1 solution.
In brief, according to data obtained from SEM and ICP analysis, we concluded that the structure of MIL-101(Fe) particles is unstable in a pH 5.8 solution. Cui et al. investigated the catalytic activity of Prussian Blue-modified MIL-101(Fe) and compared their results with those of unmodified MIL-101(Fe) [58]. They reported losing the catalytic activity of unmodified MIL-101(Fe) in acidic conditions. This data also supports our results.
3.4 Rheological analysis
Understanding rheological properties is crucial as it reveals the material’s internal structure, optimizes processing conditions for wound dressings, and elucidates the relationship between components and their functions [59, 60]. In our study, rheological analysis of the hydrogels containing MIL-101(Fe) particles without PC (30 and 50 M), was performed in the frequency sweep test to investigate the contribution of MIL-101(Fe) particles to hydrogel structure. G′ represents the elastic portion (solid-like behavior), G″ represents the viscous portion (liquid-like behavior; deformation energy dissipated and subsequently lost by the sample during shear) of the viscoelastic behavior [26]. Damping factor (tan δ) and complex viscosity (η*) were calculated for each point. Then, the rheological data G′, G″, tan δ, and η* were plotted versus frequency (Fig. 7). The results were compared with those of neat pectin hydrogel without MIL-101(Fe) particles. The analysis employed the ANOVA method complemented by Tukey’s test to evaluate results.

Rheological analysis of the hydrogels; a modules G′ and G″, b damping factor, c complex viscosity, d photographs of the self-recovering process, and e the tensile of hydrogels on the surface of different postural fingers
The G′ and G″ curves demonstrated a consistent and elastic gel structure across all samples at varying frequencies, as reported in literature [20]. Tukey’s analysis revealed no significant difference between the G′ and tan δ values at 0.1–1000 rad/s for the hydrogels, highlighting the consistency of the hydrogel (p value > 0.05). Since the G′ for all samples is higher than their G″ (Fig. 7a) and there is no crossover between storage and loss moduli with G′ and G″ being slightly frequency-dependent, the gels are interconnected, confirming the predominance of elastic character over viscous character. This behavior is also observed in other polysaccharide hydrogels [61].
Tan δ is an essential indicator in understanding the stiffness and network architecture of hydrogels. The tan δ of the hydrogels are lower than 1 (Fig. 7b). For practical applications, if tan δ > 100, a material is called ideally viscous, and if tan δ < 0.01, a material is called ideally elastic. If tan δ is between these two values, a material is called viscoelastic. Tan δ of all hydrogels are between 0.12 and 0.58 which is almost 12–58 times higher than the lower limit 0.01 and almost 172–833 times lower than the upper limit 100. This indicates a predominant elastic rather than viscous behavior in the hydrogels, suggesting a typical gel-like behavior [16, 62, 63].
In our investigation, the pure pectin hydrogel displayed a notably low tan δ value. This observation can be explained by the absence of MIL-101(Fe) particles, which enter between the pectin chains and cause the chains to move away from each other, thus reducing the interaction between the chains. On the other hand, the 30 M sample exhibited a tan δ value almost 4.8 times higher than the 50 M sample (p value = 0.000). This significant difference implies a more flexible and less rigid structure in the 30 M sample, presumably due to a lower MIL-101(Fe) particle concentration. In the case of the 50 M sample, a higher concentration of MIL-101(Fe) particles results in greater spacing between the pectin chains. This increased spacing diminishes the internal heterogeneity of the matrix, which correlates with a lower damping factor indicative of increased stiffness. However no significant difference (p value = 0.943) was observed between tan δ of neat pectin hydrogel and hydrogel with 50 mg MOF (50 M). Although a high amount of MOF particles can lead to the decomposition of hydrogel crosslinking, the secondary bonds between the MOF particles and pectin chains help maintain the polymeric structure. In this way the stiffnes of the hydrogel was increased. Similarly, DSC and XRD analyses were revealed the increasing crystallinity with increasing MOF content to 50 mg. These dynamics are similarly reflected in the drug release results from the study.
The complex viscosity data were analyzed to assess the structure of the hydrogels. The results demonstrate a linear decrease in complex viscosity corresponding to frequency on a double logarithmic scale, marked by a significantly steep slope exceeding 0.94, indicating pronounced shear-thinning behavior in the formulations (Fig. 7c) [20, 59, 64].
The 3 M hydrogel gave the highest viscosity, while the other two hydrogels showed the same viscosity profile. The linear relationship between log \(\eta\) and log \(\omega\) in Fig. 7a suggests a power law, which was used to evaluate the data from the frequency sweep analysis for a numerical comparison between the samples, as shown in Eq. (6) [65].
Here, k* (Pa sn*) is the dynamic consistency index representing the fluid’s resistance to flow, \(\omega\) represents the shear rate (s-1), and n* is the dynamic power law factor. For n* = 1, the system is viscous and η* is constant, while for n* = 0, the system is elastic and η* decreases with increasing ω. The shear thinning region of the flow curves were fitted to Power law model with good correlation and R2 for each fitted equation was upper than 0.99. The model parameters for 30, 50 M and pectin hydrogels are listed in Table 2. The k* value of 30 M was 3 times higher than that of 50 M and pectin hydrogels, while almost the same k* value was calculated for the 50 M and pectin hydrogels. This result also explains why the 30 M hydrogel resists the drug molecules to come out of the matrix compared to the 50 M and therefore, shows less drug release behavior. The n* value was obtained at almost zero over the entire frequency range for all hydrogels, which shows elastic behavior. n value is reduced when the MIL-101 particles were added. Physically, this means that the shear sensitivity of the hydrogel was increased. It is, however noted that when the MIL-101 particles loading was increased to 50 M, most probably due to the clustering of the particles, the shear sensitivity of the hydrogel was increased [66].
The ability of the hydrogels to exhibit self-recovery behavior is due to their shear-thinning properties, which involve dual dynamic bonds such as hydrogen bonds and an adequate number of crosslinking bridges. These bonds allow the hydrogel to flow under shear stress and self-recover once the stress is removed [67]. The results indicate a self-recovery capacity of the hydrogels which is a benefit of the dynamic reversible hydrogen bonding and crosslinking in the structures as demonstrated by previous studies in the existing literature [68]. This phenomenon facilitates the alignment of polymer chains with the flow direction, enhancing the self-recovery and remolding capabilities of the structures.
To examine the self-recovery ability of the hydrogels when a crack occurs, macroscopic visual analysis was applied (Fig. 6d). For this test, two pieces of hydrogel stained with different colors were brought into contact without applying external force. The results of this visual in situ self-recovery test showed that the cut hydrogel was completely healed within 10 min, with the contact surface fusing as observed by the formation of a diffusion layer. A wound dressing with self-recovery capability can repair cracks, breakages, or mechanical damage once they occur, restoring the original performance or minimizing the loss of dressing properties [69, 70]. This property is crucial in preventing wound inflammation.
As shown in Fig. 7e, pectin-MOF hydrogels exhibit dynamic adaptability to finger movements, owing to their shear thinning properties [71, 72]. This feature enables them to endure diverse body movements and deformations at various angles in different rendering them for the wounds in different geometries [62, 73, 74].
3.5 Wound-dressing properties
To demonstrate the contribution of MIL-101(Fe) particles to the structural integrity, hydrophilicity, and in vitro wound-dressing properties of the hydrogel composites, the basic wound-dressing properties of the hydrogel—prepared by adding MIL-101(Fe) particles (without drug) to the pectin matrix (30 M)—were investigated according to the literature [75, 76]. All results were compared with those of a commercial product, KALTOSTAT® Calcium Sodium Alginate dressing. Also, a neat pectin hydrogel was prepared without MIL-101(Fe) particles and the drug as a comparison sample. The wound-dressing properties of these samples are presented in Table 3.
Comparing the commercial wound dressing and 30 M given dehydration rate and fluid handling capacity together, 30 M provided a more suitable medium than a commercial product because the ratio of dehydration rate to fluid handling capacity is almost 19 and 1.5 g min−1 for KALTOSTAT® and 30 M, respectively. A low dehydration rate to fluid handling capacity ratio indicates that water molecules will be trapped more into the hydrogel, so a more humid medium will occur, which is an advantage for wound healing. Neat pectin hydrogel also has a low ratio of dehydration rate to fluid handling capacity, but not as low as 30 M.
Unlike commercial dressings, the 30 M hydrogel was not dispersed in a salt solution (Table 3). This property is important for maintaining its integrity for painless wound-dressing removal.
The 30 M hydrogel provided a more acidic environment that could serve as a barrier to bacteria for the wound site compared to the commercial product and neat pectin hydrogel. Generally, pH decreases during wound healing [77].
All samples’ mass per unit area is almost the same order of magnitude. The thickness of the pectin-based samples was almost 16 times lower than that of the commercial dressing, which provided a more comfortable dressing material.
According to Shore A hardness and flexibility test results, 30 M hydrogel showed a profile similar to highly flexible rubbers/elastomers/softer plastics like neat pectin hydrogel. These results also support the comfortable usage of 30 M hydrogel.
The wetting properties of the pectin hydrogels prepared in two different amounts of the nanoparticle addition (30 and 50 M) were determined by measuring their contact angles by direct image analysis. After adding them into the pectin matrix, the contact angle of the pectin hydrogel was increased from 50° to 90–92° (Fig. 8). Hydrophilic pectin hydrogel became more hydrophobic material after adding MIL-101(Fe) particles. So, its surface free energy decreased. Generally, this property supports protein adsorption of a biomaterial, so faster wound healing is observed [78].

Contact angle of the hydrogels
3.6 In vitro cell viability assay analysis and wound-healing model
The films’ cytocompatibility was assessed using a WST-1 cell proliferation assay using human dermal fibroblast cells. Neat pectin hydrogel and MIL-101(Fe)-added hydrogel showed no cytotoxicity compared to the control group (Fig. 9a). In the cell viability analysis performed with the WST-1 test (fibroblast cells), the number of pectin film cells increased to 110. In contrast, the number of cells increased to 118 with the contribution of MIL-101Fe.

a After treatment of PCS-201-012 human dermal fibroblast cells with the hydrogels for 24 h, cell viability of the cells was determined by WST-1 assay and b wound-healing assay of the hydrogels
The effect of hydrogels on fibroblast wound-healing ability was also investigated. After 24 h, the widths of the recovered areas in the hydrogels were the same as in the control group, as shown in Fig. 9b. This shows that hydrogels were non-toxic and did not change fibroblast cell migration ability.
4 Conclusion
In this study, drug-loaded MIL-101Fe-pectin hydrogels were prepared as a wound dressing. PC was chosen as a model drug. After loading PC to MIL-101Fe particles at different pH, they were dispersed into the pectin matrix by an ionotropic gelation method. We found that to obtain a controlled drug release in our system, the pH range of the PC solution should be between 8.2 and 9.8 in the drug-loading step.
Incorporating MIL-101(Fe) particles into the pectin matrix provided the controlled release of PC, relieving pain during wound healing. As the surface free energy of the hydrogel decreases, it will support protein adsorption to the hydrogel surface, and thus, faster wound healing will be observed. According to rheological analysis, the hydrogels showed elastic behavior. They provided a moist and acidic environment that will support wound healing. Since their structural integrity is intact, they will be strong enough to protect the wound from external effects. We have also shown that pectin composites increase cell viability in wound healing and that fibroblast cells do not deleterious affect cell migration.
In summary, we successfully prepared new hydrogel composites by dispersing drug-loaded MIL-101(Fe) particles in the pectin matrix, and promising results were obtained to design a drug delivery system as a candidate for wound-dressing applications. After examining the effects of the wound dressing containing drug-loaded particles on cells through in vitro and in vivo studies, we believe that the newly developed material has potential for wound-healing applications.
Data availability
All the materials used in the manuscript are commercially available; however, Herbstreith & Fox Company (Germany) donated LM pectin. The data required for the results and conclusion are included in the manuscript; however, the corresponding author will provide raw data upon reasonable request.
References
W. Chen, Y. Zhu, Z. Zhang, Y. Gao, W. Liu, Q. Borjihan, Y. Qu, Y. Zhang, Y. Zhang, Y.J. Wang, L. Zhang, A. Dong, Chem. Eng. J. (2020). https://doi.org/10.1016/j.cej.2019.122238
M.A. Taemeh, A. Shiravandi, M.A. Korayem, H. Daemi, Carbohydr. Polym. (2020). https://doi.org/10.1016/j.carbpol.2019.115419
F. Zhang, F. Zhou, S. Qi, B. Shu, J. Wu, Front. Bioeng. Biotech (2019). https://doi.org/10.3389/fbioe.2019.00342
A. Bal-Öztürk, G. Torkay, N. İdil, B. Özkahraman, Z. Özbaş, Polym. Bull. (2024). https://doi.org/10.1007/s00289-023-04763-z.
D. Yilmaz-Aykut, G. Torkay, A. Kasgoz, S.R. Shin, A. Bal-Ozturk, H. Deligoz, J. Biomed. Mater. Res. B Appl. Biomater. (2023). https://doi.org/10.1002/jbm.b.35295
G. Yaşayan, G. Karaca, Z.P. Akgüner, A. Bal Öztürk, Int. J. Polym. Mater. (2021). https://doi.org/10.1080/00914037.2020.1740993.
Ö. Z. Güner Yılmaz et al., ACS Omega (2024). https://doi.org/10.1021/acsomega.3c03619
H. Liu, C. Wang, Y. Qin, Z. Wang, F. Yang, Z. Li, J. Wang, RSC Adv. 8, 7533 (2018). https://doi.org/10.1039/C7RA13510F
K. Yadav et al., Med. Drug Discov. (2024). https://doi.org/10.1016/j.medidd.2024.100183
M. Li et al., ACS Appl. Mater. Interfaces (2017). https://doi.org/10.1021/acsami.7b04428
T. Inan, D. Dalgakiran, O. Kurkcuoglu, F.S. Güner, J. Polym. Res. (2021). https://doi.org/10.1007/s10965-021-02740-6
B. Eroglu, D. Dalgakiran, T. Inan, O. Kurkcuoglu, F.S. Güner, J. Polym. Res. (2018). https://doi.org/10.1007/s10965-018-1647-7
J. Long, A.E. Etxeberria, A.V. Nand, C.R. Bunt, S. Ray, A. Seyfoddin, Mater. Sci. Eng. C (2019). https://doi.org/10.1016/j.msec.2019.109873
O.Z. Guner, C. Cam, B. Arabacioglu Kocaaga, S. Batirel, F.S. Guner, J. Appl. Poly. Sci. (2018). https://doi.org/10.1002/app.46731
B. Kocaaga, O. Kurkcuoglu, M. Tatlier, S. Batirel, F.S. Guner, J. Appl. Polym. Sci. (2019). https://doi.org/10.1002/app.47640
B. Kocaaga, F. S. Guner, O. Kurkcuoglu, Mater. Today Commun. (2022). https://doi.org/10.1016/j.mtcomm.2022.103268.
W. Zhang, P. Xu, H. Zhang, Trends Food. Sci. Technol. (2015). https://doi.org/10.1016/j.tifs.2015.04.001
S. Bashir, M. Hina, J. Iqbal, A.H. Rajpar, M.A. Mujtaba, N.A. Alghamdi, S. Wageh, K. Ramesh, S. Ramesh, Polymers 12(11), 2702 (2020). https://doi.org/10.3390/polym12112702
E. Sarioglu, B. Arabacioglu Kocaaga, D. Turan, S. Batirel, F.S. Guner, J. Appl. Polym. Sci. (2019). https://doi.org/10.1002/app.48155
K. Song et al., Carbohydr. Polym. (2023). https://doi.org/10.1016/j.carbpol.2022.120272
C.T. Hsieh, K. Ariga, L.K. Shrestha, S.H. Hsu, J. Biol. Macromol. (2021). https://doi.org/10.1021/acs.biomac.0c00920
K.M.L. Taylor-Pashow, J.D. Rocca, Z. Xie, S. Tran, W. Lin, J. Am. Chem. Soc. (2009). https://doi.org/10.1021/ja906198y
G. Kumar, A. Kant, M. Kumar, D.T. Masram, Inorgan. Chim. Acta (2019). https://doi.org/10.1016/j.ica.2019.119036
B.A. Lakshmi, S. Kim, Mater. Sci. Eng. C (2019). https://doi.org/10.1016/j.msec.2019.110091
P. Horcajada, T. Chalati, C. Serre, B. Gillet, C. Sebrie, T. Baati, J.F. Eubank, D. Heurtaux, P. Clayette, C. Kreuz, J. Chang, Y. Hwang, V. Marsaud, P. Bories, L. Cynober, S. Gil, G. Ferey, P. Couvreur, R. Gref, Nat. Mater. (2010). https://doi.org/10.1038/nmat2608
M.A. Haydar, H.R. Abid, B. Sunderland, S. Wang, Drug. Des. Devel. Ther. (2017). https://doi.org/10.2147/DDDT.S145716
S. Keskin, S. Kizilel, Ind. Eng. Chem. Res. (2011). https://doi.org/10.1021/ie101312k
J. Gordon, H. Kazemian, S. Rohani, Mater Sci. Eng. C. Mater. Biol. Appl. (2015). https://doi.org/10.1016/j.msec.2014.11.046
X.G. Wang, Z. Dong, H. Cheng, S. Wan, W. Chen, M. Zou, J. Huo, H. Denga, X. Zhang, Nanoscale (2015). https://doi.org/10.1039/C5NR04045K
S.D. Taherzadeh, J. Soleimannejad, A. Tarlani, J. Nanomater. (2017). https://doi.org/10.3390/nano10122296
G. Wyszogrodzka, P. Dorozynski, B. Gil, W.J. Roth, M. Strzempek, B. Marszałek, W.P. Węglarz, E. Menaszek, W. Strzempek, P. Kulinowski, Pharm. Res. (2018). https://doi.org/10.3390/pharmaceutics11120687
J.M. Almasi, V. Zelenak, P. Palatai, E. Benova, A. Zelenakova, Inorg. Chem. Commun. (2018). https://doi.org/10.1039/C7CC09730A
A.C. McKinlay, B. Xiao, D.S. Wragg, P.S. Wheatley, I.L. Megson, R.E. Morris, J. Am. Chem. Soc. (2008). https://doi.org/10.1021/ja801997r
M.R. Ramezani, Z. Ansari-Asl, E. Hoveizi, A.R. Kiasat, Mater. Chem. Phys. (2019). https://doi.org/10.1007/s12221-020-9523-6
S. Shakya, Y. He, X. Ren, T. Guo, A. Maharjan, T. Luo, T. Wang, R. Dhakwa, B. Regmi, H. Li, R. Gref, J. Zhang, Small 15, 1901065 (2019)
S.H. Shen, Y.S. Wu, Y.C. Liu, D.C. Wu, Int. J. Nanomed. (2017). https://doi.org/10.2147/IJN.S132780
P. Sriamorsak, J. Nunthanid, J. Microencapsul. 16, 303 (1999)
M. Uzun, S.C. Anand, T. Shah, J. Biomed. Eng. Technol. (2013). https://doi.org/10.12691/jbet-1-1-1
BS EN 12127:1998, Textiles Fabrics Determination of Mass per Unit Area using Small Samples (British Standard Institute, 1998). ISBN 0 580 29132 4
BS EN ISO 9073-2:1997, Textiles, Test Methods for Nonwovens, Part 2: Determination of Thickness (British Standard Institute, 1997). ISBN 0 580 26632 X
D2240-97, Standard Test Method for Rubber Property—Durometer Hardness (American Society for Testing and Materials)
D522-93a, Standard Test Methods for Mandrel Bend Test of Attached Organic Coatings (American Society for Testing and Materials)
BS EN 13726-1:2002, Test Methods for Primary Wound Dressings. Aspect of absorbency (British Standard Institute, BSI, 2002). ISBN 0 580 39510 3
BS EN 13726-2:2001, Test Methods for Primary Wound Dressings. Part 1: Aspect of Absorbency, Section 3.6, Dispersion Characteristics (British Standard Institute, 2001)
A. Akcal, S. Karsidag, K. Yildiz, N. Yesiloglu, M.A.A. Akcal, F. Kabukcuoglu, Arch. Clin. Exp. Surg. 4, 41–45 (2015). https://doi.org/10.5455/aces.20140606054447
N.V. Maksimchuk, K.A. Kovalenko, V.P. Fedin, O.A. Kholdeeva, Chem. Comm. (2012). https://doi.org/10.1039/C2CC31877F
Q. Xie, Y. Li, Z. Lv, H. Zhou, X. Yang, J. Chen, H. Guo, Sci. Rep. (2017). https://doi.org/10.1038/s41598-017-03526-x
M.M. Mostafavi, F. Movahedi, Appl. Organomet. Chem. (2018). https://doi.org/10.1002/aoc.4529
A. Rampino, M. Borgogna, B. Bellich, P. Blasi, F. Virgilio, A. Cesàro, Eur. J. Pharm. Sci. 84, 37–45 (2016). https://doi.org/10.1016/j.ejps.2016.01.004
A. Fulias, I. Ledet, G. Vlase, C. Popoiu, A. Heghes, M. Bilanin, T. Vlase, D. Gheorgheosu, M. Craina, S. Ardelean, D. Ferechide, O. Margineanand, L. Mos, Chem. Cent. J. 7, 1 (2013)
M. Cai et al., RSC Adv. (2020). https://doi.org/10.1039/d0ra06106a
S. Saghazadeh et al., Adv. Drug Deliv. Rev. (2018). https://doi.org/10.1016/j.addr.2018.04.008
Z. Özbaş, G. Torkay, A. Bal-Öztürk, B. Özkahraman, Chem. Pap. (2022). https://doi.org/10.1007/s11696-022-02426-3
T. Nalini, S. Khaleel Basha, A. Mohamed Sadiq, V. Sugantha Kumari, Polym. Bull. (2023). https://doi.org/10.1007/s00289-022-04094-5
A. Benny, S.D. Kalathiparambil Rajendra Pai, D. Pinheiro, S.J. Chundattu, Results Chem. (2023). https://doi.org/10.1016/j.rechem.2024.101414R
K. Alavijeh, K. Akhbari, Inorg. Chem. (2020). https://doi.org/10.1021/acs.inorgchem.9b02756
M. Keramatinia, B. Ramezanzadeh, M. Mahdavian, Surf. Coat. Technol. (2024). https://doi.org/10.1016/j.surfcoat.2024.130578
F. Cui, Q. Deng, L. Sun, RSC Adv. (2015). https://doi.org/10.1039/C5RA18589K
A. Basu, J. Lindh, E. Alander, M. Strømme, N. Ferraz, Carbohydr. Polym. (2017). https://doi.org/10.1016/j.carbpol.2017.06.073
P. Dorishetty, R. Balu, S.S. Athukoralalage, T.L. Greaves, J. Mata, L. Campo, N. Saha, A.C.W. Zannettino, N.K. Dutta, N.R. Choudhury, ACS Sustain. Chem. Eng. (2020). https://doi.org/10.1021/acssuschemeng.9b05317
M. Cofelice, M.C. Messia, E. Marconi, F. Cuomo, F. Lopez, Food Hydrocoll. (2023). https://doi.org/10.1016/j.foodhyd.2023.108768
B. Kocaaga, O. Kurkcuoglu, M. Tatlier, G. Dinler-Doganay, S. Batirel, F.S. Guner, Polym. J. https://doi.org/10.3390/polym14030460
Z. Güner, B. Kocaaga, S. Batirel, O. Kurkcuoglu, F.S. Güner, Int. J. Polym. Mater. (2020). https://doi.org/10.1080/00914037.2020.1760272
P. Alaee, M. Kamkar, M. Arjmand, Langmuir (2022). https://doi.org/10.1021/acs.langmuir.2c00591
P. Suurs, H. Brand, R. Have, W.F. Daamen, Food Hydrocoll. (2023). https://doi.org/10.1016/j.foodhyd.2023.108595
X.L. Xie et al., Polym. J. (2004). https://doi.org/10.1016/j.polymer.2004.07.045
G.U. Ruiz-Esparza, X. Wang, X. Zhang et al., Nano-Micro Lett. (2021). https://doi.org/10.1007/s40820-021-00712-5
M.U. Özkaynak et al., J. Mol. Liq. (2024). https://doi.org/10.1016/j.molliq.2023.123705
A. Zhang, Y. Liu, D. Qin, M. Sun, T. Wang, X. ChenInt, J. Biol. Macromol. (2020). https://doi.org/10.1016/j.ijbiomac.2020.08.109
A. Kumar et al., Mater Today Sustain. (2023). https://doi.org/10.1016/j.mtsust.2023.100431
Y. Liang, Z. Li, Y. Huang, R. Yu, B. Guo (2021). https://doi.org/10.1021/acsnano.1c00204
K. Sahajpal, S. Shekhar, A. Kumar, B. Sharma, J. Mater. Chem. B. (2022). https://doi.org/10.1039/d2tb00077f
A.I. Cernencu, A.I. Dinu, I.C. Stancu, A. Lungu, H. Iovu, Biotechnol. Bioeng. (2022). https://doi.org/10.1002/bit.28020
H. Qin, T. Zhang, N. Li, H.P. Cong, S.H. Yu, Nat. Commun. (2019). https://doi.org/10.1038/s41467-019-10243-8
P. Zahedi, I. Rezaeian, S.O. Ranaei-Siadat, S.H. Jafari, P. Supaphol, Polym. Adv. Technol. (2010). https://doi.org/10.1002/pat.1625
N. Mao, S. Russell, J. Text. Prog. (2004). https://doi.org/10.1533/jotp.2005.36.4.1
L.A. Schneider, A. Korber, S. Grabbe, J. Dissemond, Arch. Dermatol. Res. (2007). https://doi.org/10.1007/s00403-006-0713-x
K.C. Dee, D.A. Puleo, R. Bizios, An Introduction to Tissue-Biomaterial Interactions (Wiley-Liss, 2002), p. 161. ISBN: 978-0-471-25394-5
Funding
Open access funding provided by the Scientific and Technological Research Council of Türkiye (TÜBİTAK). This work was funded by The Scientific and Technological Research Council of Turkey (TUBITAK) with Project Number 115M439.
Author information
Authors and Affiliations
Contributions
Dr. BK, Dr. AS, and Dr. FSG conceptualized and designed the study. GB, IAA, and MAM performed the experiments. Dr. SB performed in vitro cell viability and wound healing studies. All the authors have approved the manuscript for submission and publication.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Ethical approval and consent to participate
Not applicable.
Consent for publication
All authors agreed with the content and consented to submit the manuscript.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Kocaaga, B., Bagimsiz, G., Alev, I.A. et al. Fabrication of MIL-101(Fe)-embedded biopolymeric films and their biomedical applications. Macromol. Res. (2024). https://doi.org/10.1007/s13233-024-00305-2
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s13233-024-00305-2